
Abstract
Objective
Hepatocellular carcinoma (HCC) is a common cancer with a high mortality rate; the molecular mechanism involved in HCC remain unclear. We aimed to provide insight into HCC induced with HepG2 cells and identify genes and pathways associated with HCC, as well as potential therapeutic targets.
Methods
Dataset GSE72581 was downloaded from the Gene Expression Omnibus, including samples from mice injected in liver parenchyma with HepG2 cells, and from mice injected with cells from patient tumor explants. Differentially expressed genes (DEGs) between the two groups of mice were analyzed. Then, gene ontology and Kyoto Encyclopedia of Gene and Genomes pathway enrichment analyses were performed. The MCODE plug-in in Cytoscape was applied to create a protein–protein interaction (PPI) network of DEGs.
Results
We identified 1,405 DEGs (479 upregulated and 926 downregulated genes), which were enriched in complement and coagulation cascades, peroxisome proliferator-activated receptor signaling pathway, and extracellular matrix–receptor interaction. The top 4 modules and top 20 hub genes were identified from the PPI network, and associations with overall survival were determined using Kaplan–Meier analysis.
Conclusion
This preclinical study provided data on molecular targets in HCC that could be useful in the clinical treatment of HCC.
Keywords
Introduction
Hepatocellular carcinoma (HCC) is the fifth most frequent tumor in men and the ninth most frequent in women worldwide, with approximately 500,000 and 200,000 new cases per year, respectively. 1 Because of its insidious onset, imperceptible symptoms in early stages, and poor prognosis, HCC is the second most common cause of cancer-related death in the world, making its clinical treatment challenging. 2 Results from research using HCC cell lines are often not useful in clinical studies, and we hypothesize that this is because of differences between tumors from preclinical samples injected with HCC cell lines and those injected with tumor cells from patient tumor explants. Thus, further investigations into the different molecular pathophysiology of tumors from preclinical samples induced with HCC cell lines and patient tumor explants are necessary to provide more data for effective treatment.
The latest research has shown that actin gamma smooth muscle 2 (ACTG2) boosts the metastatic potential of HCC cells in a Notch homolog 1 (Notch1)-dependent manner. 3 In HCC, double-stranded RNA-dependent protein kinase (PKR) act as a tumor suppressor by inhibiting hepatitis C virus replication. However, PKR also acts as a tumor promoter through enhancement of cancer cell growth by mediating MAPK or signal transducer and activator of transcription (STAT) pathways in patients with cirrhosis. 4 One study showed, by analyzing cell lines, genetically modified mice, and HCC tissues, that Yes-associated protein (YAP) cooperates with forkhead box protein M1 (FOXM1) to contribute to chromosome instability; agents that disrupt this pathway might be developed as treatments for liver cancer. 5 These studies demonstrate that a better understanding of the mechanisms underlying HCC are of great importance to its clinical treatment. Further studies are necessary to elucidate other potential mechanisms and investigate target genes involved in different forms of induced HCC in preclinical studies.
In this study, we downloaded array data of GSE72981 from Gene Expression Omnibus (GEO) to confirm the similarities and differences of tumors from mice injected with HepG2 cells and those injected with tumor cells from patient tumor explants. We analyzed the differentially expressed genes (DEGs) using a biological informatics approach to provide further insight into HCC induced with HepG2 cells versus cells from patient tumor explants.
Materials and methods
Microarray data
The gene expression profile of GSE72981 was downloaded from the GEO database (http://www.ncbi.nlm.nih.gov/geo/), which was based on the GPL570 platform ([HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array). The GSE72981 dataset contained 30 samples derived from severe combined immunodeficiency (SCID) mice, including eight mice that were subcutaneously (ectopically) injected into the flank and orthotopically into liver parenchyma with HepG2 cell lines, and 22 that were subcutaneously (ectopically) injected into the flank and orthotopically into liver parenchyma with tumor cells from patient tumor explants.
Identification of DEGs
The limma package 6 was applied to analyze DEGs between SCID mice that were injected with HepG2 cell lines and SCID mice that were injected with tumor cells from patient tumor explants. The P-values of DEGs were calculated using a t-test in R (https://www.R-project.org/) with the limma package. An adjusted P-value <0.05 and |logFC| >2 (where FC = fold change) were set as the cut-off criteria. In total, 1,405 DEGs were found, including 479 upregulated genes and 926 downregulated genes; the 20 genes with the highest degree of connectivity were selected as hub genes. A volcano plot of DEG expression was built in R using the ggplot2 package.
GO and KEGG pathway enrichment analysis of DEGs
Gene ontology analysis (GO) is a commonly used method to annotate genes and gene products and to identify molecular function, biological process, and cellular component attributes for high-throughput genomic or transcriptomic data.7,8 KEGG is a collection of databases used for systematic analysis of gene functions and for associating related gene sets with their pathways. 9 GO annotation (P < 0.01, q < 0.05) and KEGG pathway (P < 0.05) enrichment analyses were conducted for DEGs in R with the clusterProfiler package. 10
Integration of PPI network and module analysis
The online Search Tool for the Retrieval of Interacting Genes (STRING) 11 database (https://string-db.org/) was used to evaluate interactive relationships among DEGs regarding the predicted and experimental interactions of proteins. Interactions of protein pairs in the database are presented with a confidence score. In this study, DEGs were mapped into PPIs, and a confidence score of >0.4 and maximum number of interactors = 0 were used as the cut-off values. The top 20 genes with degree of connectivity >55 were selected as hub genes. Then, PPI networks were constructed using the Cytoscape software (https://cytoscape.org/). The Molecular Complex Detection (MCODE) plug-in in Cytoscape was used to detect significant modules in the PPI network. The criteria were as follows: degree cutoff = 2, node score cutoff = 0.2, k-core = 2, and maximum depth = 100. Top modules from the PPI network were identified using the MCODE plug-in as those with a score of >6.0. Moreover, KEGG pathway enrichment analysis was performed for DEGs in the modules by using DAVID (https://david.ncifcrf.gov/). P < 0.05 and false discovery rate <0.05 were considered to indicate significant differences.
Survival analysis of hub genes
Kaplan–Meier plotter (KM plotter, http://kmplot.com/analysis/) was used to assess the effect of 54,675 genes on survival using 10,461 cancer samples, including 5,143 breast, 1,816 ovarian, 2,437 lung, and 1,065 gastric cancer patients with a mean follow-up of 69, 40, 49, and 33 months, respectively. 12 Relapse-free survival and OS information was based on GEO (Affymetrix microarrays only), the European Genome-Phenome Archive (EGA; https://ega-archive.org/) and The Cancer Genome Atlas (TCGA; https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga) databases. Hazard ratios with 95% confidence intervals and log rank P-values were calculated and displayed in plots.
Ethics
All data in this paper were obtained from the GEO database (http://www.ncbi.nlm.nih.gov/geo/). Ethical permission was deemed unnecessary for this study.
Results
Identification of DEGs
An adjusted P-value < 0.05 and |logFC| >2 were set as cut-off criteria for DEGs. A total of 1,405 DEGs were identified after analysis of GSE72581, including 479 upregulated genes and 926 downregulated genes. The top 20 upregulated and downregulated DEGs are shown in the heat map in Figure 1.

Volcano map of DEGs. Red represents upregulated genes, green represents downregulated genes, and black indicates genes that were not differentially expressed. The names of the top 20 upregulated and downregulated DEGs are shown. DEG, differentially expressed gene.
GO term and KEGG pathway enrichment analysis
To gain a better understanding of the DEGs, GO terms and KEGG pathway enrichment of the DEGs were analyzed in R with the clusterProfiler package. In cellular components, the DEGs were particularly enriched in endoplasmic reticulum lumen and blood microparticle (Table 1). For biological processes, the DEGs were enriched in lipid homeostasis and regulation of lipid metabolic process (Table 1). For molecular functions, the DEGs were enriched in steroid hormone receptor activity, aldo-keto reductase (NADP) activity, and organic acid transmembrane transporter activity (Table 1). As shown in Figure 2, most of the significantly enriched pathways analyzed by KEGG included complement and coagulation cascades, peroxisome proliferator-activated receptor (PPAR) signaling pathway, and extracellular matrix (ECM)–receptor interaction.
Gene ontology analysis of DEGs.

Term includes the identification number of GO term; count indicates the number of genes enriched in GO terms. DEG, differentially expressed gene; CC, cell component; BP, biological process; MF, molecular function.
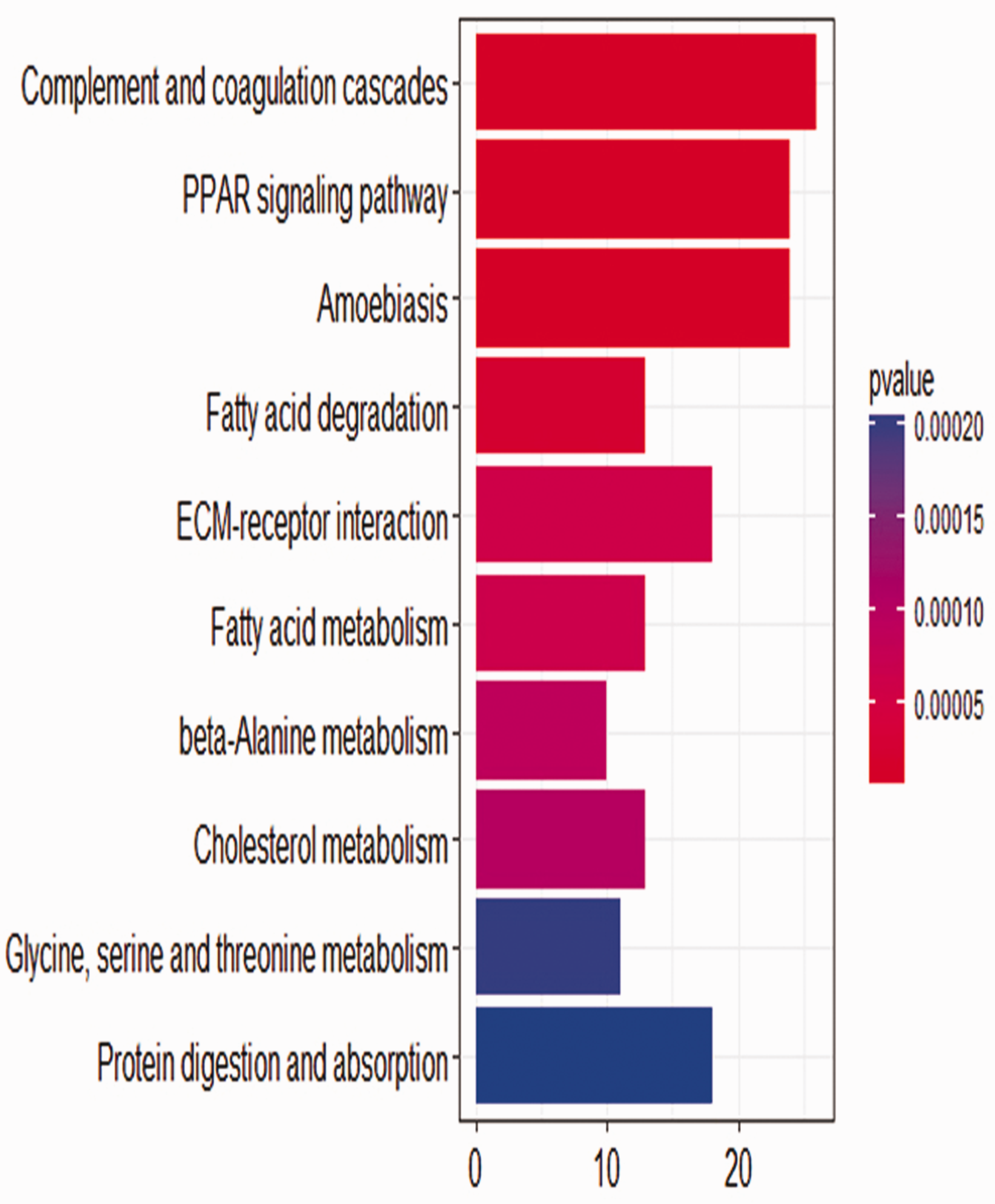
The KEGG pathway of DEGs. KEGG, Kyoto Encyclopedia of Genes and Genomes; DEG, differentially expressed gene; PPAR, peroxisome proliferator-activated receptor; ECM, extracellular matrix.
Hub genes and module screening from PPI network
Based on the information in the STRING database, PPI networks were made of DEGs having a combined score >0.4 (Figure 3). The top four modules (modules 1, 2, 3, and 4), with score >6, were detected by MCODE in Cytoscape (Figure 4). Pathway analysis of genes in each module was performed using DAVID (Table 2). The genes in modules 1 to 4 were mainly associated with chemokine signaling pathway, protein digestion and absorption, and complement and coagulation cascades. The top 20 genes with degrees of connectivity >55 were selected as hub genes; these included ALB, EGF, IL6, F2, FGF2, CDH1, PLG, AGT, APOB, FN1, HGF, TF, ALDH1A1, PTGS2, EHHADH, NTS, APOA1, EDN1, INSR, and AMBP.

Protein–protein interaction network of differentially expressed genes.

Top four modules from the PPI network. Module 1: MCODE score = 20.087; module 2: MCODE score = 11.818; module 3: MCODE score = 9.75, and module 4: MCODE score= 6.067. PPI, protein–protein interaction; MCODE, Molecular Complex Detection plug-in.
KEGG pathway analysis of DEGs in different modules.

KEGG, Kyoto Encyclopedia of Genes and Genomes; DEG, differentially expressed gene; FDR, false discovery rate.
KM plotter and expression of hub genes
Prognostic information for the 20 hub genes was freely available in http://kmplot.com/analysis/. We found that increased expression of alpha-1-microglobulin/bikunin precursor (AMBP) (hazard ratio 0.55, 95% confidence interval: 0.39–0.78; P = 7.3 × 10–4) was associated with worse OS for liver cancer patients (Figure 5).
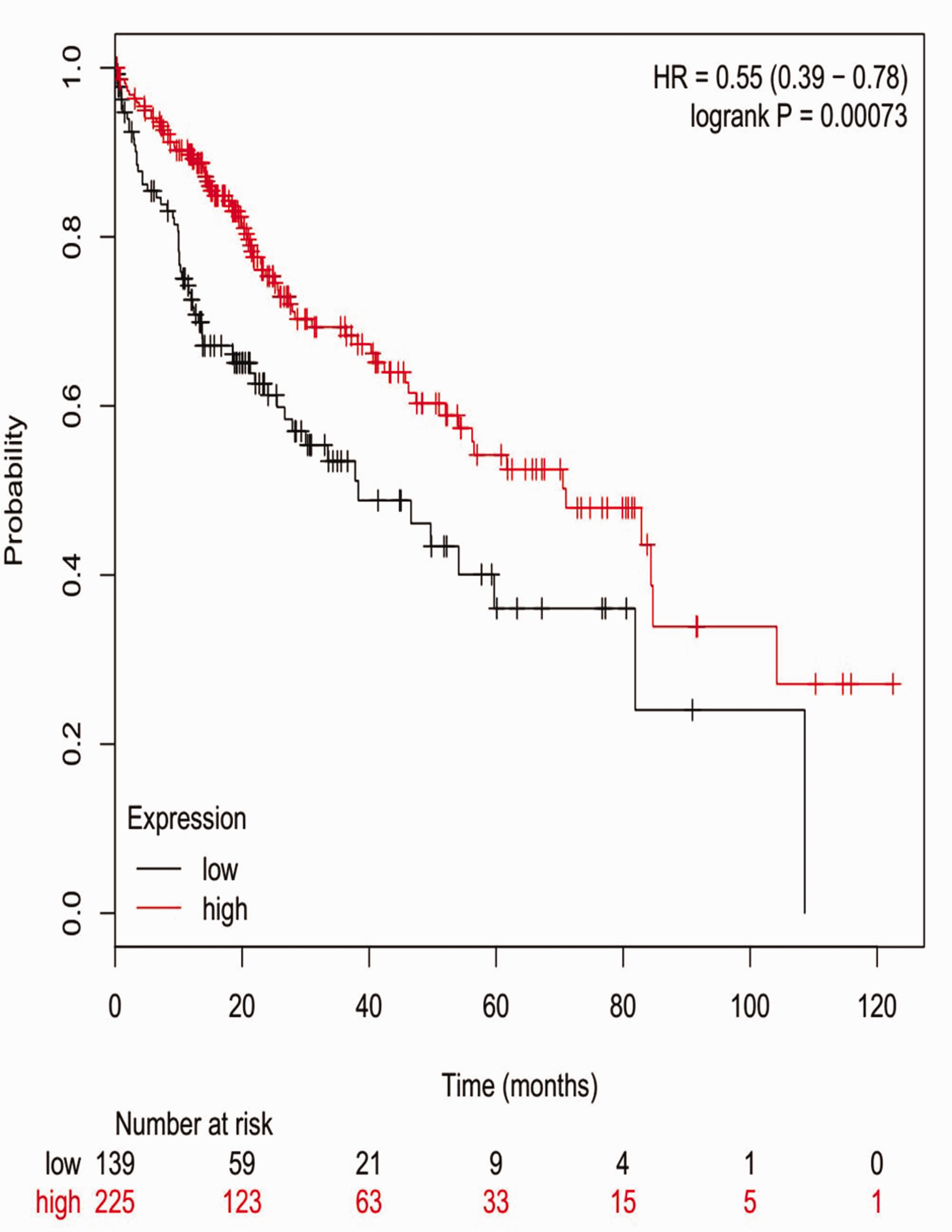
Prognostic value of expression level of AMBP. AMBP, alpha-1-microglobulin/bikunin precursor; HR, hazard ratio.
Discussion
HCC is not only one of the most common cancers worldwide, but it is also associated with high mortality due to its limited therapeutic options.13,14 Many research studies have been conducted on HCC but drugs or targeted gene therapies found to be successful in HCC cell line studies (i.e., preclinical studies) are often not useful in clinical studies.
In the present study, we analyzed dataset GSE72981 from the GEO database, which included samples from tumors of mice that were injected with HepG2 cell lines and of mice that were injected with tumor cells from patient tumor explants. We identified 1,405 DEGs, including 479 upregulated genes and 926 downregulated genes. GO analysis showed that these DEGs were mainly involved in lipid homeostasis and regulation of lipid metabolic processes, which is consistent with previous studies showing that abnormal regulation of lipid metabolism in the liver leads to HCC.15–18 The KEGG pathways of DEGs included complement and coagulation cascades, PPAR signaling pathway, and ECM–receptor interaction. Research has demonstrated that loss of liver cellular features due to reduced PPAR signaling in the early stages of HCC and PPAR-γ agonists are associated with lower risk and improved prognosis of HCC.19,20
We analyzed the PPI network of DEGs. Module analysis of the PPI network revealed that the chemokine signaling pathway was the most significant pathway in module 1, and the development of tumors in mice injected with HepG2 cells was associated with neuroactive ligand–receptor interaction, ECM–receptor interaction, and the PI3K-AKT signaling pathway. Our results were consistent with previous studies.21–23 For instance, Kanglaite, a Chinese medicine for treating HCC, was shown to reverse multidrug resistance in HCC by inducing apoptosis and cell cycle arrest via the PI3K/AKT pathway, and NLRX1 acted as a tumor suppressor in HCC by inducing apoptosis, promoting senescence, and decreasing invasiveness by repressing the PI3K-AKT signaling pathway.24–26 We identified 20 hub genes with a high degree of connectivity in the PPI network: ALB, EGF, IL6, F2, FGF2, CDH1, PLG, AGT, APOB, FN1, HGF, TF, ALDH1A1, PTGS2, EHHADH, NTS, APOA1, EDN1, INSR, and AMBP. Specifically, a negative correlation was identified between expression of AMBP and OS of liver cancer. Evidence has shown that a high level of serum AMBP is associated with a poor response to paclitaxel-capecitabine chemotherapy in patients with advanced gastric cancer. 27 AMBP has also been confirmed to be differentially expressed in seven liver cancer cell lines and 17 HCC tissues. 28 AMBP and the other DEGs may be considered novel hepatitis B virus-related HCC signature genes. 29 However, little research has been done on the function and mechanism of AMBP in HCC.
Conclusion
In this bioinformatics analysis, we identified that AMBP, as well as the chemokine signaling pathway and neuroactive ligand–receptor interactions, may be important in the development of HCC. However, the role of these pathways and AMBP in HCC remains enigmatic. Future preclinical studies in HCC using the HepG2 cell should focus on these pathways and AMBP. Our results provide further insight into HCC induced with HepG2 cells and highlight potential key genes and pathways involved in diagnosis and prognosis of HCC, as well as potential drug targets.
Footnotes
Declaration of conflicting interest
The authors declare that there is no conflict of interest.
Funding
This work was supported by the grant from Yan’an University (the Start-up fund of Yan'an University, 205040136) and Guided project of Yan’an University (205100114); the Shanxi Provincial Department of Education plan special project research (18JK0863); the Shanxi Provincial Department of Education plan special project research (18JK0869); and the Shaanxi Provincial Department of Science and Technology project (2020JQ-800). This work was also supported by Shandong Province Universities’ Development Plan for Youth Innovation Teams (2019-9-202, 201 and 2019KJK013), Scientific Innovation Team of Shandong University of Traditional Chinese Medicine, National Nature Science Foundation of China (81903948), Shandong Provincial Natural Science Foundation, China (ZR2019BH027), and Shandong Provincial University Scientific Research Project (J18KZ014).