
Abstract
Objective
This study aimed to systematically elucidate the role of gut microbial metabolites in the development and progression of ovarian cancer.
Methods
Public databases, including GutMgene, were used to screen and integrate gut microbial metabolite–target genes with ovarian cancer–related genes, ultimately identifying 59 key intersection genes. A gut microbiota–metabolite–gene regulatory network was constructed, and Gene Ontology and Kyoto Encyclopedia of Genes and Genomes enrichment analyses, protein–protein interaction network analysis, and evaluations of the drug-likeness and toxicity of key metabolites were performed.
Results
A total of 72 key genes associated with immune regulation were identified. Enrichment analyses demonstrated that these genes were significantly involved in immune-related processes, including T cell activation and the Toll-like receptor signaling pathway. Protein–protein interaction network analysis identified five core genes: STAT3, IL6, TNF, AKT1, and TP53. Drug-likeness analysis suggested that metabolites such as butyric acid and indole-3-propionic acid exhibit potential drug-like properties.
Conclusion
Gut microbiota–derived metabolites may influence ovarian cancer progression and the immune microenvironment by regulating core genes such as TP53 and AKT1 and pathways including Toll-like receptor signaling. These findings provide a potential basis for microbiota-targeted interventions in ovarian cancer.
Introduction
Ovarian cancer is a common malignant tumor of the female reproductive system. Although its incidence is lower than that of cervical cancer and endometrial cancer, its mortality rate ranks first among gynecological malignancies. Owing to atypical early symptoms and the lack of effective screening methods, most patients are diagnosed at an advanced stage, resulting in poor treatment efficacy and a high recurrence rate, which seriously threaten women’s life and health. Currently, the standard treatment for ovarian cancer includes cytoreductive surgery combined with platinum- and paclitaxel-based chemotherapy. However, drug resistance and recurrence remain major clinical challenges. Therefore, in-depth exploration of the molecular mechanisms underlying ovarian cancer initiation and progression as well as the identification of new biomarkers and therapeutic targets are of great clinical significance.1,2 In recent years, with advances in microbiome research, increasing attention has been paid to the role of intestinal flora in tumor occurrence and development. Intestinal flora participates in host metabolism, immune regulation, and barrier maintenance, and its metabolites can remotely regulate host physiological and pathological processes through the gut–organ axis.3,4 Studies have demonstrated that intestinal flora imbalance is closely associated with the development of various tumors, such as colorectal cancer, liver cancer, and breast cancer. However, the mechanisms underlying its role in ovarian cancer remain unclear. Gut microbial metabolites, such as short-chain fatty acids (SCFAs), bile acids, and polyamines, can reach distal organs through the bloodstream and influence gene expression, signaling pathways, and the tumor microenvironment, thereby contributing to tumor initiation and progression. Advances in bioinformatics have provided powerful tools for systematically analyzing the complex relationships among microbiota, metabolites, genes, and diseases.5–7 By integrating multi-omics data with public databases, multilevel molecular regulatory networks can be constructed, key genes and pathways can be identified, and underlying molecular mechanisms can be elucidated. At present, bioinformatics-based studies have explored the associations between gut microbiota and tumors, but most of them have focused on gastrointestinal malignancies. Research on ovarian cancer remains relatively limited, particularly with respect to the systematic screening of target genes and the construction of regulatory networks at the metabolite level. From a biological perspective, gut microbial metabolites may act on ovarian tissue through blood circulation, immune regulation, and endocrine pathways, participating in processes such as local immune microenvironment remodeling, cell proliferation, and apoptosis regulation, thereby influencing ovarian cancer development. Compared with previous studies that mainly focused on the role of local flora in gastrointestinal tumors or endometriosis, this study systematically reveals the long-range regulatory network of gut-derived metabolites in ovarian cancer using multi-omics integration and network pharmacology approaches. It highlights the potential significance of the gut–ovarian axis in the pathophysiology of ovarian cancer and provides new perspectives and supporting data for mechanistic studies and therapeutic target discovery.
Methods
Gut microbiome data acquisition and metabolite target gene prediction and integration
First, gut microbial data were obtained from the GutMgene database (accessed in March 2025; version v2.0), including information on gut microbial metabolites, gut microbial genes, and gut microbial metabolite–related genes. The simplified molecular input line entry system (SMILES) structures of gut microbial metabolites were retrieved. Next, target genes for each metabolite were predicted using the SwissTargetPrediction (STP) database (http://www.swisstargetprediction.ch/, version 2024.04) and merged. The filtering criterion was genes with a predicted probability greater than 0.1, with higher values indicating a greater likelihood of being metabolite target genes. In addition, the similarity ensemble approach (SEA) was used to further predict metabolite target genes by associating proteins based on chemical similarity among ligands. Human data were prioritized for subsequent analyses. Significance was evaluated such that smaller p-values indicated a higher likelihood of a gene being a metabolite target gene. Target genes predicted using the SEA were also merged. Finally, target genes predicted by STP and SEA were integrated to obtain the final set of metabolite target genes.
Construction of an ovarian cancer–related gene set and screening of key intersection genes
The GeneCards database (https://www.genecards.org, version 5.17, accessed in April 2025) was used to identify ovarian cancer–related genes. Genes with scores greater than 10 were selected. Next, the online Mendelian inheritance in man (OMIM) database (https://omim.org, accessed in April 2025) was used to retrieve ovarian cancer–related genes. Finally, the comparative toxicogenomics database (CTD) (https://ctdbase.org, version 2024.12, accessed in April 2025) was used to search for ovarian cancer–related genes. Genes from these three databases were combined to create the final disease-related gene set. The ‘ggvenn’ package in R software (version 4.3.2) was used to generate a Venn diagram illustrating the intersection of genes across databases. The total number of genes was determined by summing the values in the diagram. The intersection of metabolite target genes, gut bacteria target genes, and ovarian cancer–related genes was then determined to identify common genes. A Venn diagram was used to visualize the intersection of these three datasets.
Construction of pathway regulatory networks and protein interaction networks
A gut bacteria regulatory network was constructed, which linked gut bacteria to genes and genes to disease, aiming to analyze genes associated with gut bacteria regulation of ovarian cancer. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses were performed to identify the functions and pathways in which the intersection genes were enriched. Intersection genes were those common to metabolite target genes, gut bacteria target genes, and ovarian cancer–related genes. The filtering criteria were as follows: pvalueFilter = 0.05, p.adjustFilter = 0.05. Next, a pathway regulatory network was constructed. Genes associated with regulatory pathways were analyzed, and network relationships, node attributes, and pathway and gene lists were generated. These files were then visualized using Cytoscape (version 3.10.2) software. A protein interaction network of the intersection genes was constructed using the Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) database (https://string-db.org, version 12.0), with a scoring threshold of 0.4. Genes were ranked according to the number of connected nodes, and the core genes of the network were identified (the five genes with the highest number of nodes were selected as the core genes). Finally, the protein interaction network was visualized using Cytoscape (version 3.10.2) software.
“Gut bacteria–metabolite–gene” regulatory network and drug-likeness and toxicity analysis
A regulatory network was constructed, which linked gut bacteria to substrates, substrates to metabolites, and metabolites to core genes. This network was visualized using Cytoscape software (version 3.10.2) to observe how gut bacteria, through substrates and metabolites, influence gene expression and thereby impact the development and progression of ovarian cancer. SMILES structures of the metabolites were obtained and subjected to drug-likeness and toxicity analyses. Drug-likeness analysis was performed using the SwissADME website (http://www.swissadme.ch/, accessed in May 2025). The filtering parameters for this analysis were as follows: molecular weight less than 500, number of hydrogen bond donors less than 5, number of hydrogen bond acceptors less than 10, lipid–water partition coefficient less than 4.15, number of Lipinski rule violations less than 1, bioavailability score greater than 0.1, and topological polar surface area less than 140. Toxicity analysis was performed using the ADMETlab website (https://admetlab3.scbdd.com/server/screening, version 3.0, accessed in May 2025). The toxicity index ranges from 0 to 1 and is divided into three risk levels: 0–0.3, low risk; 0.3–0.7, medium risk; and 0.7–1, high risk.
Results
Screening of metabolite target genes and ovarian cancer–related genes and identification of key intersection genes
The target genes predicted by the STP and SEA databases were combined to obtain the final set of metabolite target genes (Figure 1(a)). The ovarian cancer–related genes identified from the GeneCards, OMIM, and CTD databases were combined to obtain the final set of disease-related genes (Figure 1(b)). Figure 2 shows the intersection of genes from each database, and the final number of genes was determined by summing the values in the figures. Common genes were identified by combining the metabolite target genes, gut bacteria target genes, and ovarian cancer–related genes (Figure 1(c)). Within the gut bacteria regulatory network, the study found that gut bacteria regulate the development and progression of ovarian cancer through 59 genes, including HDAC5, IGFBP6, HDAC4, MAPK8, and VDR (Figure 1(d)).

(a) Union of metabolite target genes; (b) Union of ovarian cancer–related genes; (c) Common genes obtained by intersecting metabolite target genes, intestinal bacteria target genes, and ovarian cancer–related genes and (d) 59 genes regulating ovarian cancer. GO: Gene Ontology; KEGG: Kyoto Encyclopedia of Genes and Genomes.
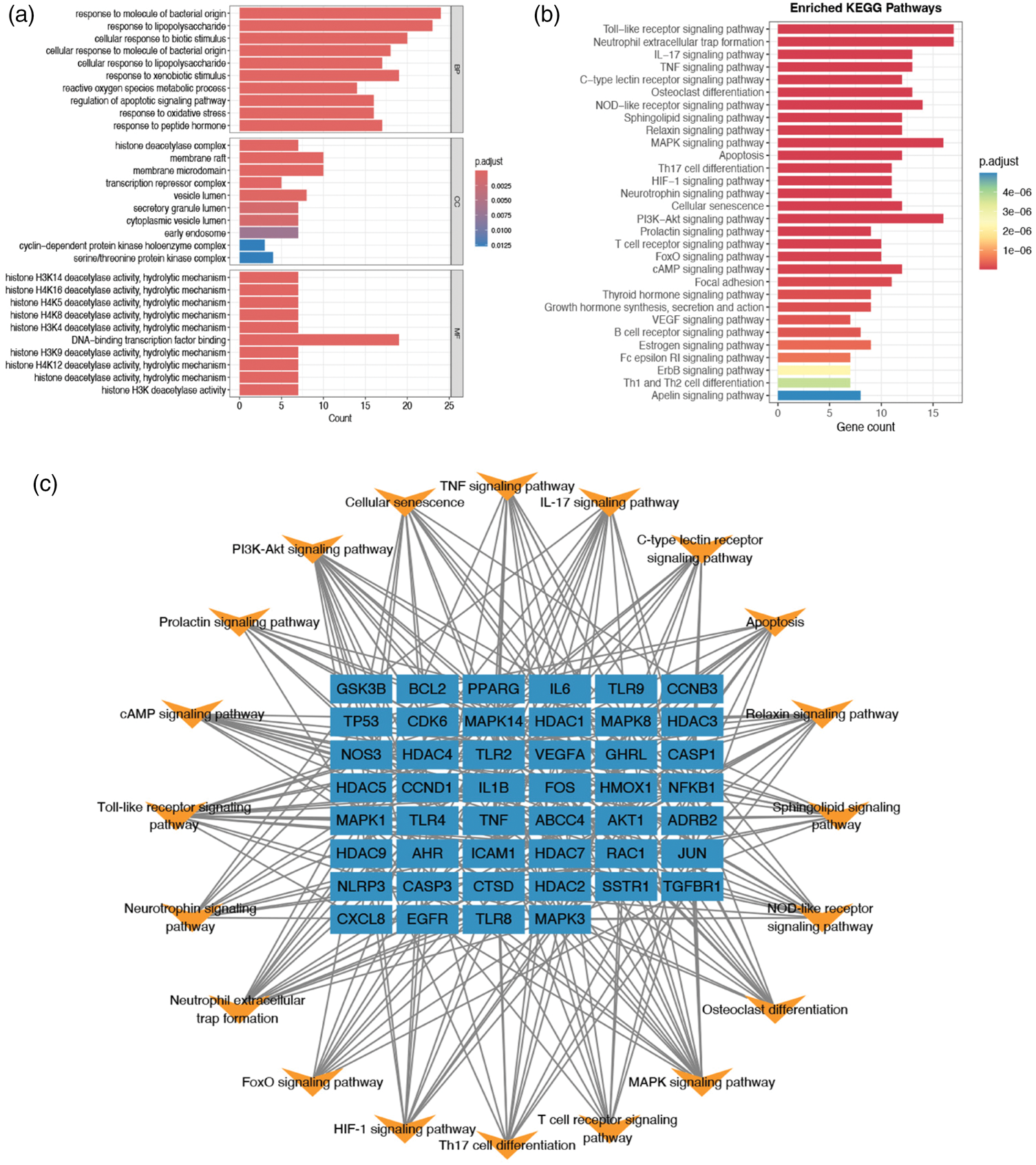
(a) GO enrichment analysis results, (b) KEGG enrichment analysis results and (c) Pathway regulatory network. GO: Gene Ontology; KEGG: Kyoto Encyclopedia of Genes and Genomes.
GO and KEGG enrichment and pathway regulatory network analyses
GO enrichment analysis (Figure 2(a)) revealed that the intersection genes were most significantly enriched in the response to molecule of bacterial origin and response to lipopolysaccharide (LPS) functions. KEGG enrichment analysis showed that the intersection genes were most significantly enriched in the Toll-like receptor (TLR) signaling pathway and the neutrophil extracellular trap (NET) formation pathway (the enrichment fractions were 8.65 and 6.92, respectively, and the p-values after false discovery rate correction were both <0.001) (Figure 2(b)). A pathway regulatory network constructed using Cytoscape software revealed that genes such as GSK3B, BCL2, PPARG, interlekin-6 (IL6), and TLR9 were involved in pathway regulation (Figure 2(c)). These pathways are highly correlated with the formation and regulation of the immune microenvironment in ovarian cancer. Sustained activation of the TLR pathway can promote tumor-associated inflammation, while NET formation has been reported to be associated with ovarian cancer metastasis and an immunosuppressive microenvironment, suggesting that gut microbiota metabolites may influence ovarian cancer progression by regulating these key immune pathways.
Protein interaction network and core gene identification
The protein interaction network for the intersection genes was constructed (Figure 3(a)). Connected nodes or protein links indicate a protein–protein interaction relationship between two genes. The bars in Figure 3(b) show the five core genes with the highest number of node connections in the protein interaction network. These core genes are TP53, AKT1, IL6, tumor necrosis factor (TNF), and IL1B. Network topology analysis showed that the connectivity and betweenness centrality of these five core genes were significantly higher than the network average, indicating that they occupy a pivotal position in the regulatory network. Figure 3(c) shows a correlation analysis of the protein interaction network visualized using Cytoscape. Figure 4 presents a regulatory network diagram illustrating the relationships between gut bacteria and substrates, substrates and metabolites, and metabolites and core genes, visualizing how gut bacteria influence gene expression through substrates and metabolites, thereby affecting the development and progression of ovarian cancer.
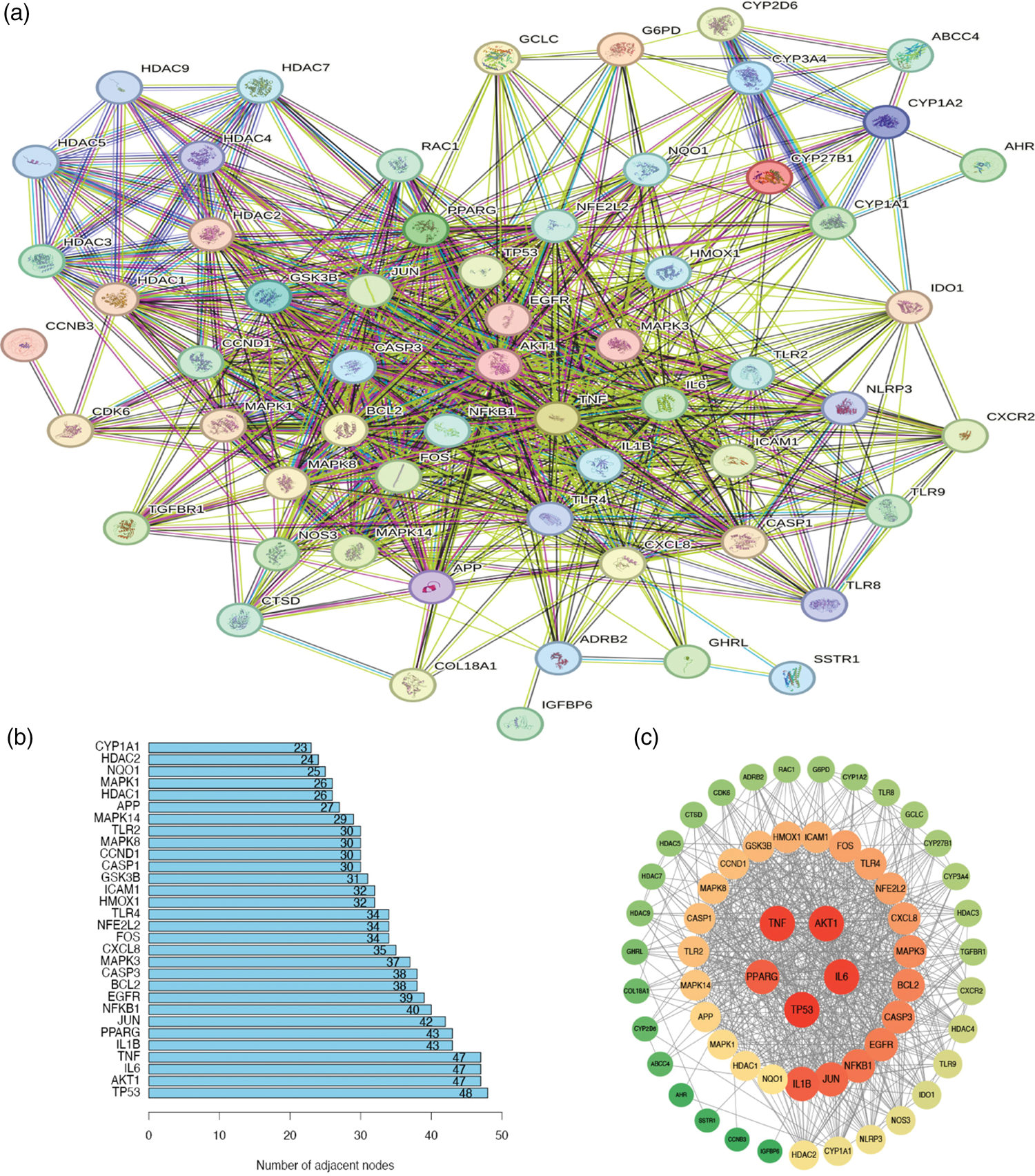
(a) Protein interaction network of intersection genes, (b) Number of connections in protein interaction network nodes and (c) Protein interaction network graph.

Regulatory network diagram showing the correspondence between intestinal bacteria and substrates, substrates and metabolites, and metabolites and core genes.
Analysis of metabolite drug-like properties and toxicity
For drug-like property analysis, metabolites were filtered using the R software packages ‘dplyr’ and ‘tidyr’ to identify those with high drug-like potential. These metabolites include icaritin, doconexent, (10-[(acetyloxy)methyl]-9-anthryl)methyl acetate, arachidonic acid, and diosmetin. Among these, arachidonic acid is a precursor of inflammatory mediators such as prostaglandins and leukotrienes, while diosmetin exhibits known anti-inflammatory activity, suggesting that they may exert effects by modulating the local immune-inflammatory response in the ovary. In the toxicity analysis, doconexent, (10-[(scetyloxy)methyl]-9-anthryl)methyl acetate, apigenin, kaempferol, and diosmetin showed the highest carcinogenicity. This indicates that certain microbial metabolites may have a “double-edged sword” effect in immune regulation, and their proinflammatory or anti-inflammatory effects may affect the risk of tumor development in different contexts. Future research should combine in vivo and in vitro models to further verify their safety and contextual effects.
Discussion
This study systematically explored the potential associations and regulatory networks between gut microbiota, its metabolites, and the development and progression of ovarian cancer by integrating bioinformatics and network pharmacology approaches. We successfully constructed a multilayered regulatory network of “gut microbiota–substrate–metabolite–core gene–pathway–disease,” identifying key core genes, functional pathways, and metabolites with potential research value. This provides new data clues and theoretical hypotheses for a deeper understanding of the role of gut microbiota in ovarian cancer.
Through multi-database joint analysis, we identified 59 key genes that may play a bridging role in the association between gut microbiota and ovarian cancer. Protein–protein interaction network analysis further identified five core genes: TP53, AKT1, IL6, TNF, and IL1B. The roles of these genes in tumor biology have been widely confirmed.8–10 TP53 is the most well-known tumor suppressor gene, and its mutation or loss of function is an important driver of various cancers, including high-grade serous ovarian cancer. AKT1 is a key node in the PI3K/AKT signaling pathway, involved in cell proliferation, apoptosis resistance, and chemotherapeutic resistance. IL6, TNF-α, and IL1β are important inflammatory factors that play critical roles in tumor microenvironment formation, immune escape, and angiogenesis.11,12 Our analysis suggests that changes in gut microbiota are associated with the regulation of this core group of cancer-related genes, particularly in areas such as tumor suppression and inflammatory immune responses, although the specific causal regulatory relationships require experimental validation.
GO and KEGG enrichment analyses revealed potential biological mechanisms. Intersecting genes were significantly enriched in GO functional terms such as “response to molecule of bacterial origin” and “response to lipopolysaccharide,” suggesting that bacterial-derived components, such as LPS, may be important mediators of gut microbiota regulation of the host.13,14 Correspondingly, KEGG pathway analysis revealed the most significant pathways, “Toll-like receptor signaling pathway” and “neutrophil extracellular trap formation.” The TLR pathway is central to recognizing pathogen-associated molecular patterns and initiating innate immune responses. Its sustained activation can lead to a chronic inflammatory state, which has been shown to be a key factor in promoting the development and progression of ovarian cancer.15–17 NET formation is an antimicrobial defense mechanism of neutrophils, and recent studies have found that NETs can promote ovarian cancer metastasis and peritoneal implantation.18–20 Based on these findings, we propose a conceptual model of the gut–ovarian axis: the gut microbiota may remotely influence the local immune microenvironment of the ovary via pathways such as blood circulation through its metabolites (e.g. SCFAs and bile acids) or translocated bacterial components (e.g. LPS). Potential mechanisms include activating the TLR signaling pathway in ovarian tissue or infiltrating immune cells, inducing a chronic inflammatory state; regulating the function of immune cells such as neutrophils, affecting NET formation; and subsequently, through interactions with key inflammatory factors such as IL-6 and TNF-α and key oncogenes such as TP53 and AKT1, jointly shaping a microenvironment conducive to tumor growth, metastasis, and immune escape. This hypothetical model provides a clear direction for subsequent mechanistic research.21,22
At the metabolite level, the regulatory network constructed herein demonstrates how gut bacteria influence core genes through specific substrates and metabolites, such as arachidonic acid and diosmetin. Drug-likeness analysis identified metabolites with promising druggability, such as icaritin and arachidonic acid.23–25 Arachidonic acid, a precursor of prostaglandins and leukotrienes, has complex metabolic pathways involved in inflammation and cancer, providing a rationale for its potential as an intervention target.26,27 However, toxicity analysis revealed that some metabolites, such as doconexent and apigenin, have high predicted carcinogenic risk, suggesting that caution is warranted when utilizing these metabolites due to their potential double-edged sword effects. Future research should focus on further exploring their specific mechanisms of action and structure–activity relationships.28–30 This study has several limitations that need to be considered when interpreting the results. First, this is a bioinformatics analysis based on a public database, essentially a computer simulation prediction. All discovered gene, pathway, and metabolite associations require rigorous functional verification and mechanistic confirmation through subsequent in vitro and in vivo experiments. Second, the databases used in this study have inherent incompleteness and potential biases, which may result in missing genes, interactions, or metabolites that have not yet been included. Additionally, different databases may follow distinct nomenclature and annotation standards. Third, this study could not precisely identify which specific gut bacterial species and metabolites dominate the abovementioned regulatory processes; the extreme complexity of the gut microbiome makes causal inference challenging. Fourth, the analysis is mainly based on gene sets at the tissue or cellular level, without integrating individual patient gut microbiota composition, metabolomics, or clinicopathological characteristics for association verification, thus limiting the generalizability of the conclusions.31,32 Furthermore, the composition of the gut microbiome is highly complex, and this study has not yet precisely defined which bacterial species and metabolites dominate the aforementioned regulatory processes.
In summary, through bioinformatics and network pharmacology analyses, we preliminarily constructed a regulatory network of “gut microbiota–metabolites–genes–pathways” and proposed a potential mechanistic model in which gut microbial metabolites may contribute to ovarian cancer development by regulating genes such as TP53 and AKT1 and through pathways such as TLR signaling and NET formation. These results provide preliminary molecular insights and analytical frameworks for the “gut–ovarian axis” hypothesis as well as research directions and a foundation for further exploration of gut microbiota and their metabolites as potential biomarkers or intervention targets for ovarian cancer.
Footnotes
Acknowledgments
We sincerely thank the GutMgene, STP, SEA, GeneCards, OMIM, CTD, STRING, SwissADME, and ADMETlab databases for providing free access to their valuable data, which were indispensable for the completion of this study.
Author contributions
All authors have read and approved the final manuscript and are responsible for the completeness of the research, the accuracy of the data analysis, and academic integrity. Liu Yinghong: Contributed to the study design and data analysis, including data collection, bioinformatics analysis, network construction, visualization, and drafting of the manuscript. Yang Xiaojuan: Contributed to the overall study conception and design, provided methodology guidance, interpreted results, critically revised the manuscript, and served as the corresponding author responsible for manuscript submission, communication during peer review, and coordination.
Data availability statement
The datasets used in this study are available in online databases and comprise public data.
Declaration of conflicting interests
The authors declare that there is no conflict of interest.
Ethical review and consent to participate
Not applicable.
Funding
This study did not receive any funding.