
Abstract
Background
Coronary artery disease (CAD) is the leading cause of mortality worldwide. We aimed to screen out potential gene signatures and construct a diagnostic model for CAD.
Method
We downloaded two mRNA profiles, GSE66360 and GSE60993, and performed analyses of differential expression, gene ontology terms, and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. The STRING database was used to identify protein–protein interactions (PPI). PPI network visualization and screening out of key genes were performed using Cytoscape software. Finally, a diagnostic model was constructed.
Results
A total of 2127 differentially expressed genes (DEGs) were identified in GSE66360, and 527 DEGs in GSE60993. Of the 153 DEGs from both datasets that showed differential expression between CAD patients and controls, 471 biological process terms, 35 cellular component terms, 17 molecular function terms, and 49 KEGG pathways were significantly enriched. The top 20 key genes in the PPI network were identified, and a diagnostic model constructed from five optimal genes that could efficiently separate CAD patients from controls.
Conclusion
We identified several potential biomarkers for CAD and built a logistic regression model that will provide a valuable reference for future clinical diagnoses and guide therapeutic strategies.
Keywords
Introduction
Cardiovascular disease (CVD) is the leading cause of death worldwide, and coronary artery disease (CAD) is considered one of the most serious CVDs.1,2 CAD is the predominant cause of ischemic heart disease, which often leads to myocardial infarction and death. 3 Coronary heart disease is always caused by reduced blood flow in the heart muscle and the accumulation of fat and plaques in the heart’s arteries. 4
Globally, the total number of deaths from CAD increased by 4.1% to 55.8 million between 2005 and 2015, although age-standardized mortality rates fell by 17.0% through prevention and treatment strategies established from growing knowledge of basic CAD pathophysiology. 5 Treatment strategies for patients with complex CAD often include percutaneous coronary intervention or coronary artery bypass grafting. 6 However, mortality and rehospitalization rates for CAD patients remain high, 7 so the identification and development of specific gene signatures in the early diagnosis of CAD is crucial to improve treatment.
A number of noninvasive diagnostic approaches have been applied to the early diagnosis of CAD, of which exercise electrocardiography is the most studied and least accurate for female patients. However, electrocardiography combined with imaging techniques such as echocardiography or nuclear single photon emission computed tomography can improve the accuracy of diagnosis. 8 Additionally, a series of key genes has been identified to aid CAD diagnosis through the analysis of expression profiles. For instance, circulating miR-765 and miR-149 were identified as potential noninvasive diagnostic biomarkers for geriatric CAD patients. 9 Zhang et al. 10 showed that several microRNAs and long non-coding RNAs influence the progression of CAD by regulating the function of vascular endothelial cells, smooth muscle cells, and macrophages, as well as vascular inflammation and metabolism. These studies suggested that noninvasive diagnostic approaches have important diagnostic value in CAD.
In this study, we downloaded the mRNA profiles of blood samples from CAD patients and healthy controls, then analyzed differential gene expression between the two groups to explore the diagnostic signatures of potential key genes. A protein–protein interaction (PPI) network was constructed based on 153 differentially expressed genes (DEGs) that showed significant expression differences. Twenty key genes were screened out, and five genes closely associated with CAD progression were further optimized and used to build a logistic regression model with good diagnostic value for CAD.
Materials and methods
Data collection
mRNA profiles GSE66360 11 and GSE60993 12 were downloaded from the Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/). GSE66360 included blood samples from 49 CAD patients and 50 healthy controls, and mRNA profiles were detected using the Affymetrix Human Genome U133 Plus 2.0 Array (Thermo Fisher Scientific, Santa Clara, CA, USA). GSE60993 included blood samples from 26 CAD patients and seven healthy controls, and mRNA profiles were examined by a HumanWG-6 v3.0 gene expression beadchip (Illumina, San Diego, CA, USA).
Differential expression analysis
The probes of mRNA profiles were stripped when expression values were 0 in more than 50% of the samples, and remaining data were standardized using the robust multi-array method. Differential expression analysis was performed based on the limma function package of R language, 13 with |log 2 (fold change [FC])| > 1 and false discovery rate (FDR) ≤ 0.05 as significant thresholds to screen differentially expressed probes.
Functional enrichment analysis
Gene ontology (GO) analysis (including biological process [BP], molecular function [MF], and cellular component [CC]) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis were carried out based on the clusterProfiler function package of R language. 14 P < 0.05 was used as the threshold to screen significantly enriched GO terms and KEGG pathways.
Protein–protein interaction (PPI) networks
The STRING database (https://string-db.org/version 11.0) analyzes and predicts functional connections and interactions of proteins. 15 PPI interactions with a confidence score of ≥ 0.4 were retained after identification by the STRING database. The PPI network was visualized using Cytoscape software (https://cytoscape.org/, version 3.7.2), 16 and key genes of the PPI network were screened by the cytoHubba plug-in of Cytoscape software based on the Maximal Clique Centrality (MCC) algorithm.
Construction of the logistic regression model
The glm function in R language 17 was used to construct a multivariate logistic regression model with the expression value of target genes as the continuous prediction variable and the sample type as the categorical response value (disease or not). Variables were then further screened using the stepwise regression method, and used to reconstruct the model. The model calculated the P value of each variable, and variables with P < 0.05 were used to construct the final model. Generally, points with a COOK distance > 0.5 affect the accuracy of the model. The area under the curve (AUC) value represents the quality of the model, with a larger AUC value representing a better model.
Results
Identification of DEGs
The mRNA profiles from GSE66360 and GSE60993 databases were standardized, and no obvious change was seen in the data deviation of each sample (Fig. S1). A total of 2127 DEGs (925 upregulated and 1202 downregulated) were identified in CAD patients compared with controls from the GSE66360 dataset (Figure 1a). DEG expression differed between patients and controls (Figure 1b). A total of 527 DEGs (333 upregulated and 194 downregulated) were identified in CAD patients compared with controls from the GSE60993 dataset (Figure 1c). DEG expression again differed between patients and controls (Figure 1d). Moreover, 153 DEGs from the two datasets simultaneously exhibited notable differences between CAD patients and controls (Figure 1e).

Identification of DEGs. (a) Volcano plot of DEGs between CAD patients and healthy controls from the GSE66360 dataset. The horizontal axis shows Log2(FC) and the vertical axis shows –Log10(FDR). Red points represent up-regulated genes, blue points represent down-regulated genes, and black points indicate no significant difference. (b) Heatmap of DEGs between CAD patients and healthy controls from the GSE66360 dataset. The horizontal axis shows genes and the vertical axis shows samples. Red indicates high expression and blue indicates low expression. (c) Volcano plot of DEGs between CAD patients and healthy controls from the GSE60993 data set. The horizontal axis shows Log2 FC and the vertical axis shows –Log10 (FDR). Red points represent up-regulated genes and blue points represent down-regulated genes. (d) Heatmap of DEGs between CAD patients and healthy controls from the GSE60993 dataset. The horizontal axis shows genes and the vertical axis shows samples. Red indicates high expression and blue indicates low expression. (e) Venn diagram of DEGs.
Functional and pathway enrichment analysis
GO and KEGG pathway analyses were performed on the 153 DEGs from the two datasets showing significant differences between CAD patients and controls. We identified 471 significantly enriched BP terms including neutrophil-mediated immunity, T cell activation, and leukocyte differentiation and migration (P < 0.05); 35 significantly enriched CC terms including specific granule, tertiary granule, and secretory granule (P < 0.05); and 17 significantly enriched MF terms including protein heterodimerization activity, carbohydrate binding, and signaling pattern recognition receptor activity. We also detected 49 significantly enriched KEGG pathways including tuberculosis, hematopoietic cell lineage, phagosome, and human immunodeficiency virus 1 infection. The full list of significantly enriched GO terms and KEGG pathways is shown in Table S1. The top 15 most significantly enriched BP terms are shown in Figure 2a, the top 15 most significantly enriched CC terms in Figure 2b, the top 15 most significantly enriched MF terms in Figure 2c, and the top 15 most significantly enriched KEGG pathways in Figure 2d.
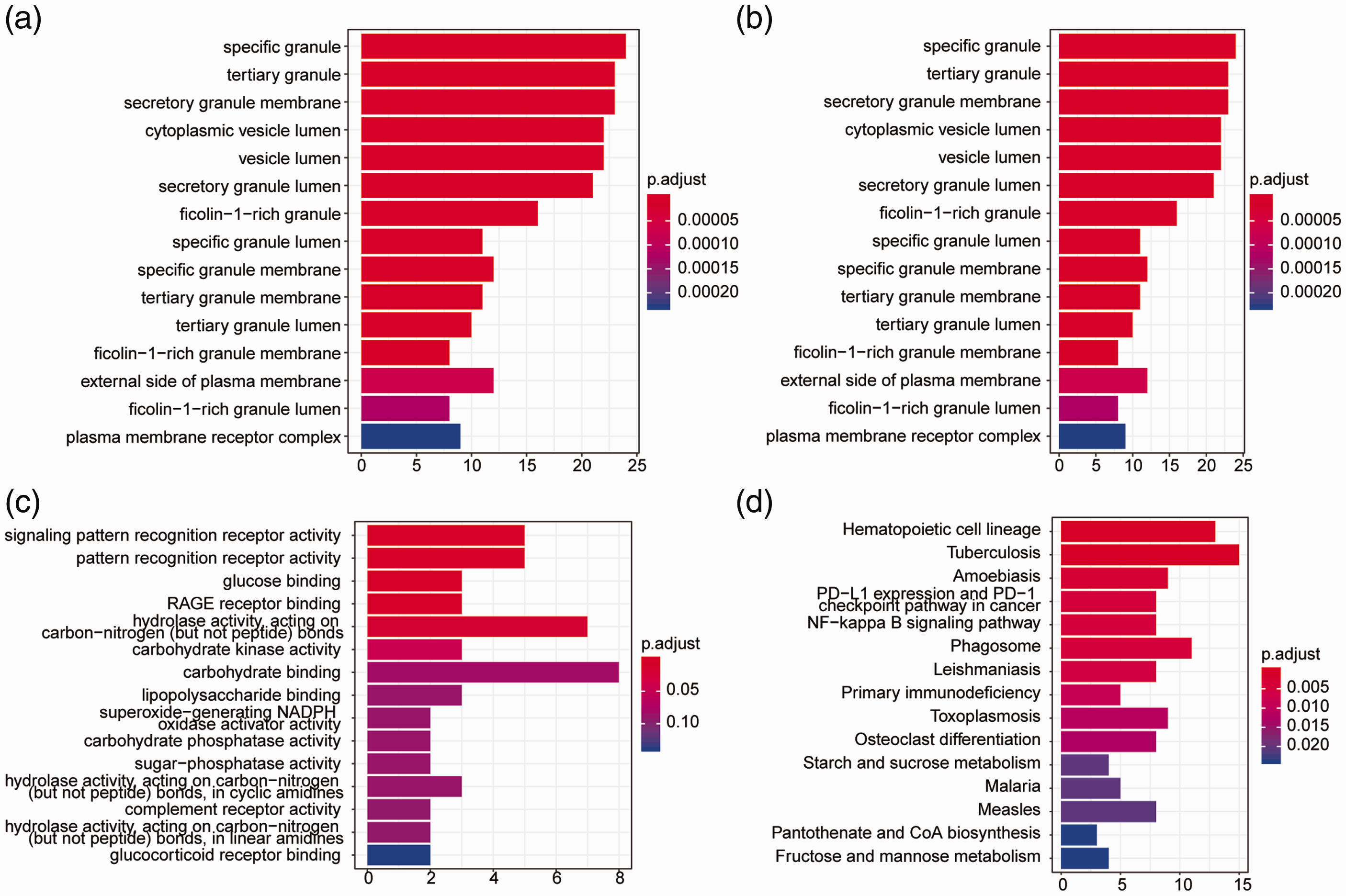
Enrichment of GO terms and KEGG pathways based on 153 DEGs. (a) The 15 most significantly enriched BP terms. (b) The 15 most significantly enriched CC terms. (c) The 15 most significantly enriched MF terms. (d) The 15 most significantly enriched KEGG pathways. The horizontal axis shows the number of enriched genes and the vertical axis shows the corresponding biological process or KEGG pathway. Longer bars represent more enriched genes. Colors represent the P value.
Key genes in PPI networks
Functional connections and interactions of proteins were predicted by the STRING database, and interactions between protein pairs with a confidence score of ≥ 0.4 were selected. The PPI network was visualized using Cytoscape software (Figure 3a) and shown to contain 125 nodes, with a maximum node degree of 47 and minimum node degree of 1. The integrin alpha M gene had the maximum node degree. CytoHubba plug-in was used to screen key genes in the PPI network according to the MCC algorithm, and the top 20 key genes by score are shown in Table S2. The subnetwork composed of these 20 genes is shown in Figure 3b.

Construction of the PPI network. (a) PPI network of shared genes. Each dot represents a node. importance of the gene in the network is reflected by the degree of the node which is represented by the number of lines connected to the dot (node). A thicker line reflects a stronger interaction between two nodes. Red points represent up-regulated genes and blue points represent down-regulated genes. (b) The network of key genes selected using the Maximal Clique Centrality algorithm. Important genes are shown in darker colors.
The logistic regression diagnostic model
We combined the expression value of 20 key genes from GSE66360 and GSE60993 datasets, and removed the batch effect using the sva package 18 of R language. We randomly selected two-thirds of the samples as the training set, and the remaining samples as the testing set (Table S3). Logistic regression model 1 was constructed with the expression value of all 20 key genes as the continuous type prediction variable and the sample type (disease or not) as the categorical response variable. Eight genes including CLEC4D, CLEC5A, HP, LCN2, MMP9, SLC2A3, SLPI, and TIMP2 were screened out from the 20 predicted to be closely related to CAD progression.
Logistic regression model 2 was then constructed with these eight genes as a variable; the detailed parameters of this model are shown in Table 1. An odds ratio (OR) value > 1 indicated that genes with high expression promoted the occurrence of CAD, whereas an OR value < 1 indicated that genes with high expression inhibited the occurrence of CAD. P values of CLEC4D, HP, LCN2, MMP9, and TIMP2 were less than 0.05, suggesting that these genes contributed markedly to the model. Finally, logistic regression model 3 was constructed from these five genes. Sample GSM1620895 (with a COOK distance > 0.5) was found to have little impact on the accuracy of the model (Figure 4a). The accuracy of the model was evaluated by the receiver operating characteristic (ROC) curve; AUC values in the training set and testing set were 0.9295 and 0.8674, respectively (Figure 4b), suggesting that the model constructed by these five genes had a good diagnostic value in CAD.
Model interpretation of the logistic regression model 2.

SE, standard error; OR, odds ratio; CI, confidence interval.

Construction of the logistic regression diagnostic model. (a) The logistic regression diagnostic model. Red dashed line indicates the COOK distance. (b) The ROC curve. The horizontal axis represents the false positive rate and the vertical axis represents the true positive rate.
Discussion
While CVD has traditionally been considered a disease of western society, its global incidence is on the rise and it is currently more prevalent in low and middle income countries in Asia and Africa. 19 CAD is a multifactorial inherited disorder associated with at least three major risk factors: hypertension, diabetes mellitus, and obesity. 20 Although gene expression analysis has had a great impact on the identification and development of biomarkers in the cardiovascular field, current risk prediction models only provide a rough estimation of individual risk. 21 Therefore, the identification and development of new biomarkers for CVD risk prediction is still an urgent need.
In recent decades, several genes have been identified or predicted to closely participate in the occurrence and development of CAD. For instance, the intercellular adhesion molecule 1 gene polymorphism rs5498 was correlated with a decreased risk of myocardial infarction and may reduce the risk of CAD. 22 Additionally, gene knockout of transforming growth factor-β, its receptors, and downstream signaling proteins demonstrated the importance of this pleiotropic cytokine during vasculogenesis and the maintenance of vascular homeostasis. 23 A meta-analysis suggested that receptor for advanced glycation end products gene polymorphism Gly82Ser was associated with an increased risk of CAD, especially in Chinese populations, 24 while another meta-analysis indicated that the T allele of the 5,10-methylenetetrahydrofolate reductase gene rs1801133 polymorphism was a risk factor for CAD and that this was partly mediated by abnormal lipid levels. 25 Moreover, Hou et al. 26 reported that the interleukin-6 gene –174G/C polymorphism C allele was associated with increased CAD risk in Caucasians. Nevertheless, early diagnosis and treatment of CAD is still difficult, so the use of informatics approaches may help prioritize molecules that are biologically and functionally related to CAD, which will aid diagnosis and treatment.
Herein, a network based on the PPI was constructed with 153 DEGs that showed significant expression differences between CAD patients and controls in the two GEO datasets. Twenty key genes were selected (Table S2), of which five optimal genes (CLEC4D, HP, LCN2, MMP9, and TIMP2) were predicted to be closely related to the development of CAD. Although the role of CLEC4D in CAD remains unclear, necrotic cell sensor CLEC4E was identified to promote a proatherogenic macrophage phenotype through activation of the unfolded protein response. 27 Graves et al. 28 reported that HP was not only an important antioxidant in vascular inflammation and atherosclerosis, but also an enhancer of inflammation in cardiac transplants. LCN2 plays a pivotal role in processes involved in atherogenesis by promoting the polarization and migration of monocytic cells and the development of macrophages towards foam cells. 29 Wang et al. 30 found that the expression of MMP9 was significantly upregulated in CAD samples compared with controls, and participated in the progression of CAD. These reports confirm that the biomarkers we predicted are functionally similar to known CAD risk factors, providing a theoretical basis for the subsequent logistic regression diagnostic model. The accuracy of this model was evaluated by the ROC curve, identifying AUC values of 0.9295 in the training set and 0.8674 in the testing set.
Conclusion
Our logistic regression model was shown to have a good diagnostic value in CAD. Our study therefore provides new insights into the discovery of diagnostic biomarkers in CAD which could aid early clinical diagnosis and guide therapeutic strategies. Our results should be verified using a larger sample size.
Supplemental Material
sj-pdf-1-imr-10.1177_0300060520979856 - Supplemental material for Bioinformatics analysis identifies potential diagnostic signatures for coronary artery disease
Supplemental material, sj-pdf-1-imr-10.1177_0300060520979856 for Bioinformatics analysis identifies potential diagnostic signatures for coronary artery disease by Dong Zhang, Liying Guan and Xiaoming Li in Journal of International Medical Research
Supplemental Material
sj-xlsx-2-imr-10.1177_0300060520979856 - Supplemental material for Bioinformatics analysis identifies potential diagnostic signatures for coronary artery disease
Supplemental material, sj-xlsx-2-imr-10.1177_0300060520979856 for Bioinformatics analysis identifies potential diagnostic signatures for coronary artery disease by Dong Zhang, Liying Guan and Xiaoming Li in Journal of International Medical Research
Supplemental Material
sj-xlsx-3-imr-10.1177_0300060520979856 - Supplemental material for Bioinformatics analysis identifies potential diagnostic signatures for coronary artery disease
Supplemental material, sj-xlsx-3-imr-10.1177_0300060520979856 for Bioinformatics analysis identifies potential diagnostic signatures for coronary artery disease by Dong Zhang, Liying Guan and Xiaoming Li in Journal of International Medical Research
Supplemental Material
sj-xlsx-4-imr-10.1177_0300060520979856 - Supplemental material for Bioinformatics analysis identifies potential diagnostic signatures for coronary artery disease
Supplemental material, sj-xlsx-4-imr-10.1177_0300060520979856 for Bioinformatics analysis identifies potential diagnostic signatures for coronary artery disease by Dong Zhang, Liying Guan and Xiaoming Li in Journal of International Medical Research
Footnotes
Data availability
Declaration of conflicting interest
The authors declare that there is no conflict of interest.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Supplemental Material
Supplementary material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.