
Abstract
Objective
To identify male infertility-related long non-coding (lnc)RNAs and an lncRNA-related competing endogenous (ce)RNA network.
Methods
Expression data including 13 normospermic and eight teratozoospermic samples from postmortem donors were downloaded from the GEO database (GSE6872). The limma R package was used to discriminate dysregulated lncRNA and micro (m)RNA profiles. Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses of differentially expressed (DE) mRNAs were performed using the clusterProfiler R package. The ceRNA network of dysregulated genes was visualized by Cytoscape.
Results
A total of 101 DE lncRNAs and 1722 mRNAs were identified as male infertility-specific RNAs with thresholds of |log2FoldChange| >2.0 and adjusted P-value <0.05. GO and KEGG pathways were analyzed for DE mRNAs. Gene set enrichment analysis revealed that DE genes were enriched in embryonic skeletal system development and cytokine–cytokine receptor interactions. A ceRNA network was constructed with 26 key lncRNAs, 33 microRNAs, and 133 mRNAs. DE lncRNAs in male sterility were mainly associated with transferring phosphorus-containing groups and complexes of histone methyltransferases, methyltransferases, PcG proteins, and serine/threonine protein kinases.
Conclusion
This provides a novel perspective to study lncRNA-related ceRNA networks in male infertility and assist in identifying new potential biomarkers for diagnostic purposes.
Keywords
Introduction
Male infertility is one of the most serious diseases of the male reproductive system, and is typically caused by the disturbance of spermatogenesis. 1 , 2 It can seriously affect the quality of life and relationships of affected patients. 2 Clinically, it is thought to be associated with environmental pollution, unhealthy living habits, a lack of sexual knowledge, and reproductive system diseases. 1 , 3 , 4 However, because the exact pathogenesis is unknown, there are a lack of clear treatment targets and effective treatment measures. 5 , 6 Therefore, there is an urgent need for basic research into the molecular mechanism of male infertility to identify new biomarkers and therapeutic targets.
High-throughput technology has been extensively used to investigate gene expression in male infertility, providing a novel method to identify important genes and explore the disease. 7 , 8 Long non-coding (lnc)RNAs are a type of non-coding RNA, exceeding 200 nucleotides in length, which lack coding ability and are an important part of all non-coding transcripts. 9 , 10 LncRNAs regulate the translation of genes by binding micro (mi)RNAs, 11 , 12 but although miRNAs have attracted extensive attention for their role in a variety of physiological and pathophysiological processes, the role of lncRNAs has not been fully elucidated. 13 LncRNAs are involved in several biological processes in cancer development such as cell proliferation, invasion, and metastasis, and they play key roles in the modulation of tumor biology. Previous work showed that lncRNAs function in the physiology and pathology of immune system regulation, and some have been used as therapeutic targets or potential prognostic biomarkers of cancer. 14 , 15 However, little is known about their involvement in male infertility.
Competing endogenous RNAs (ceRNAs) are a complex post-transcriptional regulatory network in which lncRNAs, mRNAs, and other RNAs function as natural miRNA sponges to suppress target mRNA functions by sharing one or more miRNA response elements. LncRNAs acting as ceRNAs regulate gene-encoding protein levels and participate in the regulation of cell biology by competing with miRNAs. 16 , 17 This study investigated the lncRNA-related ceRNA network in male infertility. First, we downloaded and reannotated the expression profile of male infertility from the Gene Expression Omnibus (GEO) database and identified differentially expressed genes and lncRNAs. Then we mapped them to the global lncRNA–mRNA network to obtain a male infertility lncRNA gene network. Genes associated with male infertility-related lncRNAs underwent pathway enrichment analysis by the Database for Annotation, Visualization and Integrated Discovery tool to predict the lncRNA functions.
Methods
Data sources and processing
Gene expression profiling arrays were systematically searched using the GEO database (http://www.ncbi.nlm.nih.gov/geo) update to October, 2019, with keywords including “Male Sterility” [MeSH] OR “Male Infertility” [MeSH] OR “Male Subfertility” [MeSH]. Genes showing significant differential expression between normospermic and teratozoospermic groups were identified as follows: 1.2 times the expression level in 20% of the total samples was greater than the median expression level of the total samples; or the variance of expression in every sample was higher than the median. Subsequently, log2 transformation and Z correction were performed to normalize the expression value of each gene. We applied the R limma package to compute differentially expressed lncRNAs and mRNAs based on normalized mRNA and lncRNA expression data. 18 Bayes test was used to select differentially expressed lncRNAs and mRNAs; a change ≥2 fold and a P value cutoff of 0.05 were defined as statistically significant.
Gene ontology and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis of differentially expressed genes
Genes and gene products were annotated by gene ontology (GO) analysis, which is a common approach to explore characteristic biological attributes for transcriptome data or high-throughput genomes. KEGG pathway analysis serves as a knowledge base for analyzing gene functions, linkage between genomic information, and higher-order functional information. Synthetically, mapping a set of genes to the related biological annotation is an important basis for gene functional analysis of any high-throughput data. KEGG pathway analysis and GO enrichment were conducted using the clusterProfiler package to analyze different mRNAs at the functional level. 19 P<0.05 was considered to be statistically significant.
Integration of ceRNA network construction
Differentially expressed mRNAs, miRNAs, and lncRNAs were selected to construct a ceRNA regulatory network. Differentially expressed lncRNAs–miRNAs were predicted by the miRDB database, 20 miRTarBase database, 21 and TargetScan database. 22 Then the regulatory ceRNA network was constructed using ceRNA network construction.
Results
Identification of differentially expressed lncRNAs in male sterility
To analyze lncRNAs showing differential expression between normospermic and teratozoospermic individuals, we located and manually created 19 publicly available male sterility datasets. Expression data including 13 normospermic samples and eight teratozoospermic samples from postmortem donors were downloaded from the GEO database (GSE6872), and the probe was re-annotated using Bowtie based on the GENCODE release annotation for lncRNA. Using P<0.05 and [logFC]>2 as cut-off criteria, a total of 101 differentially expressed lncRNAs and 1722 differentially expressed mRNAs were found to be significantly dysregulated in the GSE6872 dataset. 23 Compared with the normospermic group, 68 lncRNAs were upregulated and 33 lncRNAs were downregulated in the teratozoospermic group. The differentially expressed lncRNAs associated with male sterility are shown in Figure 1a and b.

Genes and lncRNAs associated with male infertility showing differential expression between teratozoospermic and normospermic samples. (a) Volcano plots of differentially expressed lncRNAs in male infertility; black dots represent non-differentially expressed genes. (b) Heatmap showing hierarchical clustering of differentially expressed lncRNAs in male infertility. Horizontal axis shows sample names in the GSE6872 dataset. Right vertical axis shows lncRNA names; each column represents a sample and each row represents a specific lncRNA. |log2FC| >2 and adjusted P<0.05. Red indicates upregulation and green indicates downregulation of expression.
Next, GO biological processes and KEGG pathways were used to elucidate the potential biological functions of differentially expressed mRNAs in male sterility. All of the 1754 identified differentially expressed mRNAs were screened by gene set enrichment analysis (GSEA), and 180 important enrichments were identified by GO and KEGG analysis. As shown in Figure 2a, the top GO pathways from enrichment analysis of the differentially expressed mRNAs in male sterility were mainly associated with nicotinamide adenine dinucleotide (NADH), including structural constituents of ribosomes, oxidoreductase activity, acting on NADH, electron transfer activity, and NADH dehydrogenase activity. The top KEGG pathways from enrichment analysis of the differentially expressed mRNAs in male sterility were mainly associated with Huntington disease, thermogenesis, non-alcoholic fatty liver disease, ribosomes, and Alzheimer’s disease (Figure 2b).
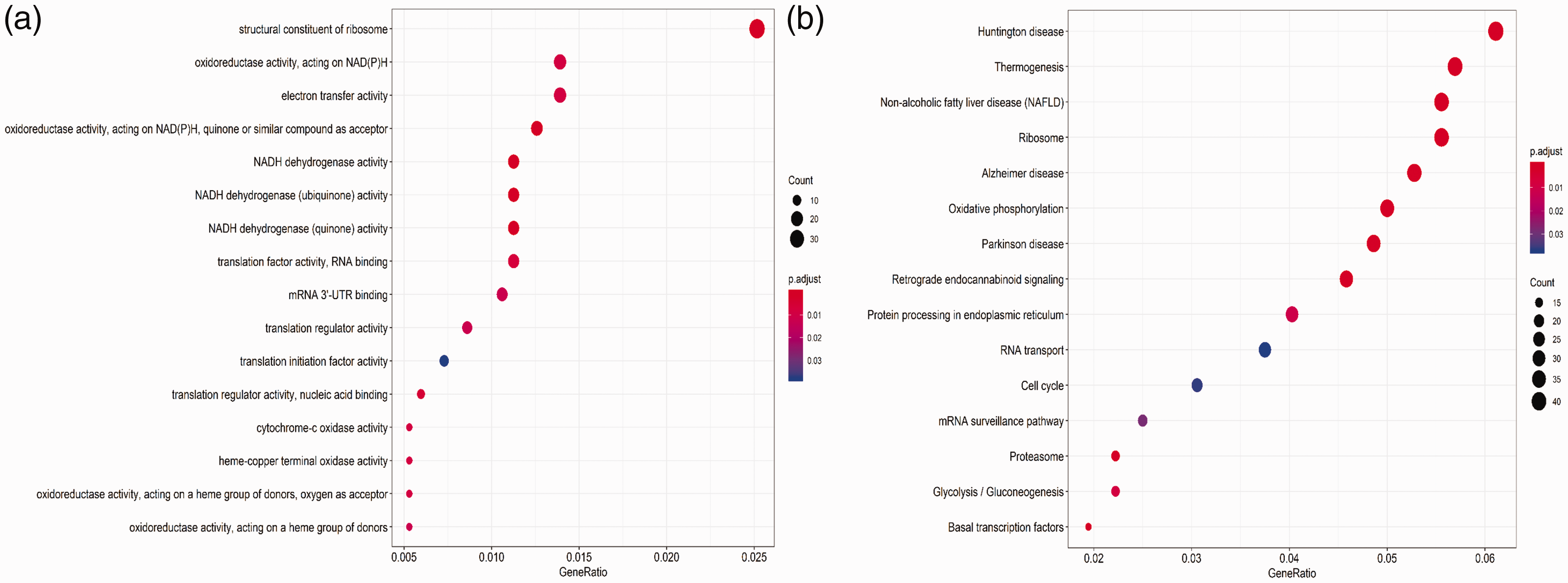
Functional enrichment analysis of differentially expressed mRNAs between normospermic and teratozoospermic groups. (a) Gene ontology (GO) analysis. (b) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis. The bubble size is directly proportional to the number of mRNAs. The adjusted P-value is represented by color, with redder bubbles representing smaller adjusted P-values
Gene set enrichment analysis
GSEA was used to elucidate the key signaling pathways of high- or low-expressing genes in male sterility. GSEA GO analysis revealed that, compared with low-expressing genes, high-expressing genes in male sterility were mainly enriched in embryonic skeletal system development, endocrine system development, G protein-coupled receptor activity, the positive regulation of cell division, and the regulation of synapse assembly (Figure 3a). GSEA KEGG analysis revealed that, compared with low-expressing genes, high-expressing genes in male sterility were mainly enriched in cytokine receptor interaction, neuroactive ligand receptor interaction, the phospholipase D signaling pathway, REP1 signaling pathway, and RAS signaling pathway (Figure 3b).
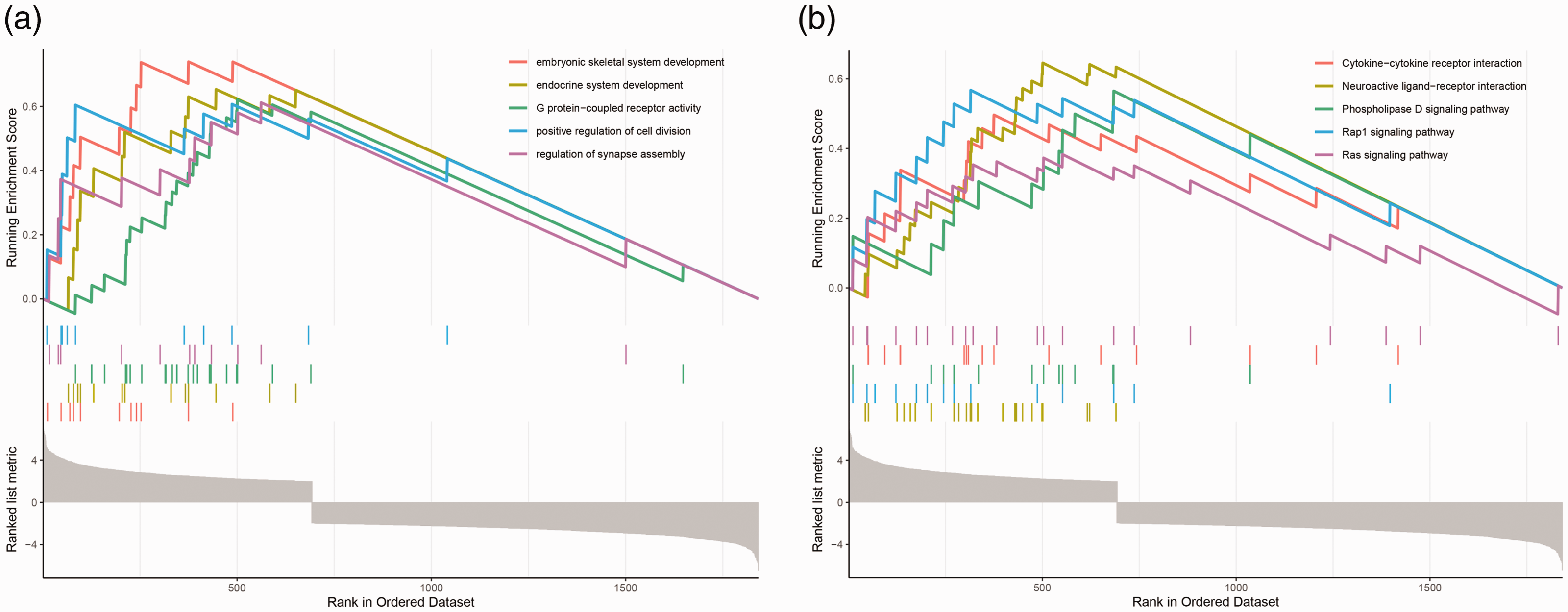
Gene set enrichment analysis of differentially expressed mRNAs between normospermic and teratozoospermic groups. (a) Gene set enrichment analysis using GO. (b) Gene set enrichment analysis using KEGG.
Functional enrichment analysis of differentially expressed lncRNAs
To elucidate the function of differentially expressed lncRNAs, we used the miRcode database to predict miRNA-targeted lncRNAs, and mapped target miRNAs into TargetScan, miRDB, and miRTarBase databases to identify target mRNAs. A total of 1621 interactions between 26 lncRNAs and 206 miRNAs were identified using the miRcode database, and 1034 interactions between 206 miRNAs and 134 mRNAs were also identified. As shown in Figure 4a, 134 mRNAs were predicted to be associated with differentially expressed lncRNAs in male sterility. The top GO pathways from enrichment analysis of the 134 predicted mRNAs were mainly associated with transferring phosphorus-containing groups, the histone methyltransferase complex, methyltransferase complex, PcG protein complex, and serine/threonine protein kinase complex (Figure 4b).
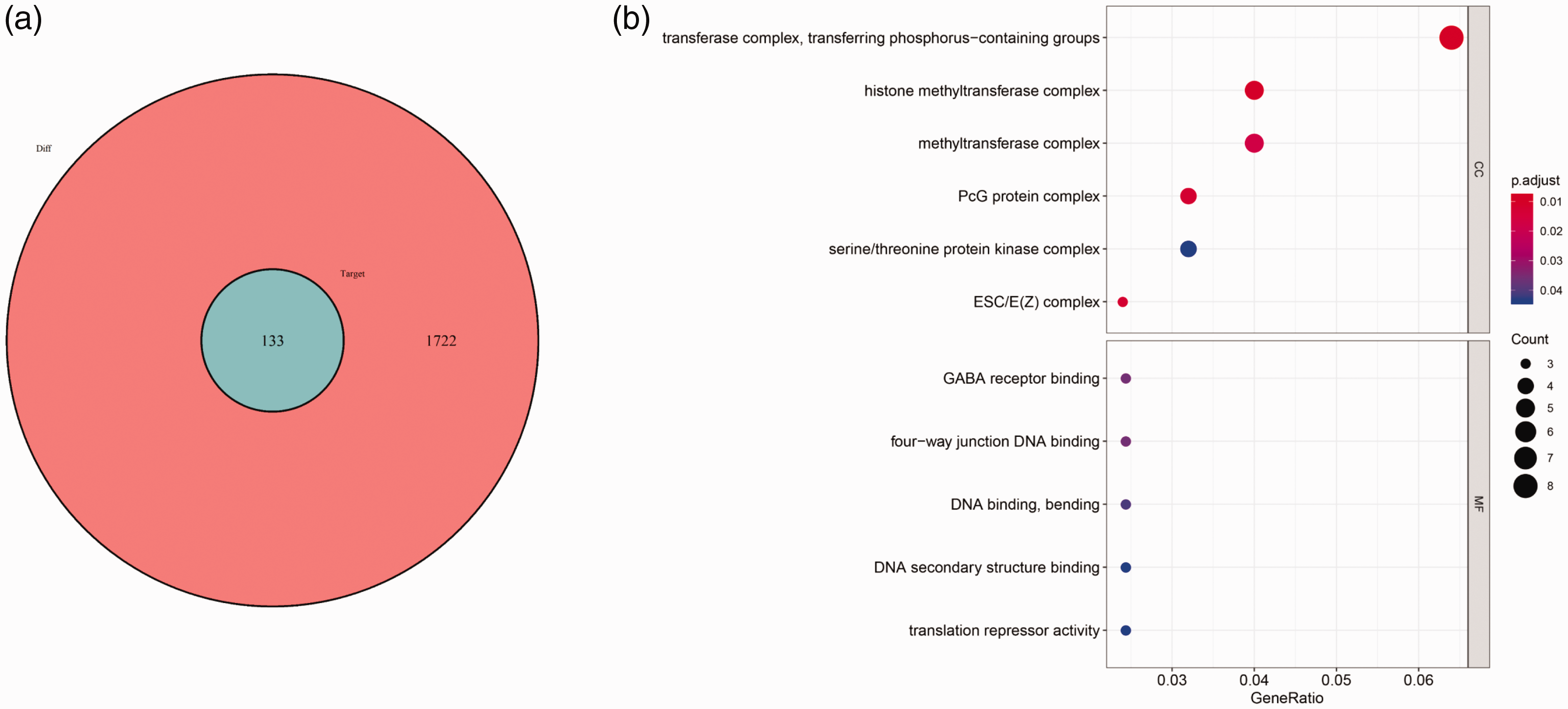
Mutual mRNAs between differentially expressed and predicted mRNAs, and their GO analysis. (a) Venn diagram of differentially expressed mRNAs and predicted mRNAs. Red circle shows the 1722 dysregulated mRNAs; blue circle shows the 134 target mRNAs of dysregulated lncRNAs. (b) GO enrichment analysis of mRNAs involved in the competing endogenous RNA network.
Construction of ceRNA network in male sterility
To further elucidate the mechanisms of these differentially expressed lncRNAs in male sterility, a ceRNA network was constructed based on the above data. Consequently, the ceRNA network was shown to involve 26 differentially expressed lncRNAs (Table 1), 33 miRNAs targeting the 26 lncRNAs (Table 2), and 133 mRNAs targeted by the 33 miRNAs. Within the ceRNA network, 12 lncRNAs and 87 mRNAs were downregulated, while 14 lncRNAs and 46 mRNAs were upregulated. These findings indicate that differentially expressed lncRNAs indirectly interact with mRNAs through miRNAs in male infertility (Figure 5).
Differentially expressed lncRNAs involved in the ceRNA network.

lncRNA, long non-coding RNA; ceRNA, competing endogenous RNA; Adj., adjusted.
miRNAs targeting male infertility-specific lncRNAs in the ceRNA network.

lncRNA, long non-coding RNA; ceRNA, competing endogenous RNA; miR/miRNA, micro RNA.

Competing endogenous RNA network in male infertility. Red rhombuses represent lncRNAs, green triangles represent miRNAs, and blue dots represent mRNAs. Gray lines indicate lncRNA–miRNA–mRNA interactions.
Discussion
Research into complex diseases is often difficult because of the involvement of multiple genetic and environmental factors. Differential co-expression analysis can be used to explain the molecular mechanism of such diseases, and for model analysis following the merger of co-expression interaction networks to provide theoretical support for disease research. 24
The global incidence of male infertility has recently been increasing,1,2 so it is crucial to understand its pathogenesis and identify clear treatment targets and effective treatment measures. With the development of bioinformatics, the effect of lncRNAs on growth and development has gradually become apparent, and they have been shown to participate in the occurrence and development of diseases through a variety of regulatory mechanisms. Several studies on male reproduction have confirmed that lncRNAs are associated with spermatogenesis. For example, Wen et al. 25 systematically identified the testis-specific expression of lncRNAs in a Drosophila melanogaster model, and confirmed their important roles in spermatogenesis. Hu et al. 26 found that lncRNA Gm2044 is highly expressed in spermatocytes and inhibits translation by interacting with mRNA Utfl. Moreover, Wichman et al. 27 identified the slight imbalance of lncRNAs as a novel mechanism for a decreased sperm count or infertility. However, there are few reports about the relationship between lncRNAs and male infertility, and it is not clear whether lncRNAs function as ceRNAs in this context.
Here, we identified 101 lncRNAs and 1754 mRNAs as differentially expressed between normospermic and teratozoospermic groups based on the GSE6872 dataset, including 33 downregulated and 68 upregulated genes. Subsequently, we explored the role of lncRNAs in the development of male infertility by constructing a ceRNA regulatory network and investigating its potential mechanisms through functional enrichment analysis. The differentially expressed lncRNAs identified were mainly associated with transferring phosphorus-containing groups, and complexes of histone methyltransferases, methyltransferases, PcG proteins, and serine/threonine protein kinases.
Previous lncRNA work has largely focused on cancer, 28 but this study adopted the ceRNA theory to integrate into the lncRNA–mRNA network, and predicted lncRNA functions through analyzing mRNAs that compete with lncRNAs. This is a useful strategy and basis for lncRNA functional research. Additionally, differentially expressed lncRNAs and mRNAs were obtained from male infertility-related expression profile data to construct a male infertility-specific lncRNA–mRNA network which clearly explains the competitive relationship between lncRNAs and mRNAs in the pathogenesis of male infertility. Network analysis revealed that some lncRNAs regulate multiple genes and play an important regulatory role, while others only compete with some genes for miRNAs, indicating the specificity of their regulation.
In summary, we identified lncRNAs associated with male infertility using bioinformatics methods, and constructed a ceRNA network that regulates the differentially expressed lncRNAs in male infertility. This approach is a useful method for identifying disease-related lncRNAs and is conducive to an improved understanding of the underlying molecular mechanisms of male infertility. Our findings should aid the discovery of new male infertility drugs and therapeutic targets and provide more theoretical support for its prevention and treatment.
Footnotes
Data availability
All datasets generated for this study are included in the manuscript.
Declaration of conflicting interest
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by a grant from the National Natural Science Foundation of China (81260172).