
Abstract
Collateral blood vessels are unique, naturally occurring endogenous bypass vessels that provide alternative pathways for oxygen delivery in obstructive arterial conditions and diseases. Surprisingly however, the capacity of the collateral circulation to provide protection varies greatly among individuals, resulting in a significant fraction having poor collateral circulation in their tissues. We recently reviewed evidence that the presence of naturally-occurring polymorphisms in genes that determine the number and diameter of collaterals that form during development (ie, genetic background), is a major contributor to this variation. The purpose of this review is to summarize current understanding of the other determinants of collateral blood flow, drawing on both animal and human studies. These include the level of smooth muscle tone in collaterals, hemodynamic forces, how collaterals form during development (collaterogenesis), de novo formation of additional new collaterals during adulthood, loss of collaterals with aging and cardiovascular risk factor presence (rarefaction), and collateral remodeling (structural lumen enlargement). We also review emerging evidence that collaterals not only provide protection in ischemic conditions but may also serve a physiological function in healthy individuals. Primary focus is on studies conducted in brain, however relevant findings in other tissues are also reviewed, as are questions for future investigation.
Introduction
Collaterals are arteriole-to-arteriole anastomoses that are present in most tissues and cross-connect a fraction of the outermost branches of adjacent arterial trees.1 –3 A second type of collateral, collateral arteries, 3 are artery-to-artery anastomoses that provide alternative routes around large structures, eg, elbows, knees, and base of the brain. When large-artery obstruction occurs, for example in acute ischemic stroke (AIS), the major determinants of the severity of tissue injury are the location and duration of occlusion, sensitivity of the tissue to hypoxia/ischemia, and amount of collateral blood flow. The primary determinants of the latter are the number and diameter (ie, “extent”) of the native, pre-existing collaterals present prior to obstruction. In the absence of spontaneous or therapeutic recanalization, they provide the sole source of perfusion. Collaterals also confer protection in slowly-developing athero-stenotic diseases.
Despite their unique properties3 –7 and clinical importance,1 –21 much less is known about the basic biology of collaterals, compared to blood vessels of the general arterial-capillary-venous circulation. Recently however, they have become a focus of both basic and clinical investigation.3 –21 This extends in part from findings that collateral score and other imaging-based estimations of collateral flow within an individual’s tissues varies widely among subjects, for example following AIS where it correlates inversely with early core size, infarct expansion, risk of hemorrhagic transformation, early neurological deterioration, and directly with functional outcome and efficacy of thrombolysis and thrombectomy.8 –18 The variation is such that a large percentage of humans and other mammalian species exhibit significant deficiency in collateral blood flow.8 –20
We recently reviewed studies showing that the primary cause of this deficiency, at least in mice, is the presence of naturally occurring deleterious polymorphisms in genes and genetic loci important in the signaling pathway that drives collaterogenesis—a unique angiogenic process that occurs during development. 22 The result, in those who harbor such variants, is reduced or sparse-to-absent collaterals in their brain, heart and other tissues. Also reviewed were studies examining whether sexual dimorphism contributes to differences in collateral abundance, genetic-dependent variation in collaterals in other species including those that have evolved at high altitude, and recently-developed genetic mouse models of sparse, intermediate, and abundant collateral extent in their tissues.
Besides genetic background, a number of other factors contribute to variation in collateral blood flow (Figure 1). The purpose of this review is to summarize current understanding of these additional determinants, drawing on animal and humans studies published primarily in the last fifteen years. Particular attention is given to collaterals in brain and also, where reports are available, other tissues since such comparisons strengthen conclusions. We also review evidence supporting the recent hypothesis22 –25 that collaterals may serve a physiological function in the absence of arterial obstruction. Details of many of the studies are provided to aid in assessing the strength of the findings and conclusions, and questions for future investigation are also discussed.
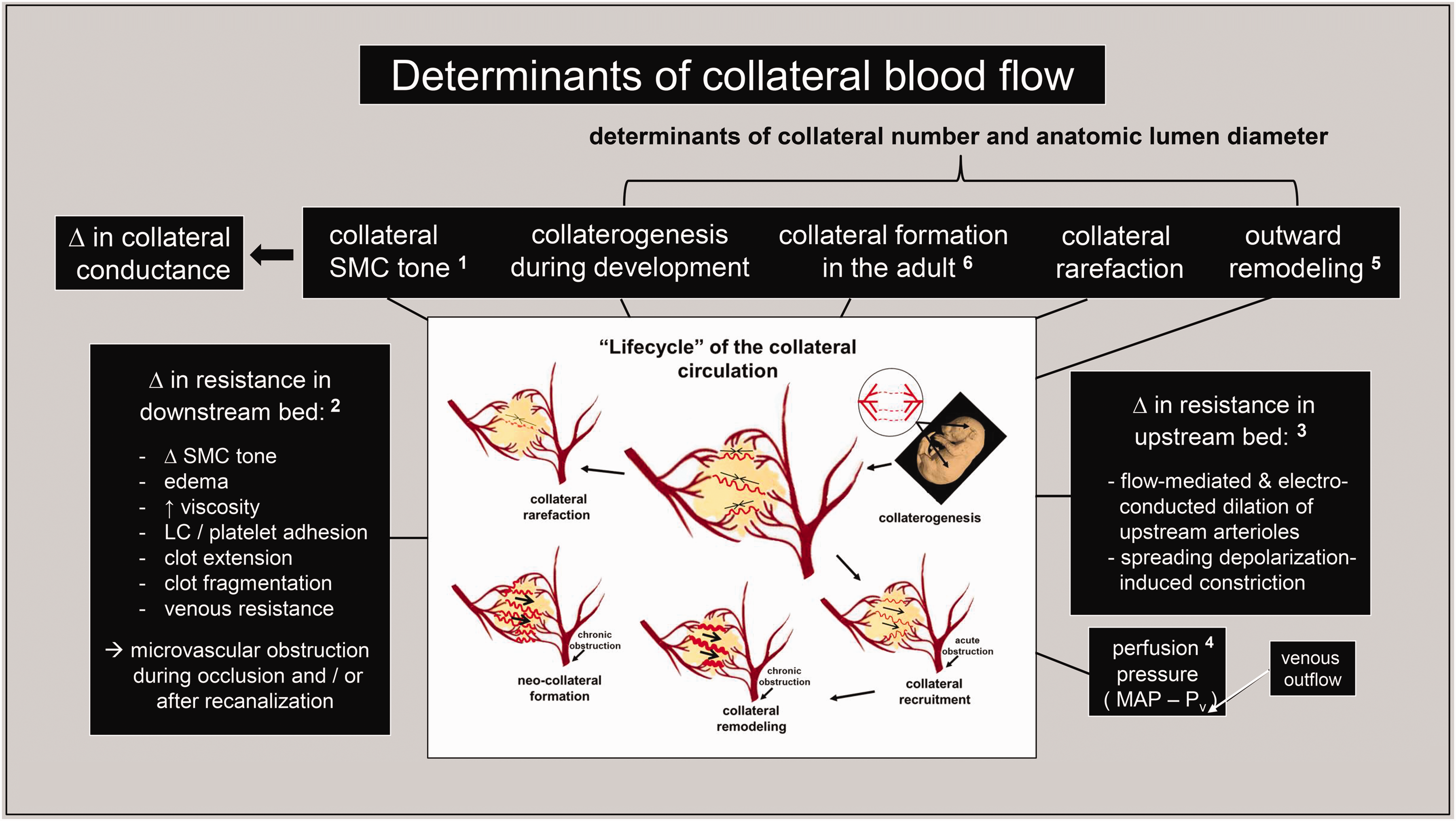
Collateral vascular smooth muscle: basal tone, minimal myogenic reactivity, no innervation
Reports vary regarding collateral tone at baseline. For example, Jiang and coworkers 26 observed in C57BL/6 (B6) mice in a CSF-suffused open window preparation that M2-MCA ligation was followed immediately by similar 30–45% increases in diameter of pial collaterals and nearby terminal arterioles (TAs). However, interpretation vis-à-vis the amount of tone present prior to occlusion is complicated because, in addition to the effect of anesthesia and surgery—and although the suffusate’s reservoir was equilibrated with 95% N2 and 5% CO2—PO2 within the window (which was not measured) may have been elevated above in vivo levels, resulting in SMC contraction. Beard et al27,28 found that collateral diameter increased 14% shortly after MCA filament occlusion in rats with acutely implanted closed cranial windows. However, filament presence causes large hemispheric infarctions from occlusion of lenticulostriate arteries, which can lead to edema-induced extravascular compression and thus complicate conclusions about tone. Filament presence also obstructs orthograde flow in the ipsilateral A1-ACA and anterior choroidal artery, 29 resulting—depending on anatomic status of the ACom—in reduced pressure in the ACA tree and thus ACA→MCA collateral flow. Status of the ACom (ie, its presence and conformation) may, to judge from what is seen for PCom status, evidence variation among individuals, even those with identical genetic background. 30 Ma and colleagues 31 reported that pial collateral diameter decreased ∼5% after bilateral carotid occlusion plus distal M1-MCA ligation in rats with acute closed cranial windows. However, inflammation induced by window emplacement can alter SMC contraction, and ACom/PCom variation are also a factor with this model of AIS. Recently, using methods that better preserve physiological conditions, Binder and coworkers 32 found in B6 mice with windows implanted 1–2 weeks earlier that pial collateral diameter increased 50% when examined 30 minutes after obstruction of the proximal M2-MCA by intravascular microinjection of thrombin. This suggests that collaterals have considerable SMC tone at baseline.
Chan et al 33 reported that pial collaterals, isolated in vitro from 4.5 months-old rats and pressurized to in vivo values without flow, showed little myogenic tone (∼8%) compared to terminal arterioles (TAs, ∼40%). Notably, no myogenic constriction occurred on graded increases in pressure, in distinction to its expected occurrence seen in TAs. Collaterals from 12 months-old rats (∼33 human-year-equivalence, per the 2–3.5 y life-span of Wistar rats 34 ) had increased myogenic tone at baseline (∼30%) but remained unresponsive to increases in pressure. Pharmacological inhibition of eNOS caused greater constriction of collaterals than TAs (∼24% versus 10%), and collaterals trended toward more robust dilation to a nitric oxide (NO) mimetic. Interestingly, collaterals isolated from rats with hypertension evidenced myogenic tone at baseline similar to TAs (both ∼50%) 33 that was linked to renin-angiotensin mechanisms, 5 and constriction induced by pharmacological inhibition of eNOS was similar for collaterals and TAs from hypertensive rats.
If the above findings predict in vivo they suggest that, compared to TAs: collateral tone at baseline has greater dependence on eNOS/NO, collaterals from young-adult mice have little or no myogenic reactivity, and the latter becomes evident with early middle-age or hypertension. Acquisition of myogenic reactivity would be expected to impair, whereas absence of it and greater dependence on eNOS/NO would aid, the protective functions of collaterals during arterial obstruction (discussed below). These findings are consistent with other evidence that collaterals have unique features: 4 Minimal myogenic responsiveness is favored by PaO2 being predictably low in the watershed regions where collaterals reside;23,35,36 the latter promotes SMC relaxation and may underlie the higher levels of expression of KLF2/KL4-driven eNOS/NO in collaterals compared to TAs. 4 This is supported by the aforementioned observation 33 that constriction in response to inhibition of eNOS was greater for pial collaterals than TAs. In vitro studies from the Cipolla group 37 also demonstrated that collaterals, like arterioles and arteries, evidence flow-mediated dilation (FMD) that involves shear-sensitive ion channel→eNOS/NO signaling and other mechanisms. Interestingly, FMD of collaterals was impaired by hypertension and restored by inhibitors of reactive oxygen species.
Also in distinction to pial arteries and arterioles, collaterals lack innervation and do not respond to norepinephrine in vitro (although they constrict on depolarization or calcium influx; whether VSM cells of collaterals and distal pial arterioles share similar receptor profiles is not known).5,33 These additional specializations may prevent collaterals from constricting during sympatho-excitation, which would otherwise compromise their functions to: 1) maintain patency and perfusion of arterioles that branch from them, ie, “penetrating arterioles” (PAs) in the case of pial collaterals, despite their low and disturbed blood flow at baseline,4,27,28 2) dilate and thus provide unrestrained retrograde perfusion if arterial obstruction occurs downstream from the collateral network, and 3) physiologically, to help optimize local blood flow to meet local oxygen demand with changes in regional neuronal activity (neurovascular coupling38 –40), as was recently proposed (see section ‘Collaterals may serve a physiological function’).22 –25 Regarding the former two functions, reduced myogenic responsiveness would ensure that increased circumferential wall stress in collaterals caused by their convergent flow at baseline,4,41 –43 wherein kinetic energy is converted to potential energy—a unique situation within the arterial circulation—elicits no or minimal myogenic constriction. And that, upon arterial obstruction (eg, following MCAO) the following sequence also would not elicit myogenic constriction: obstruction→pressure drop→autoregulatory dilation of the dependent tree→recruitment of collateral flow→inhibition of tone by FMD (via NO and, potentially, other shear-sensitive dilator signals) in the collaterals and their anastomosed distal arterioles within the ACA and PCA trees→little/no myogenic constriction to any increase in collateral intralumenal pressure→maintained collateral dilation→unimpeded collateral flow.
Perfusion pressure and upstream and downstream determinants of collateral blood flow
The topics below will be discussed briefly, given space limitations and the extensive available literature.
Perfusion pressure
Basic and clinical studies have examined the effect of manipulating arterial pressure using pharmacologic agents, orthostatic interventions, and vascular mechanical devices on CBF, collateral perfusion, and functional outcome in AIS.44
–49 For example, Li and colleagues
49
reported that prehospital in-ambulance reduction of blood pressure to 130–140 mmHg associated with reduced risk of poor outcome (90-day mRS) among patients with hemorrhagic stroke (OR 0.75). The opposite was observed with AIS (OR 1.30). Studies such as this that test treatments capable of increasing collateral flow in AIS stroke before thrombectomy—are informative because perfusion of the territory at risk is solely dependent on collateral flow if arterial obstruction is complete. Venous outflow and pressure also affect collateral flow, as expected from hemodynamic considerations.
50
Upstream resistance
Flow-mediated dilation is mediated by multiple mechanosensors of fluid shear stress and their effector pathways that reside primarily in endothelial cells (ECs) but also in SMCs.37,54 The sensors signal predominantly through eNOS/NO, as well as via other effectors including mechanical and electrical cell-cell coupling of ECs and SMCs. The latter can proceed both retrograde and orthograde to promote up- and down-stream dilation. Although in vivo study of FMD of collaterals, per se, is complicated by the difficulty in controlling interacting factors, it is likely that FMD mitigates basal tone in collaterals and any subsequent myogenic constriction following acute obstruction. Also, metabolic and myogenic dilation within the dependent tree would, by reducing downstream resistance seen by the anastomosed terminal arterioles of the unobstructed trees (eg, ACA and PCA trees), cause them to also undergo FMD and contribute to increased collateral perfusion pressure and flow to the obstructed tree. Support comes from findings in B6 mice that distal arterioles of the ACA and PCA trees imaged by open cranial window dilated 32%,
26
and that blood flow increased in the ACA and PCA territories imaged by closed cranial window immediately after M2-MCAO.
32
Also, ACA flow (ultrasound) and infarct size correlated, directly and indirectly respectively, with collateral score in patients with AIS.55,56 Aging, hypertension, diabetes, and other cardiovascular risk factors (CVRFs) are known to impair FMD of arteries and arterioles, in large part through reduced activity of eNOS/NO.37,39,40,54
Downstream resistance
Unlike upstream mechanisms, which predictably play a smaller role in collateral blood flow post-occlusion than collateral extent and downstream mechanisms, factors affecting the latter have received significant investigation.5,32,39,40,44,45 When occlusion occurs, SMC tone is reduced in the dependent tree by metabolic and myogenic mechanisms, resulting in rapid dilation and induction of retrograde flow across the collateral network. Various factors can oppose or restrict this downstream autoregulatory response and/or progress, with time, to increase downstream resistance. Prominent among them are: release of autacoid constrictors from activated platelets, leukocytes, and glial cells caused by ischemia or other conditions that oppose dilators that are also released from the latter two cell types;58
–60 other vasoconstrictor mechanisms,
5
extravasation of RBCs at micro-hemorrhagic sites that elicits hemoglobin-induced constriction;
61
increased permeability of the endothelium leading to edema, reduction in transmural pressure, and thus extravascular compression;39,40,62,63 increased intracranial pressure that also promotes the latter;27,28 increased viscosity that accompanies altered blood rheology caused by increased capillary filtration, low velocity of blood flow, and other mechanisms;64,65 and promotion of platelet and leukocyte adhesion caused by ischemia and reduced velocity.66
–68 As well, clot extension and fragmentation can obstruct branches downstream from the initial obstruction.
69
Importantly, induction or exacerbation of many of the above factors/mechanisms can occur following recanalization (ie, ischemia-reperfusion injury), including generation of reactive oxygen species and release of platelet/leukocyte inflammatory and vasoconstriction mediators,39,40,66
–68 leading to reduced collateral flow (“collateral failure”). Venous resistance is also capable of influencing collateral flow.50,70
The latter question has recently begun to be addressed: Binder and coworkers 32 subjected collateral-rich B6 mice, B6.Rabep2–/− (knockout) mice with intermediate collaterals, and collateral-poor BALB/c mice 72 to a thrombin-based model of MCAO followed by fibrinolytic treatment. B6 mice evidenced good cerebral autoregulation, gradual reperfusion on thrombolysis, and smaller infarctions. BALB/c mice experienced collapse of distal arterial segments and, on recanalization, deleterious hyperemia and hemorrhage, and high mortality. The authors extended these findings to stroke patients undergoing thrombectomy. Those with poor collaterals evidenced rapid reperfusion, increased incidence of hemorrhagic transformation, and poor recovery. Kanoke et al 51 also used segmental vascular imaging of B6 and BALB/c mice subjected to a different model of MCAO to provide additional understanding. Kemps et al 73 reported that low-frequency electromagnetic stimulation, which augmented Akt-NO signaling, reduced infarct volume following permanent MCA occlusion (pMCAO) in B6 but not BALB/c mice, suggesting involvement of increased collateral flow induced by one or more of the mechanisms shown in Figure 1. Saver et al 74 found that sphenopalatine ganglion stimulation increased CBF and NIHSS on day-7 in patients with large vessel occlusion (LVO) AIS within 24 h of onset who did not receive recanalization therapies—effects which may result from dilation downstream and/or upstream of the collateral network (and collateral remodeling, see section ‘Collateral remodeling: Arterial occlusion or sustained hypoxemia induce lumen enlargement’).
Cipolla and colleagues5,75 found that a carboxyhemoglobin-based compound, Sanguinate, that facilitates delivery of oxygen to ischemic tissue and inhibits constriction of pial arteries during MCAO, did not increase core perfusion when given 30 minutes after filament occlusion but did so during reperfusion begun at 2 hours, and reduced infarct volume. Although uncertainty exists regarding mechanism (ie, Sanguinate increased arterial pressure; and measurement of flow in a region of the outer MCA tree does not differentiate orthograde MCA flow from retrograde collateral flow during reperfusion), these findings and other supportive studies have led to a phase I trial examining Sanguinate in LVO-AIS. Chan et al 76 reported that administration of an inhibitor of the fibrinolysis inhibitor, PAI-1, that was shown to elicit NO-dependent dilation of pial collaterals studied in vitro, increased flow in the outer MCA tree during reperfusion after filament removal, and decreased infarct volume. Similar results were obtained with rapamycin although infarct volume was unaffected. 77 Other agents that may augment collateral flow by effects upstream, downstream, and/or on the collaterals themselves, have been investigated, including NO donors, NO-independent dilators, sphenopalantine ganglion stimulation, remote ischemic conditioning, and low-dose aspirin.5,45,78,79
Collaterogenesis—formation of collaterals during development
Previous studies had shown that the extent of watershed collaterals in brain, heart, lower extremities, and other tissues vary substantially in different strains of inbred mice and cohorts with mixed genetic backgrounds (the latter modeling “outbred” humans) (Figures 2 and 3).22,42,72,80 –85 These findings led to the suggestion 80 that naturally occurring polymorphisms govern the vigor of collaterogenesis and hence contribute to the wide variation in collateral flow seen following acute arterial occlusion in humans and other species (Figure 3).19,20,22,25,80,85,86 However at the time, 80 when and how collaterals form had not yet been investigated although it had been speculated upon. 87 This may have been a consequence of their small number, compared to similarly-sized distal arterioles (0.01–0.001% in murine brain and skeletal muscle80,81,88), and difficulty in distinguishing them from other vessels in thick tissues like heart and lower extremities whose arterial and venous trees are arranged at depth in three-dimensions. A way forward was provided by recognition that the collateral network overlying the lisencephalic rodent neocortex is arranged in two dimensions and can be readily imaged. And that the underlying neocortex lacked collaterals between its penetrating arteriole (PA) trees,42,82,89 indicating that quantifying collateral extent would not be confounded by the presence of an inaccessible fraction. Following on this, Faber and colleagues42,90 –93 found that formation of pial collaterals, denoted “developmental collaterogenesis” to distinguish it from formation of additional new collaterals in adults (see section ‘De novo formation of collaterals after arterial occlusion or sustained hypoxemia’), begins in mice on embryonic day-14.5 (E14.5; birth occurs after 20–21 days gestation) by a novel arteriolar sprouting process involving a number of well-known angiogenic signaling proteins (eg, Phd2, Hif1α, VEGF-A, Flk1, Notch1) as well as novel ones uniquely required for collaterogenesis (eg, Rabep2):22,72,85 After formation of the MCA, ACA, PCA and venous trees and intervening capillary beds (the trees have not yet acquired SMCs), arterial-fated ephrin-B2-expressing ECs bud off from a small fraction of the distal-most arterioles abutting the watershed regions (Figure 4).42,92 This is followed by EC proliferation and formation of EC cords composed of a migrating filapodial-led tip cell and trailing stalk cells. The cords then cross-connect randomly to a fraction of the arterioles in the adjacent trees and undergo lumenization. This process, which was confirmed and extended recently, 94 is complete by ∼E18.5-to-postnatal day-1 (P1), occurs within the subarachnoid space, and is accompanied by attachment of the nascent collaterals to the underlying pia.42,92 The robustness of collaterogenesis depends greatly on genetic background. For example, BALB/c mice have, by post-natal day-1 (P1), formed many fewer collaterals than B6 mice (Figure 5). 42 This prominent dependence on genetic background is not shared by the angiogenesis program governing formation of the general arterial-capillary-venous vasculature, which is comparable in B6, BALB/c, and other strains of mice.42,80,81,92 While BALB/c and B6 evidence subtle differences in branch patterning during development, the latter do not associate with their difference in collaterogenesis.42,92 Consistent with this, BALB/c mice exhibit no deficits in capillary density, 92 cardiovascular performance, or reproductive fitness compared to B6 mice.
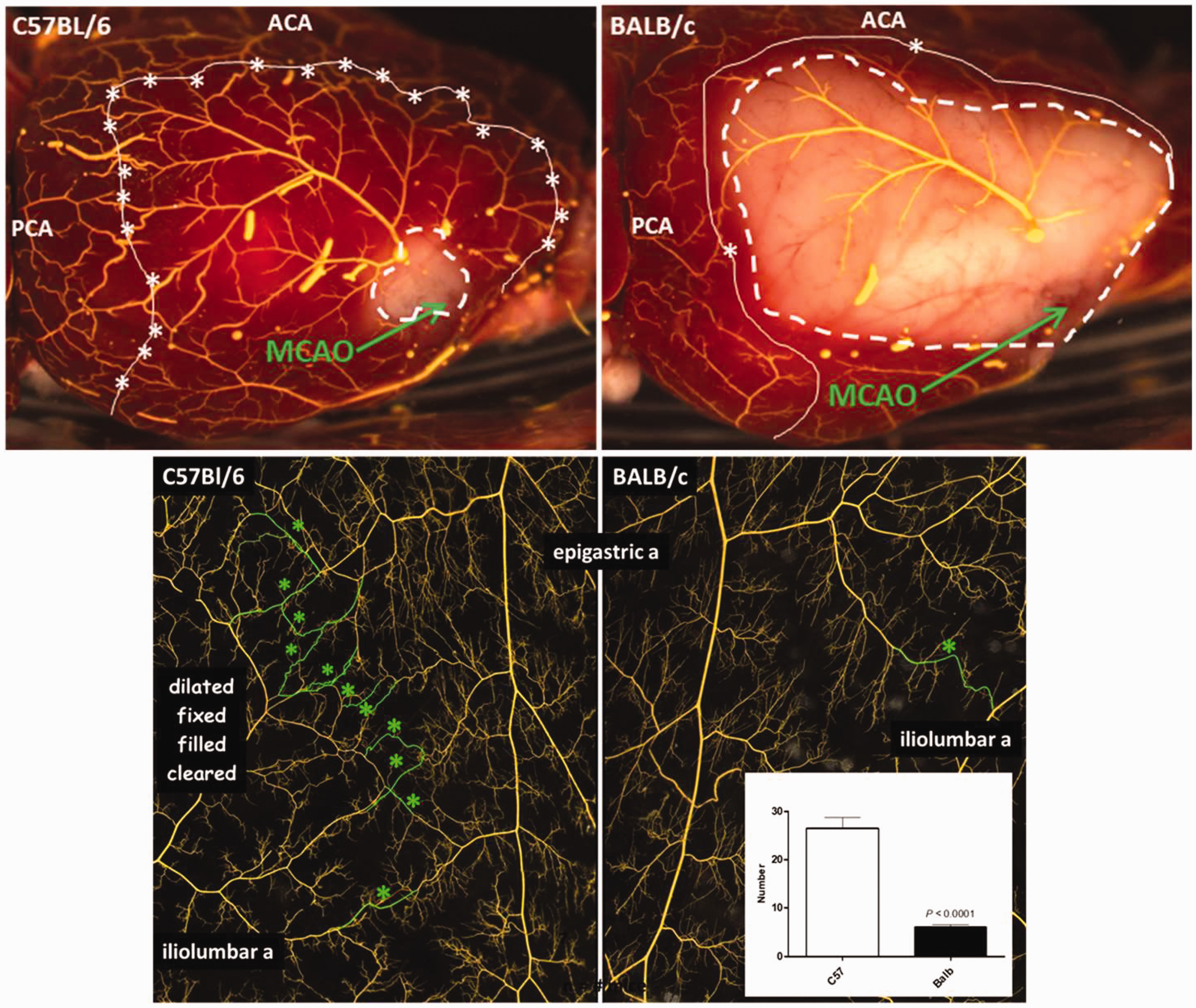
Genetic-dependent differences in collateral number (and diameter) in brain reflect similar differences in other tissues of the same strain/individual. Stars, collaterals within the watershed regions in brain (white line in upper panels) and skeletal muscle (lower panels). Upper panels: MCA tree was back-filled via the collaterals after the vasculature was maximally dilated, fixed, and filled with latex 24 h following distal M1-MCA occlusion (MCAO), followed by staining with 2,3,5-triphenyltetrazolium chloride. Adjustment of filling pressure until occasional short, large-diameter, latex-filled venous segments become evident aids confirmation of complete filling of the precapillary vasculature and identification of all collaterals. Dashed lines delineate ∼infarct limits. From literature 23 with permission. Lower panels: Collaterals pseudo-colored green; insert, average collateral number per strain in ROI shown (n = 7, 9, respectively, p < 0.0001); from literature 88 , modified.
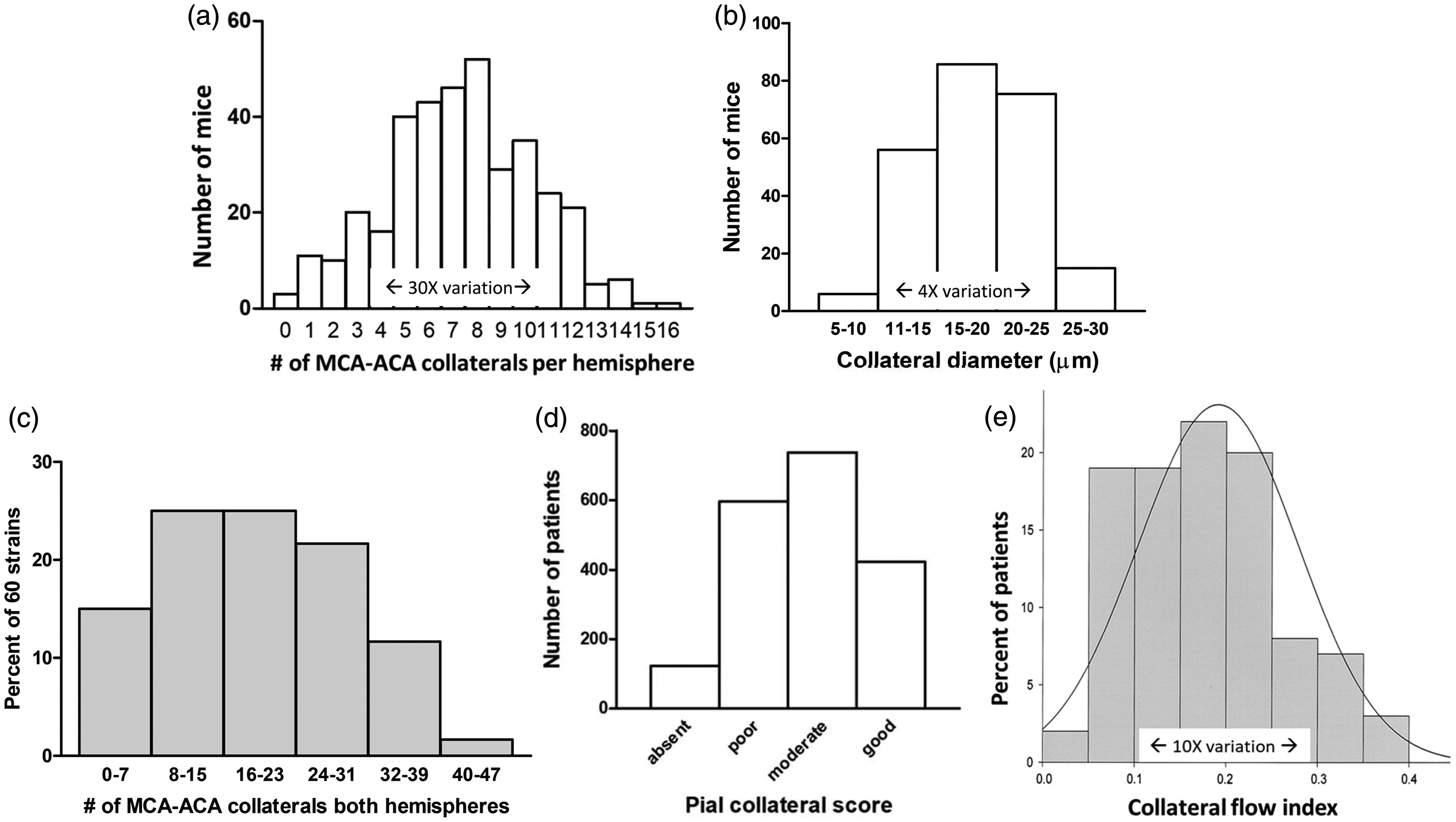
Collaterals vary widely in mice and humans due (in mice) to differences in genetic background. (a), (b) C57BL/6 x BALB/cBy (n = 243) and SWR x SJL (n = 120) F2 generation adult mice (363 total, each with different genetic background per meiotic recombination) have large differences in collateral extent; unpublished data from Refs 83, 84. (c) Pial collateral number among 60 strains of Collaborative Cross mice (number and diameter varied by 47- and 3.4-fold); from literature 85 with permission. (d) Baseline pial collateral score on single phase CTA in 1988 patients with acute LVO-AIS; data from literature 13 , 36% have poor-to-absent collaterals, and (e) Coronary collateral flow index (Pwedge- CVP/Paorta – CVP) in 100 healthy humans, ie, without coronary artery disease; from literature. 86
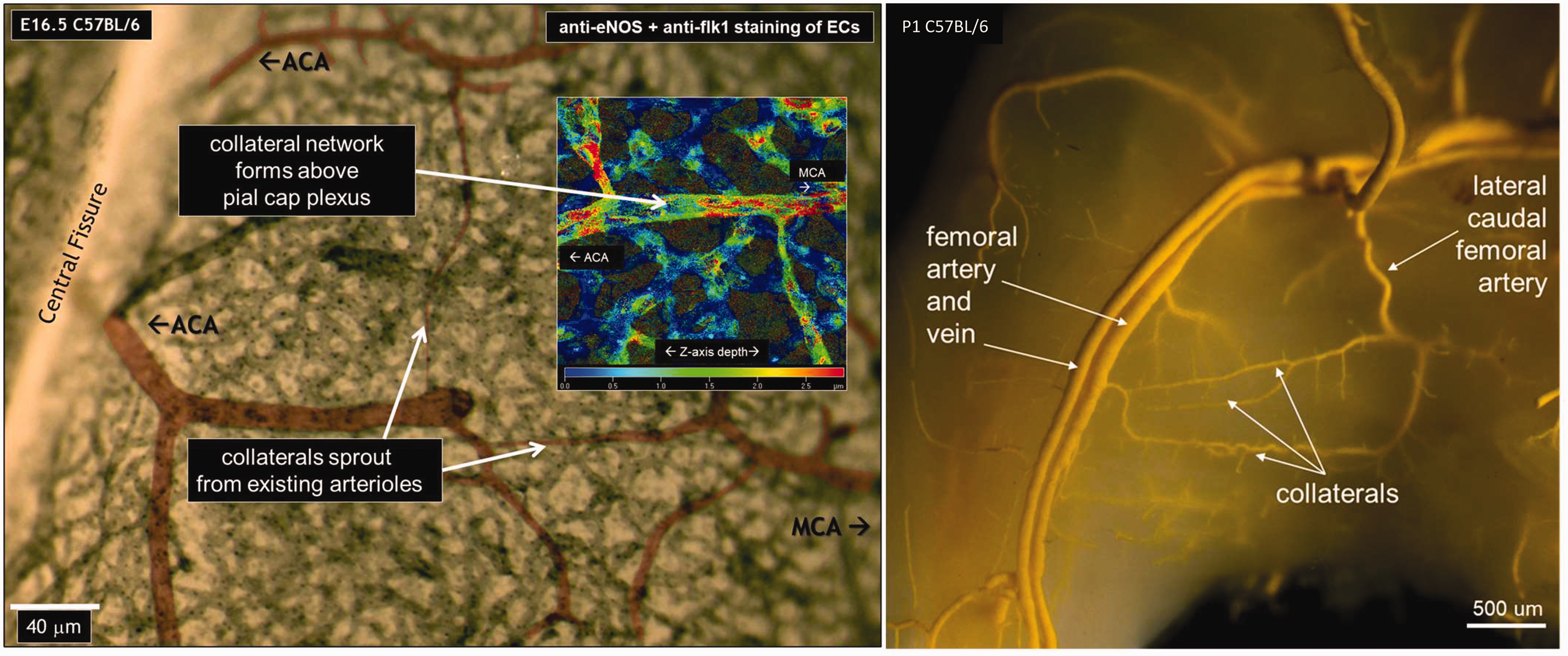
Pial collateral formation at embryonic day-16.5 (left), and presence on postnatal day-1 of fully formed large-diameter collaterals in anterior and posterior gracilis muscles (right). Left, Insert, confocal image showing that nascent collaterals form in the subarachnoid space above the capillary plexus in the embryonic pia (nascent collaterals lack tortuosity 4 ). MCA and ACA branches were artificially colorized. From literature. 92 Right, filling as described in Figure 2, with viscosity of latex and perfusion pressure adjusted to fill only large arteries and veins, followed by partial optical clearing; from literature 96 with permission.
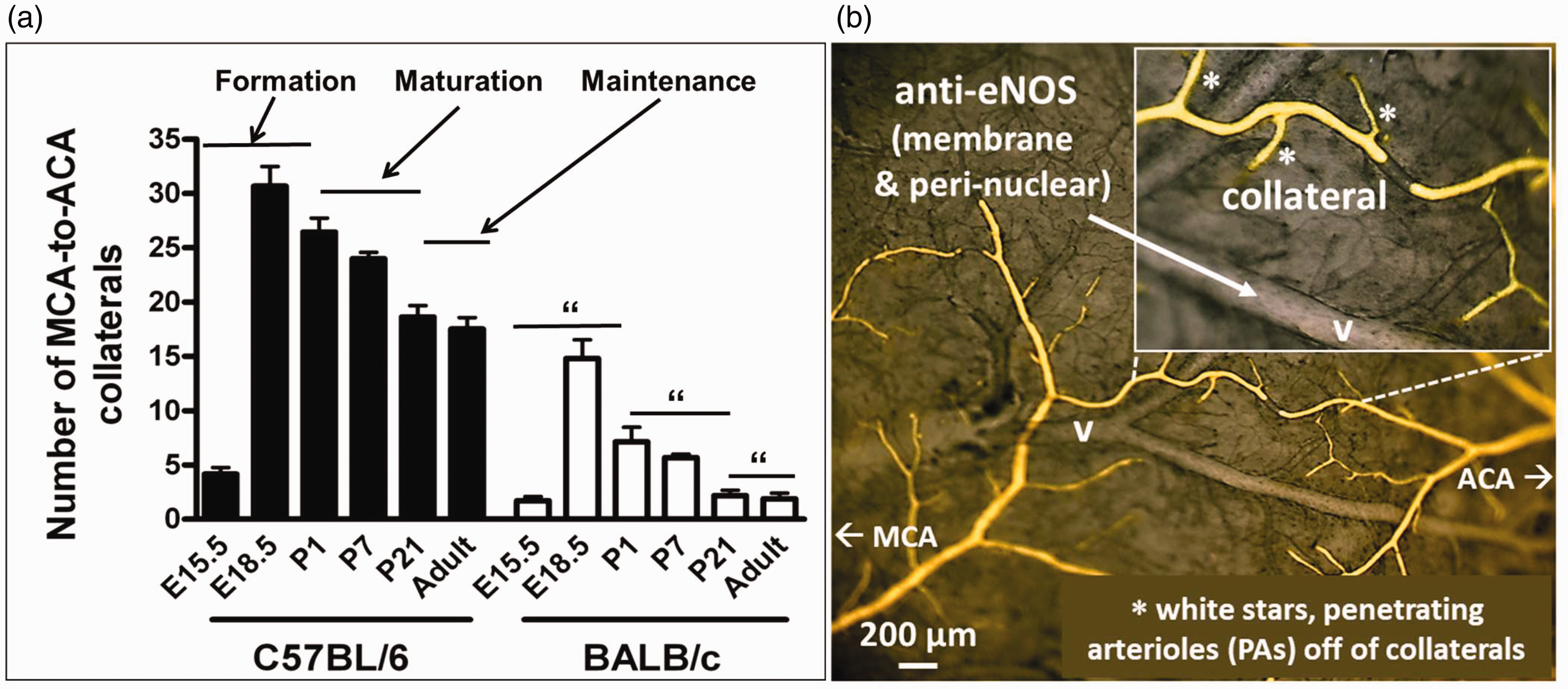
Three phases of collateral blood vessels (panel a). Formation occurs late during gestation (E, embryonic days; mouse gestation is 20–21d) and varies with genetic background. During the first 21–28 postnatal (P) days, a fraction of the nascent collaterals are pruned away, and the lumen diameter and wall thickness (data not shown) of those that remain enlarge to establish the number and anatomic diameter that are present in the 10–12 week-old young adult. During the maintenance phase, collaterals are susceptible to rarefaction caused by aging and cardiovascular risk factor presence; data from literature. 42 (b) Filling as described in Figure 2. On average, 3.2 PAs branch from each collateral irrespective of mouse strain; 81 anti-eNOS staining (gray color), unpublished figure.
Formation of watershed collaterals during development has not been studied in other tissues or species. However, Ramo and colleagues 95 examined the subcutaneous collateral located on the medial surface of the anterior and posterior gracilis muscles of mouse hindlimb. These collaterals are different from watershed collaterals: Their location, number (usually one per muscle), and lumen diameter (∼30 microns) do not differ with genetic background.80,81,88,90 They form by a different process that appears to involve remodeling of a vessel(s) of the embryonic capillary plexus into a artery-like anastomosis that is present by P1 and courses with a peripheral nerve, an arrangement that is preserved in the adult. 95 Faber et al 96 had similarly noted that, unlike watershed collaterals located at depth between the arterial trees within the underlying adductor musculature, 42 these large-diameter gracilis collaterals are fully developed by P1 (Figure 4). The above findings suggest that their formation may align more with “collateral arteries” such as those that exist around joints, within the circle of Willis (CoW), and elsewhere.3,22
Following collaterogenesis, newly formed pial collaterals undergo a process of maturation (Figure 5):42,90 –93 A fraction are pruned away and those that remain undergo lumen enlargement and continued investment with SMCs to reach, by P21-P28, the number, diameter, and wall thickness that will be present in the young-adult (10 weeks-old) (Figure 2), findings confirmed by others. 97 It is not known whether muralization involves migration of SMCs from the anastomosed terminal arterioles or differentiation of mesenchymal cells adjacent to the EC cords into SMCs. While maturation was comparable in B6 and BALB/c mice, with respect to percent pruning and increase in diameter (Figure 5), it varies with genetic background though modestly compared to collaterogenesis (H Zhang, JE Faber, unpublished data analysis): Collateral number at P1, P21, and in 10 months-old (middle-aged) adults were, for B6: 26.2, 17.8, 16.7 (36% decrease by adulthood), BALB/c: 6.8, 2.3, 2.0 (71% decrease), 42 CD1: 18.5, 18.0, 17.4 (5% decrease), 91 different CD1 sub-strain: 15.0, 10.0, 10.5 (30% decrease) 90 ; for lumen diameter (microns), B6: 14.2, 23.7, 20.0 (41% increase), BALB/c: 11.8, 19.8, 10.8 (9% decrease), CD1: 13.0, 17.5, 13.0 (no change; diameter was not obtained in the “different” CD1 sub-strain type).
The above findings, together with the fact that collateral number varies 8- and 14-fold more than diameter in mice with different genetic backgrounds and across 60 strains of adult mice with highly admixed genomes (Figure 3), indicate that genetic-dependent variation in collaterogenesis has a much greater impact on collateral extent than variation in collateral maturation. The signaling mechanisms that direct collaterogenesis are beginning to be understood, however much less is known about the determinants of collateral maturation. While shear and circumferential wall stress-dependent mechanisms likely dominate, maturation is affected by differences in the levels of VEGF-A, Dll4, Clic4, NFkB, and EphA4. 22 Whether they are similar to those that govern maturation of arteries/arterioles98,99 awaits investigation. The above studies provide a start towards understanding the processes of collaterogenesis and collateral maturation. 100
It is known that pial collaterals form in humans much earlier than in mice. This is expected given the considerably longer time for in utero development in humans. However, formation follows a similar sequence: the pial collateral network is established by 14 weeks gestation well after the cerebral artery trees have formed.see 101,102 Formation of the PCom collateral arteries of the CoW also occurs earlier, followed by substantial re-structuring as cerebral growth proceeds.see 101,102
Changes in collateral extent in adulthood
Due to space limitations, see studies cited below for additional information and references.
De novo formation of collaterals after arterial occlusion or sustained hypoxemia
It has long been assumed that collaterals cannot form in adulthood. Recent findings have challenged this assumption. Zhang et al found that additional collaterals form, de novo, following pMCAO in adult mice.23,82 This process was termed “neo-collateral formation” (NCF) to distinguish it from collateral formation during development. Collateral number increased 2-to-4-fold after pMCAO among strains with sparse native collaterals (eg, BALB/cBy; confirmed by Iwasawa et al 103 ) but not strains with abundant collaterals (eg, B6). 82 The same occurred in individuals with low-collateral number in a population of 162 B6 × BALB/cBy F2 genome-admixed mice wherein number varied by 30-fold. 23 Although B6.Rabep2−/− mice have reduced collateral number NCF failed to occur, indicating that Rabep2 is required for NCF. 23 This is in agreement with the major effect of functional alleles of Rabep2 in collaterogenesis in B6 and, by genetic inference, 20 other classical strains. 72 The authors proposed 23 that NCF occurs in collateral-poor strains because their watersheds are adjacent to their large evolving hypoxic infarctions, whereas the small infarctions of collateral-rich strains are confined proximal to the site of obstruction, well away from where collaterals reside (Figure 2), ie, that low oxygen is a primary stimulus for NCF post-stroke.
An additional factor contributing to reduced oxygen in watersheds of low-collateral mice was also proposed:
23
Besides providing alternative routes for perfusion, collaterals serve a “scaffolding” function, ie, on average, 2.3 penetrating arterioles (PAs, Figure 5) were found to branch from each collateral among 15 strains examined, irrespective of low versus intermediate versus high native collateral number.
82
Thus, strains with fewer collaterals have fewer PAs branching to the underlying watershed parenchyma. And therefore a larger percentage of flow to this region proceeds by a longer, less hemodynamically efficient route, ie, via the PA trees (that descend adjacent to the watersheds) and their interconnected capillary plexi within watershed brain. Consequentially, more oxygen is lost by pre-capillary diffusion,
104
and thus, the watershed parenchyma in low-collateral strains is at risk for greater reduction in oxygen if arterial narrowing or obstruction occur or overall CBF is compromised. Also, the to-and-fro flow present in collaterals at baseline, which results in little-to-no net flow across the collateral network,4,41
–43 favors unloading of oxygen from the collaterals’ erythrocytes, which further contributes to low watershed oxygen.35,36 The above factors may underly the susceptibility of watershed brain to strokes caused by sustained periods of low CBF.35,36,105 Consistent with the above findings in mice, Wang and coworkers
106
reported that NCF occurred after filament-induced pMCAO (30% increase in number of ACA/PCA→collaterals) in Sprague-Dawley rats (low collateral strain—∼10 ACA/PCA collaterals per hemisphere at baseline). This was enhanced and infarct volume and functional deficits reduced when bone marrow-derived mononuclear cells were administered 24 h after occlusion, in association with their localization to the border zone and incorporation into the blood vessel wall.
Formation of additional collaterals also occurs in other tissues. Zhang et al 107 observed that NCF began in B6 mice 1–2 days after ligation of the left coronary artery at midpoint, with ∼10 averaging ∼18 microns diameter having formed by day-7. Fewer, smaller-diameter collaterals formed in three other strains, confirming previous findings23,82 that NCF varies with genetic background. Collateral network conductance, infarct volume−1, and recovery of pump function followed rank-order for number of collaterals formed among the strains. 107 Gene targeting, immunohistochemistry, reporter-gene imaging, and bone marrow transfer identified involvement of myeloid cells, MCP1→CCR2, and fractalkine→CX3CR1 signaling. NCF also occurred after coronary ligation in: neonatal mouse heart, with involvement of CXCL12→CXCR4;108,109 adult mouse hindlimb with involvement of TRPC6; 110 zebrafish embryo skeletal muscle with involvement of CXCR4, myeloid cells, and nitric oxide;111,112 chick embryo yolk sac; 113 and adult mouse skeletal muscle.114,115 A capillary arterialization process was found to underlie NCF in several of the above studies,108,112,114,115 similar to that proposed by Ramo et al for formation of gracilis collaterals during development 95 and different from the pial collaterogenesis process described in Section ‘Collaterogenesis—formation of collaterals during development’.
Something akin to NCF also occurs in humans with steno-occlusive moyamoya disease, moyamoya syndrome in sickle cell anemia, and following indirect surgical revascularization in patients with chronic cerebral ischemia.116 –119 However, moyamoya collaterals can exhibit features resembling dysmorphic capillaries that provide inadequate perfusion and are subject to hemorrhage. Collaterals arising from the vasa vasorum of surgically trans-positioned arteries or from muscle fascicles are, in contrast, relatively robust. Interestingly, Marushima et al 120 found that indirect revascularization using temporalis muscle inoculated with myoblasts expressing VEGF-A improved outcome after pMCAO in mice.
Genetic studies associating SNP presence and Rentrop collateral score have been conducted in patients with coronary artery disease (see 123 for Refs); however, the inability to differentiate among differences in native collateral number, NCF, collateral remodeling, and downstream mechanisms (Figure 1) complicates mechanistic conclusions. This is also a limitation in similar investigations in braineg 121 –123 and heart.eg 19 However, such studies provide starting points, ie, targets to examine using rodent models wherein NCF can be differentiated from other determinants of collateral flow.
In support of the hypothesis that hypoxia is the proximal stimulus for NCF in obstructive disease,23
–25 new pial collaterals also form after prolonged exposure to reduced oxygen availability. Following on preliminary findings in heart,
124
Zhang et al
23
acclimated collateral-rich B6 mice by gradually reducing inspired oxygen (FIO2) over 2 weeks, followed by maintenance at 12, 10, 8.5 or 7% for 2-to-8 weeks. This resulted in a hypoxemia-dose-dependent formation of additional pial collaterals (39% increase in number), outward remodeling of the pre-existing ones (30% increase in diameter), and 50% reduction in infarct volume when pMCAO was tested six weeks after return to normoxia to allow restoration of hematocrit, capillary density and other physiological setpoints. Intriguingly, the increase in collaterals and amount of remodeling evidenced no regression at this time-point. However a subsequent experiment (H Zhang, JE Faber, unpublished) found that B6 mice, maintained for six weeks at 10% FIO2 to induce NCF and then returned to normoxia for six months, had collateral number (20.4 ± 0.5, ACA-MCA collaterals, both hemispheres) and infarct volumes 24 h after pMCAO (3.5 ± 0.7% of forebrain volume) that no longer differed from age-matched controls (20.2 ± 0.8 and 3.4 ± 0.7%; n = 10 for each age and sex-matched group). Hypoxic NCF was accompanied by increased expression of Hif1a, Hif2a, Vegfa, Rabep2, Angpt2, Tie2, and Cxcr4 in excised watershed pial tissue and abolished by genetic deletion of Rabep2 or knockdown of Vegfa, Flk1, and Cxcr4.
23
Hypoxemia-induced NCF also occurs in heart.24,124 Two-weeks exposure of B6 mice to reduced FIO2 stimulated NCF and, after seven days of return to normoxemia to allow re-acclimation of systemic parameters, reduced infarct volume following coronary ligation. Support comes from studies examining cardiac regeneration in neonatal mice.108,125 And findings that coronary conductance or Rentrop collaterals is/are prominent in humans with chronic obstructive pulmonary artery disease, sleep apnea, cyanotic heart disease, sickle cell disease, coronary artery disease, exposure to prolonged hypoxemia, in piglets kept at low FIO2, and in dogs with chronic anemia.see 23,25
Since pMCAO- and hypoxemia-induced NCF in brain were abolished in Rabep2−/− mice, the authors
23
hypothesized that NCF recapitulates aspects of developmental collaterogenesis according to the following scenario using pial collaterals as a reference point: Watershed collateral PaO2 is favored to be low at baseline because collaterals are the farthest “arterial outpost” from the ascending aorta, with its fully oxygenated blood, and substantial pre-capillary diffusion of oxygen occurs (eg, terminal arteriolar PaO2 is 35–40 mmHg at baseline
104
), and because their low and convergent to-and-fro flow favors diffusion of oxygen to the parenchyma.
4
This is consistent with watershed PO2 being lower than elsewhere.35,36,105,126
–128 Thus, autoregulatory dilation of terminal arterioles and their PAs adjacent to the watershed regions reduces their vasodilator reserve at normal FIO2 and CBF. Oxygen in the watersheds is hence near-threshold for activating pathways similar to that for developmental collaterogenesis (see section ‘Collaterogenesis—formation of collaterals during development’) if oxygen is further reduced by hypoxemia or pMCAO, leading to hypoxia→EGLN1/VEGF-A/etc→EC spouting→EC cords→lumenization→muralization→formation of additional collaterals.
22
Whether PAs sprout from newly formed collaterals was not determined.
23
If normal FIO2 is re-established, the neo-collaterals are pruned away sometime between six weeks and six months (per data given above). The above scenario for NCF in pial watersheds differs from the capillary arterialization process that underlies NCF in striated muscle.108,112,114,115
The above studies indicate that the commonly-held assumption that new collaterals cannot form in the adult is incorrect, and suggest that negative findings following arterial occlusion may have resulted from studying subjects with abundant native collaterals at baseline. Such individuals have small infarctions that are confined to the proximal portion of the occluded tree, which results in the hypoxic tissue being well away from the watersheds (Figure 2). This would not be the case with systemic hypoxemia, which readily induced NCF in B6 mice despite their abundant collaterals. 23 Of note regarding terminology, 3 it is often stated in studies that a given intervention, including exercise trainingeg, 130 and arterial occlusion, promoted collateral “formation” or “development”, yet the measured parameter(s) did not include collateral number or diameter, or distinguish among remodeling of existing collaterals, growth/extension of distal arterioles into the watershed (ie, distal muscularization 3 ), or other determinants of collateral flow (Figure 1).
Collaterals may serve a physiological function
The aforementioned findings with hypoxemia support the hypothesis that collaterals, besides providing a backup system if obstruction occurs, may also serve a physiological purpose—to optimize oxygen delivery, in particular, when oxygen availability is limiting.23 –25 This extends not only from their ability to scaffold PAs to the watershed cortex (see section ‘De novo formation of collaterals after arterial occlusion or sustained hypoxemia’) but also to facilitate control of blood flow to tissues that are perfused by more than one supply artery (Figure 6). The latter is akin to the efficient design of large electrical grids wherein regional networks, each with their own power plant/generator, are interconnected to assure optimal capacity and supply of electricity to adjacent municipalities (wherein current flow is governed by voltage drop) to meet increases in local demand in a given “neighborhood(s)”. However, a caveat exists regarding this analogy to the “vascular grid”: Resistance in the conductor(s) supplying a neighborhood’s blood flow can be modulated by neurovascular coupling and retrograde dilation via electrotonic gap-junction-conducted131,132 and FMD37,54 mechanisms, extending from the PAs that supply the region to their upstream pial arterioles and arteries (using the neocortical vasculature as reference). Accordingly, a small increase in local blood flow demand caused by a restricted increase in local neuronal activity (Figure 6, yellow circle) will cause dilation of the local PAs, reduction in local intravascular pressure (“voltage”) and a limited dilation and pressure drop in the supplying distal arteriole(s). Blood flow is thus recruited to the region by pressure drop, not only from the M1-MCA supply-artery “power (pressure-flow) source” (large red arrow) but also via collateral “connection(s)” (yellow arrow) to the adjacent A1-ACA and P1-PCA power sources (small red arrow). By comparison, a large increase in activity and blood flow demand in the primary locality (blue circle) causes a large local dilation and pressure drop therein and thus a large dilatory response extending more widely retrograde into the MCA tree than into the ACA and PCA trees. Accordingly, pressure in the pial arteriole(s)/artery(s) supplying the affected PA(s) increases more, such that collateral flow reverses and runs in the opposite direction (blue arrow). The result is an additional physiological efficiency, ie, provision of an “anticipatory” increase in blood flow to a wider region (dashed blue circle) should the latter become activated secondarily, for example, by a sensory- or cognitive-motor program initiated by neuronal activity in the primary region.
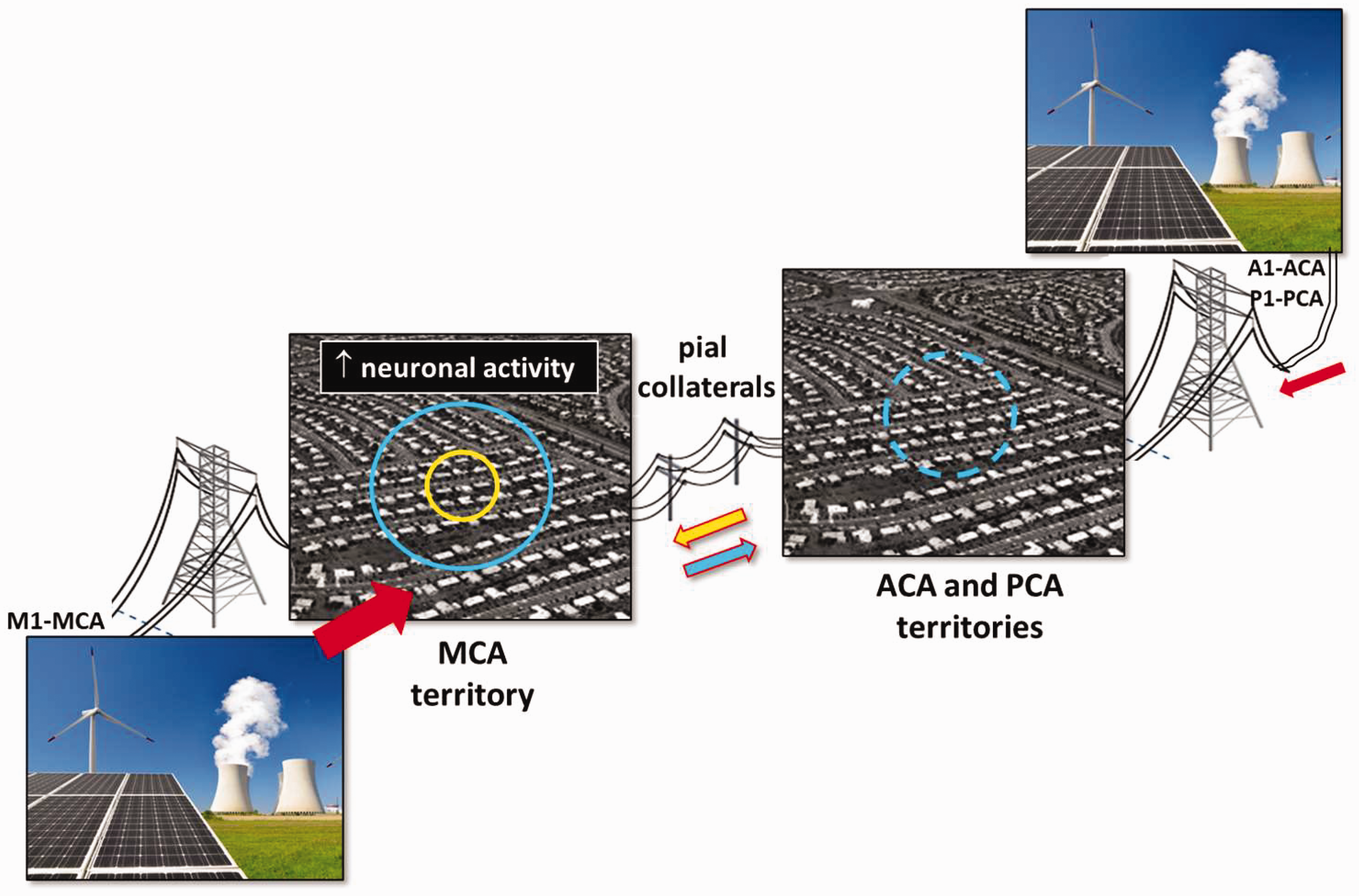
Proposed physiological function of collaterals to optimize matching of blood flow to meet local increases in oxygen demand. The analogy is to the design and function of an efficient power grid interconnecting multiple municipalities each with their own power plant, wherein current flow is governed by voltage drop, with the caveat that resistance can be modulated by the “neighborhood’s” demand via neurovascular coupling and retrograde conducted upstream dilation. Accordingly, a restricted increase in local blood flow demand caused by an increase in local neuronal activity (yellow circle) is met by recruiting blood flow from the M1-MCA arterial “power (pressure-flow) source” and also via collaterals connecting to the adjacent A1-ACA and P1-PCA power sources (yellow arrow). With a large increase in local demand (blue circle), collateral flow may proceed in the opposite direction (blue arrow), resulting in an additional physiological efficiency (dashed blue circle; see text).
Future questions
Studies are required to test this hypothesis, for example in mice with sparse versus abundant collaterals maintained under acute (and sustained) hypoxemia or anemia. Indirect support comes not only from the studies showing that sustained hypoxemia induces NCF (see section ‘De novo formation of collaterals after arterial occlusion or sustained hypoxemia’), but also recent findings that species that have evolved at high altitude have exceptionally abundant collaterals and protection from ischemic tissue injury.22,25 And from other studies examining collateral status in conditions of reduced oxygen availability or delivery to tissues (see section ‘De novo formation of collaterals after arterial occlusion or sustained hypoxemia’). Interestingly, enhanced collateral flow and reduction in infarct volume were achieved by repeated stimulation of neuronal activity within the territory at risk 133 (however, see literature 134 ). Also of note, collateral-like connections between branches within a given cerebral arterial tree, denoted “intra-tree anastomoses” (ITAs),3,25,82 form at the same time during embryogenesis as watershed collaterals, undergo the same pruning away of a fraction during postnatal maturation, and exhibit the same genetic background dependence for their abundance, eg, for skeletal muscle—abundant ITAs in B6 versus sparse in BALB/c (Figure 2), 42 suggesting that formation of collaterals and ITAs share similar mechanisms. 42 Since ITAs serve a somewhat analogous role as above for collaterals, ie, optimizing blood flow to a local region by (in the case of brain) connecting a PA(s) to more than one feed artery/arteriole, it would be of interest to know if formation of additional ITAs occurs with prolonged hypoxemia or anemia in the perinate and adult. It would also be important to ascertain if the above-proposed physiological role for collaterals (and ITAs?) only becomes evident when oxygen availability is limited and other autoregulatory mechanisms have become maximally recruited, or as well, contributes to more efficient oxygen delivery under normoxemic conditions. The latter possibility, however, is not supported (at least not fully) by the fact that the abundance of collaterals and ITAs varies widely among classical inbred and wild-derived strains and subspecies of mice that have evolved at low altitude,42,82,84 ie, that collaterals (and ITAs) may not confer enough of a fitness advantage, when oxygen availability is normal, to assure genetic selection against the survival/existence of individuals with poor abundance.22,25
Aging and cardiovascular risk factors cause rarefaction of collaterals; prevention by exercise training or increased eNOS
As discussed above, collaterals reside in a unique arterial environment of low PaO2 and low and convergent to-and-fro flow at baseline. 4 Their ECs are thus subjected to disturbed shear stress and SMCs to high circumferential wall stress.41 –43 Such conditions elsewhere in the arterial circulation promote atherogenesis, arteriogenesis, and wall hypertrophy.see 4 In addition and possibly owing to the above, proliferation and turnover of their mural cells are increased, favoring accelerated senescence and rarefaction.4,88 At the same time, collaterals have features that may mitigate these adverse conditions, including longitudinally aligned ECs despite their low/disturbed shear stress, and increased expression of eNOS, ephrin-B2, Dll4, and KLF2/4, compared to similarly-sized distal arterioles. 4
Given these conditions, collaterals have been suggested to be “at-risk” vessels within the circulation. 4 A number of studies support this notion. Faber et al 135 found that number and diameter of pial collaterals progressively decreased across 3, 16, and 24–31 months-age in B6 mice (∼equivalent to 14, 50 and 72–90 human years). This resulted in a 6- and 10-fold increase in collateral network resistance and increased infarct volume following pMCAO. A similar decline in collateral number was reported in 18 months-old B6 mice. 136 Aged cats evidenced an increase in pial collateral resistance. 137 Ma et al 71 found that collateral flow after MCAO was less and infarct volume greater in 17 versus 2.5 months-old rats. Diameter of PCom collaterals decreased in aged mice, while no change or the opposite occurred in the primary intracranial arteries.30,138 Most but not all studies in humans have also reported decreased PCom diameter with aging.see 30 In mice, age-associated collateral rarefaction was accompanied by a decrease in eNOS,135,138,139 was mimicked in young mice by genetic deletion 140 or pharmacological reduction in eNOS/NO (despite normalization of blood pressure with hydralazine), 88 prevented by eNOS overexpression, and interestingly, also prevented by running ad-libitum (ie, aerobic exercise training) from 3-to-24 months-age. 138 Infarct volume post-pMCAO followed accordingly in the above studies. Exercise training increased eNOS and SOD2, decreased markers of inflammation and oxidative stress (NFkB, d8OHdG), and decreased the cell cycle arrest marker, P16INK4a, in the vascular wall of the aged mice. 138 Running also prevented the increase in collateral tortuosity seen with aging, presumably by reducing the accelerated proliferation of their mural cells that occurs with aging.4,88 While arteries and arterioles also acquire tortuosity with advanced age, it is smaller in magnitude,see 138 and acquisition of collateral tortuosity begins, notably, several days after birth. 4 Accordingly, collateral ECs and SMCs evidence increased expression of markers of proliferation (Ki67, PDGF-B, angpt2), compared to similarly-sized arterioles, at 10 weeks-age. 4 Collateral rarefaction with aging also occurred in hindlimb in association with reduced eNOS/NO.135,139
Mice with genetic models of CVRF presence also evidence collateral rarefaction. Moore and colleagues 88 reported rarefaction, which was not affected by statin treatment, in two different models of hypertension in brain (eg, 20 and 24% decreases in pial collateral number and diameter, respectively, in mice with renal hypertension), hindlimb, and skeletal muscle, along with increased proliferation of their mural cells, tortuosity, and tissue injury after arterial occlusion. Mice with obesity, dyslipidemia, type-1 diabetes, and metabolic syndrome also sustained rarefaction. Akamatsu and coworkers 141 found that while collateral flow in type-2 diabetic mice was lower immediately after pMCAO, collateral number did not differ from non-diabetic mice (however, the vasculature may not have been maximally dilated prior to post-mortem assessment to assure all collaterals were detected). Interestingly, 6 months exposure to cigarette smoke did not cause collateral rarefaction, 88 a finding supported in humans. 142 Hypertension also decreased PCom diameter, however hyperlipidemia, metabolic syndrome, obesity, and diabetes mellitus were without effect. 30 Rarefaction of pial collaterals and increased tortuosity and infarct volume were also observed in genetic mouse models of vascular cognitive impairment (VCI) and Alzheimer’s disease (AD), in association with recruitment of peri-collateral CD11b+ myeloid cells and increased markers of inflammation, oxidative stress, and aging in their mural cells. 143 Distal arterioles, by comparison, showed no rarefaction or loss of their PAs. Consistent with the aforementioned aging studies, transgenic increase in eNOS prevented collateral rarefaction.
Studies examining whether the above findings extend to humans, which rely on scoring collateral blood flow, are complicated by differences in genetic background and possible effects of aging and CVRFs on other factors that influence collateral flow besides collateral extent (Figure 1). Variable findings are therefore not surprising: Menon and coworkers 144 found that older age (OR 1.3), metabolic syndrome (OR 3.2), and hyperuricemia (OR 1.4) independently predicted poor collaterals in patients with M1-MCA and/or ICA occlusions. Arsava et al. 145 confirmed this for older age (OR 1.9) in patients with proximal MCAO, as did Malik and colleagues. 146 Pi et al. 147 reported that older age and higher homocysteine level associated with poor collaterals in patients with chronic MCAO, while Wiegers et al 13 found that poor collaterals associated with older age (OR 0.92) but not CVRF presence. Poor collaterals also accompanied hypertension, independent of admission systolic blood pressure (OR 2.8).148,149 Nannoni and colleagues 150 found that good collaterals associated with younger age in patients with acute proximal MCAO. Recently, Heitkamp et al 151 observed that older age in LVO-AIS patients associated on admission with lower venous outflow (CTA) and hypoperfusion intensity ratio (perfusion mapping)— indexes of collateral flow proposed to be more robust 152 —but not with lower Tan collateral scores. In contrast to the above, Liebeskind et al 153 and Sperti and coworkers 154 found wide variability and no association between collateral score and CVRF presence. Lower collateral score and increased infarct volume also did not accompany the presence of type-2 diabetes in patients with acute ICA+MCA occlusion. 155
Lin and coworkers 156 reported that collateral score on admission associated with small vessel disease (SVD) score after adjusting for confounders in patients with acute ICA or MCA occlusion (OR 1.9). Similar results (OR 3.04) were obtained by Mark et al 157 and Chen et al 158 for LVO-AIS, including that ICA stenosis and female sex were independent predictors of poor collaterals (OR 0.26 and 0.63, respectively). However, association between collateral and SVD scores was not confirmed (OR 1.11, P = 0.51), 159 and collateral score did not causally link age or metabolic syndrome to white matter hyperintensities. 160 Also, Mikati et al 161 pointed out that a relationship between SVD and collateral status and functional outcome may be confounded by faster core progression mitigating against selection for late-window thrombectomy in AIS patients with pre-existing moderate-to-severe SVD. Rebchuk and colleagues 162 found that poor collaterals in LVO-AIS patients associated with leukocytosis and male sex (OR 1.1 and 1.9, respectively). Interestingly, the measured variables only explained ∼50% of the observed between-patient variability in collaterals, leading to the suggestion that the remainder may be due, in part, to genetic differences. In agreement with mouse studies showing that pMCAO recruits Willisian collaterals, whose high level of variation is in turn capable of affecting pial collateral flow, 30 Christoforidis and coworkers 163 found that higher pial collateral score associated with proximal MCA versus ICA occlusion (OR 9.3), as did lower diastolic blood pressure (OR 5.1). However age, gender, diabetes, systolic blood pressure, and blood glucose did not evidence significant association, which is consistent with another study. 164 Coronary CFI negatively correlated with peripheral artery disease and systolic blood pressure. 165 Several other studies in humans suggested hypertension adversely affects collaterals.5,33,78,88
The above studies in mice show that, although genetic differences can result in dramatic differences in native collateral extent,
22
aging and CVRF-induced rarefaction can also contribute to collateral insufficiency in brain and other tissues, and with VCI and AD in the former. Most of the above studies measuring collateral flow in humans are consistent with these findings.
Collateral remodeling: Arterial occlusion or sustained hypoxemia induce lumen enlargement
Only a small proportion of patients with LVO stroke receive intravenous thrombolysis (eg, 23% in one recent analysis 165 ) and an even smaller percentage receive thrombectomy. 166 Thus, strategies to increase collateral flow by targeting its various determinants (Figure 1) merit investigation. One of them—outward remodeling of collaterals (also termed “arteriogenesis” 3 ) induced by atherostenosis- or acute occlusion-induced increase in EC shear stress—has been studied extensively in peripheral tissues, especially in rodent hindlimb following femoral artery ligation. The mechanisms involved and potential therapies to augment it have been recently reviewed.99,167 –170 By comparison, remodeling of cerebral collaterals has received little attention,171 –175 despite the exponential relationship between collateral diameter and blood flow, and the importance of the latter on initial core volume and infarct progression.
Recent studies have begun to address this deficiency. Zhang and coworkers 82 found in B6 mice that the 20-fold increase in collateral flow and shear stress that occurs immediately after pMCAO4 induced robust pial collateral remodeling: A 50% increase in anatomic lumen diameter occurred by 36 h, with a doubling by 72 h which did not increase further with time. A maximal two-fold increase in diameter was also reported in rats.176,177 Comparable remodeling was seen in ten other mouse strains, was greater (3-fold) in three strains, and proceeded more slowly in BALB/cBy mice, indicating genetic variability. 82 PCom remodeling post-pMCAO, which was much less in magnitude, also evidenced genetic variability. 30 The above rapid rate and magnitude of pial collateral remodeling in B6 and CD1 mice were confirmed by others.178 –180 Importantly, given the high oxygen requirement of neurons, this rate is 3- to 4-fold faster than for cutaneous and hindlimb (gracilis) collaterals which require ∼21–28 days to reach maximal remodeling irrespective of genetic background.80,81,88,90,91,181 This suggests factors that determine the rate of remodeling of pial collaterals may differ, in part, from those in other tissues.99,167 –171 The authors proposed 82 that the fast pace of pial collateral remodeling could reflect that matrix proteolysis and restructuring of parenchymal tissue are required for remodeling in non-brain tissues 168 but not for pial collaterals which reside in the subarachnoid space. Also, pial collaterals are surrounded by a partial blood brain barrier and thus their remodeling may involve signals from resident perivascular myeloid cells and/or microglia instead of (or in addition to) circulating monocytes that contribute to remodeling of peripheral collaterals.167 –170 In addition, the extracellular matrix of pial collaterals, like that of cerebral arteries/arterioles, differs from that of peripheral collaterals, ie, the former have less elastin compared to the extensive elastic reticulum of peripheral collaterals that requires degradation and subsequent restoration during the remodeling process.168,182,183 In support, inhibition of lysyl oxidase, which impaired remodeling of peripheral collaterals, 183 had no effect on remodeling of pial collaterals in B6 mice seven days after pMCAO: lumen diameter of vehicle-treated mice was 41.1 ± 1.5 microns and those treated with 150 mg/kg beta-aminoproprionitrile was 39.7 ± 0.42 (n = 4/group, daily administration begun one day before pMCAO; H Zhang, J Faber unpublished findings). In humans, rapid onset of remodeling also appears to occur in spinal cord pial collaterals within 24 h, providing the rationale for staged surgical occlusion of segmental arteries to prevent or mitigate paraplegia on repair of thoracic aortic aneurisms. 184 Indirect evidence for collateral remodeling in humans also comes from findings that higher collateral scores post-stroke are seen in patients with prior cranial artery atherosclerosis. 148
Wang and coworkers 106 reported that pial collateral diameter approximately doubled when bone marrow-derived mononuclear cells were intravenously administered 24 h after filament pMCAO in rats, which is consistent with their involvement in remodeling of gracilis hindlimb collaterals.99,167 –170 However, remodeling failed to occur in the vehicle-treated group, which is at variance with the aforementioned findings in rats and mice following permanent M1-MCAO. Engagement of the CoW by filament occlusion may explain this discrepancy.
Pial collateral remodeling can also occur in the absence of arterial obstruction. Zhang et al 23 found that exposure to prolonged reduction in FIO2 caused a hypoxemia “dose-dependent” remodeling of pial collaterals. Reversal of the remodeling did not occur after return to normoxia for six weeks (but did so when examined six months thereafter; Zhang and Faber, unpublished results in Section ‘De novo formation of collaterals after arterial occlusion or sustained hypoxemia’). Importantly, infarct volume 24 h after pMCAO was 50% less when induced after return to normoxia for six weeks, consistent with remodeling remaining unchanged at this time. 23 The authors proposed that since flow in collaterals slowly oscillates to-and-fro,4,41 –43 hypoxic remodeling was induced by oscillatory shear stress and wall stress being increased by both cerebral vasodilation and the elevated viscosity (polycythemia) that accompanied the hypoxemia. They also found that hypoxic remodeling was unaffected in B6.Rabep2−/− mice, consistent with the same lack of effect on remodeling in brain and hindlimb after pMCAO and femoral artery ligation.23,185 This contrasts with the major role of Rabep2 in collaterogenesis during development22,72 and NCF formation in adults after pMCAO (see section ‘De novo formation of collaterals after arterial occlusion or sustained hypoxemia’), findings in agreement with collaterogenesis and collateral remodeling being distinct processes.
Regarding signaling, Okyere and coworkers 178 found that EphA4 negatively regulates pial collateral remodeling in mice after pMCAO by inhibiting Akt/Tie2 signaling and cell proliferation, and that inhibition of EphA4 augmented remodeling, reduced stroke volume, and improved functional outcome. Genetic deletion of EphA4 in ECs increased remodeling by 39% and 50% one and four days after pMCAO and decreased infarct volume 50%. Supportive findings were also obtained in hindlimb. 97 Involvement of VEGF-A, CLIC4, and VWF has also been reported.22,90 –93 Tian et al 180 found that rat mesenchymal stem cells (MSCs) given 6 h after pMCAO in B6 mice augmented collateral remodeling, increased CBF, reduced infarct volume, and improved functional outcome (note potential involvement in immunological effects caused by species-heterologous MSCs), in association with increased Rabep2 and VEGF-A expression and Flk1 and ERK1/2 phosphorylation in brain tissue. Whether Rabep2 is required for remodeling was not examined. However, as described above, others have found it is not, albeit in a different setting.23,185 Sugiyama and colleagues 186 reported subcutaneous G-CSF increased remodeling of pial collaterals in rats induced by common carotid occlusion (CCO), along with increased recruitment of CD68-positive cells to the cortical surface. Ischio et al 187 found that augmenting (or inhibiting) S1P→S1PR1 signaling, which is induced by increased EC shear stress, with intraperitoneally-administered agonist (or antagonist) increased (or reduced) collateral remodeling in mice following CCO, along with increasing (or reducing) CBF, reducing (or increasing) infarct volume, and increasing (or reducing) functional outcome. Jiang and coworkers 188 observed that viral administration into the lateral ventricles of the pro-angiogenic long non-coding RNA, SNHG12, in B6 mice increased collateral remodeling ∼40% at day-1, increased CBF and reduced infarct volume ∼50% at day-3, and improved functional recovery in the MCA filament ischemia (60 min)-reperfusion model of AIS. In vitro evidence found that SNHG12 increased dedifferentiation of SMCs to the synthetic proliferative phenotype, which is known to be required for remodeling of gracilis collaterals in hindlimb.167 –169 Small sample-sizes and evidence that the vessels examined included ITAs and terminal arterioles complicate these findings. Remodeling of PComs after cerebral artery occlusion reportedly involved bradykinin and connexin 40.189,190
Remodeling is impaired by CVRFs. Remodeling of pial and hindlimb collaterals in B6 mice was reduced by aging,135,138,139 hypertension, diabetes mellitus, and metabolic syndrome. 88 Yukami et al 191 observed that remodeling, which was reduced following CCO in diabetic mice in association with reduced recruitment of Mac-2+ macrophage cells to the cortical surface, was restored by TNFα inhibition. Akamatsu and coworkers 141 found that the increase in collateral flow seven days after pMCAO (plus ipsilateral carotid occlusion) was reduced in mice with diabetes mellitus, however collateral diameter was not measured to distinguish it from other mechanisms that affect collateral flow (Figure 1). Aging and CVRFs also lessen remodeling of arterioles/arteries in rodent peripheral tissues. 192 Collateral remodeling in peripheral tissues of humans may also be impaired,167 –169 although imaging limitations prevent measurement of remodeling, per se.
The above findings in animals, showing that pial collateral remodeling can be augmented and infarct volume reduced by hypoxemia, inhibition of EphA4 and TNFα, and by administration of MSCs, G-CSF, S1P, and SNHG12, provide rationale for investigating potential therapies to promote remodeling in humans.
Additional future questions
Several areas for future investigation are highlighted below, in addition to those identified in the preceding sections.
Little is known about how differences in collateral extent at baseline affect the determinant of resistance downstream of the collateral network when arterial obstruction occurs (Figure 1).32,51 The recent advent of genetically engineered strains of mice with sparse, intermediate and abundant collaterals provides a way to investigate this question. 22 These models will also aid the search for treatments to increase collateral flow immediately following occlusion and in models of “collateral failure”, ie, a decline in collateral flow post-recanalization that occurs in a fraction of patients, in stroke after recanalization. It is likely the effectiveness of many treatments may be modified by differences in collateral extent.
What we know about the mechanisms that direct collaterogenesis during development and collateral maturation after birth are limited by the small number of extant studies in mice (see section ‘Collaterogenesis—formation of collaterals during development’). Along with extensions of these studies, understanding would be furthered by development of an in vitro model, eg, using a microfluidics platform seeded with ECs, SMCs, and other cell-types that may contribute. Greater knowledge concerning collaterogenesis would facilitate investigations aimed at expanding our appreciation of the factors that induce NCF following acute arterial occlusion and prolonged hypoxemia. Both similarities and differences likely exist. Such studies would aid investigating ways to augment collateral formation in occlusive disease. They may also provide insights into the mechanisms that cause collaterals to be pruned away (rarefaction) and strategies to lessen or prevent it. Of note, new collaterals that form shortly after pMCAO can be distinguished from pre-existing collaterals by at least three markers: 1) their leakage of residual Evans blue dye that can be added to the perfusate used to wash out blood and maximally dilate the vasculature before latex infusion under pressure;see 23,85 2) their susceptibility to bulging during the latter as a consequence of their immature, weak walls, and 3) their absence of tortuosity (H Zhang, J Faber, unpublished findings).
No study has examined how collateral tone changes during the subacute, acute, and post-acute stages after arterial occlusion (with and without recanalization). This likely reflects, in part, the difficulty in addressing these questions in vivo without potential effect of a test-intervention on vessels downstream and upstream of the collaterals. Knowledge of this is important toward understanding the basis of collateral failure and developing a therapy to reverse it as well as augment collateral flow in the absence of recanalization. Whether tone is elevated in different neurological conditions or during hemorrhagic stroke also remain to be determined. Studies examining potential approaches to dilate collaterals, or increase collateral flow via targeting up- and downstream mechanisms, have recently been reviewed.5,44,45,171 –175 Limitations in animal studies could be addressed by using SMC-sparing anesthesia, thinned skull imaging, and M1 occlusion with optically-induced or thrombin injection thrombosis.eg 32 However, separating the passive effect of drop in circumferential wall stress post-occlusion from any active myogenic, metabolic, or shear stress-mediated inhibition of SMC tone would remain difficult. The same problem exists with administration of acute hypercapnia for assessment of collateral tone. Optogenetic 193 or other optically-activated methods for manipulating SMCs or ECs of single collaterals in vivo could provide an approach to address this and other questions raised in this review. New methods for wide-field transcranial perfusion mapping and angiographic and light sheet imaging will also aid future investigation.194,195
Collateral remodeling: therapeutic opportunity
The rate of recanalization is only 20–30% for thrombolysis and 70–85% for thrombectomy, and ∼50% of patients who received the latter are still left significantly disabled.9 –18 Augmenting collateral remodeling offers a promising approach. However, understanding of this process and its role in infarct progression have received little attention. At the outset of remodeling in peripheral tissues of young, healthy rodents, SMCs lose their ability to contract and change to a proliferative “synthetic” phenotype.99,167 –170,196,197 This suggests tone in cerebral collaterals is absent during the acute and subacute phases of stroke. While additional collaterals form after pMCAO, they became detectable by day-2 and required 5–7 days to reach mature diameter 23 (see section ‘De novo formation of collaterals after arterial occlusion or sustained hypoxemia’). Thus, NCF may be too slow to save substantial penumbra, although this question remains to be examined. Individual collaterals (and terminal arterioles for comparison) can be isolated from mice,4,33 allowing dissociation of their mural cells for expression profiling, immuno-detection of proteins, and probing of cellular mechanisms and other biological targets. Also, more robust indexes of collateral flow in patients with AIS, including dynamic regional scoring of pial collateral flow 48 and the multi-measure “cerebral collateral cascade” have been developed. 70 In addition, recently discussed methods for quantifying collaterals in humans may prove feasible. 22
Conclusions
Collaterals are fascinating blood vessels. Despite being unique among the vasculature’s cardinal members, ie, the arteries, arterioles, capillaries, venules and veins—regarding their formation, morphology, location, hemodynamic environment where they reside, function, susceptibility to wide genetic-dependent variation in abundance, and particular vulnerability to rarefaction by aging, cardiovascular risk factors and certain neurodegenerative conditions—their basic biology is the least understood. Understanding how conductance of the collateral network and remodeling become impaired, as well as therapies to address collateral insufficiency, is important because rescuable penumbra remains for several days after cerebral artery occlusion. And because augmenting collateral blood flow may aid angiogenesis, neurogenesis, and neuronal remodeling in the peri-infarct region and improve functional outcome. Hopefully, this review will serve researchers already invested in studying the collateral circulation and attract new investigators toward answering the many fundamental questions and translational opportunities before us.
Abbreviations
ACA: anterior cerebral artery; ACom: anterior communicating collateral artery; AIS: acute ischemic stroke; B6: C57BL/6 inbred mouse strain; CBF: cerebral blood flow; CCO: common carotid occlusion; CFI: collateral flow index; CoW: Circle of Willis; CVFR: cardiovascular risk factor; E#: Embryonic day-1, 2, etc; EC: endothelial cell; FIO2: fraction of inspired oxygen; FMD: flow-mediated dilation; ICA: Intracranial internal carotid artery; ITA: intra-tree anastomosis; LVO: large vessel occlusion (anterior cerebral circulation); MCA: middle cerebral artery; MCAO: middle cerebral artery occlusion; mRS: modified Rankin score; MSC: mesenchymal stem cell; NCF: neo-collateral formation (formation of new additional collaterals in the adult); NIHSS: NIH stroke scale; OR: odds ratio; P#: postnatal day-1, 2, etc; PA: penetrating arteriole; PCA: posterior cerebral artery; PCom: posterior communicating collateral (commissural) artery; pMCAO: permanent middle cerebral artery occlusion; SMC: smooth muscle cell; TA: terminal arteriole (distal-most pial arteriole).
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: NIH-NINDS R01 NS083633, NIH-NHLBI R01 HL111070.
Acknowledgements
The author gratefully acknowledges Hua Zhang, previous lab members, and collaborators.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.