
Abstract
Currently, successful preclinical cerebroprotective agents fail to translate effectively into clinical practice suggesting the need for a comprehensive evaluation of all aspects of brain function. Selective vulnerability refers to the specific regional response of the brain following global ischemia, with observed patterns of vulnerability attributed to the distribution of neuronal subtypes and the functions of respective brain regions. Conversely, the concept of differential vulnerability pertains to the cell-type-specific reactions to cerebral ischemia, dictated by the biological characteristics of individual cells. This review aims to explore these vulnerability hypotheses and elucidate potential underlying cellular mechanisms.
Introduction
Stroke is a leading cause of death and disability, persisting as a significant public health concern globally. In the United States, the incidence of stroke has increased 20.5% percent since 2012 impacting about 795,000 people annually. 1 Current treatment options for acute ischemic stroke are intravenous thrombolysis, endovascular treatment, or a combination of both2,3 Despite the success of recanalization in the improvement of stroke outcomes, not all patients can be treated.2,4,5 Overall, even with successful recanalization of the artery, many patients suffer long-term neurological deficits. Thus, there is considerable need for additional, adjunctive treatment for acute ischemic stroke.
To improve the recovery of injured cells and brain tissue after ischemic injury, many agents have been introduced as neuroprotectants. While these agents have demonstrated success in preclinical animal models, however, the repeated failure of neuroprotectants in clinical trials is well documented.6 –8 One potential explanation for these translational-clinical failures may be an underappreciation of variability in brain cell-type responsiveness to intervention: it may be as important, or more important, to target glia and vasculature, as to target neurons. The term cerebroprotection has been proposed to replace neuroprotection and to emphasize the need to target all brain cell types in the central nervous system. 9 The neurovascular unit (NVU) consists of neurons, astrocytes, endothelial cells, and pericytes, among other cell types (Figure 1). Previously we described the responses of these cell types to ischemic/reperfusion (I/R) injury and cerebroprotective therapies.10,11 Among the NVU components, we observed cell-type-specific responses, which we have termed differential vulnerability. 12 Understanding differential responses to treatment will prove critical to future success in designing cerebroprotective treatment.
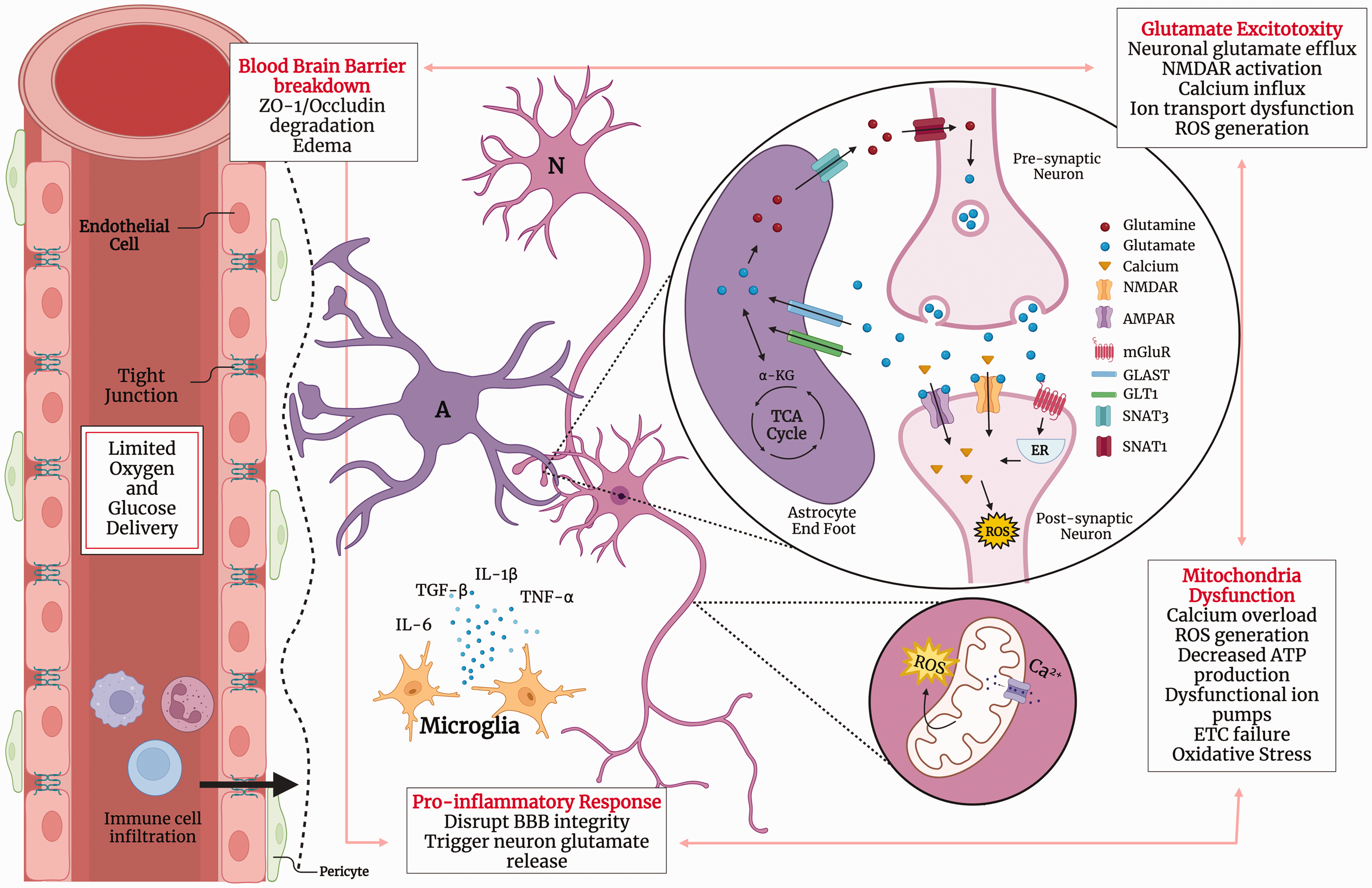
Schematic diagram of the neurovascular unit (NVU) response during cerebral ischemia. During ischemia, the limited delivery of glucose and oxygen results in energy failures that lead to downstream pathologies contributing to cell injury and death (glutamate excitotoxicity, mitochondria dysfunction, pro-inflammatory response, and blood-brain barrier breakdown). A: astrocyte; α-KG: α-Ketoglutaric acid; AMPAR: α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; ATP: Adenosine triphosphate; BBB: blood-brain barrier; Ca2+: Calcium; ETC: electron transport chain; GLAST: glutamate-aspartate transporter; GLT1: glutamate transporter-1; IL: interleukin; N: Neuron; NMDAR: N-methyl-D-aspartate receptor; ROS: reactive oxygen species; SNAT1: solute carrier family 38a member 1; SNAT3: solute carrier family 38 member 3; TGF: transforming growth factor beta; ZO-1: zonula occludens. Created with BioRender.com.
Here we aim to compare the concepts of selective vulnerability and contrast that to our newly defined phenomenon, differential vulnerability. We will discuss the inherent biological differences among the cell types and how they may dictate their responses to cerebral ischemia and its treatments. We hope to point future research towards developing cerebroprotectants that will benefit all elements of the NVU and consequently improve the likelihood of successful subsequent clinical trials.
Description: Selective vulnerability
Selective vulnerability describes the regional tissue damage profile that follows the reduction of regional cerebral blood flow (rCBF) and emphasizes that all brain regions are not equally affected by stroke. Selective vulnerability has been extensively studied and continues to influence the research on the pathology of ischemia as researchers aim to limit damage in the described vulnerable areas of the brain.
Many studies used incomplete global ischemia models to assess the tissue-specific vulnerability in forebrain ischemia. The two vessel occlusion model (2VO) is a classical model of temporary bilateral carotid occlusion with hypotension; the four vessel occlusion model (4VO) uses the temporary occlusion of both carotid arteries after permanent ligation of both vertebral arteries. These models, 2VO or 4VO, induce restricted rCBF to the neocortex, hippocampus, and striatum, so they do not evaluate regions like the thalamus, cerebellum, and brain stem. The hippocampus (CA1) demonstrates greater vulnerability, followed by neocortical layers III and V, with the CA3 and dentate gyrus being the most tolerant vulnerable region after 30 minutes 4VO 13 and transient 2VO.14 –17 By observing hippocampal neuron changes following 5 min 2VO, Kirino introduced the concept of delayed neuron death. This concepts explains that neurons in the CA1 region are most vulnerable because of the secondary injuries caused by cerebral ischemia. 17 These findings were corroborated in organotypic hippocampal slices and neocortex cultures. 18 The concept of selective vulnerability was demonstrated in a focal ischemia gerbil model through observations after progressively longer occlusion times of the left common coratid artery. 19 It is important to note that the sensitivity of these regions was evaluated by the observations only of neuronal cell death, but not other elements of the NVU.
Mechanisms underlying selective vulnerability
GABA and glutamate distribution
There are several possible mechanisms underlying selective regional vulnerability. One postulated mechanism is the heterogeneous distribution of GABAergic and glutamatergic neurons in the brain. GABA and glutamate are the principal inhibitory 20 and excitatory 21 neurotransmitters, respectively. The balance of these neurotransmitters is important for brain signaling and regulation of higher-order brain functions. When this balance is disrupted, it can give rise to a range of cerebral dysfunctions, contributing to the onset of pathologies including psychological disorders, 22 Alzheimer’s, 23 Parkinson’s Disease, 24 and epilepsy. 25
The distribution of GABA and glutamate receptors and transporters throughout the brain correlates with the pattern of vulnerability to global ischemia. Data demonstrated the tolerance of GABAergic neurons in the vulnerable region of the CA1 after 2VO occlusion. 26 The greatest distribution of GABA receptors is in the cerebellum, and the intermediate distribution is in the hippocampus and cortex. 27 The release of GABA and activation of GABA receptors is associated with improved ischemia tolerance in hippocampal slices28,29 and in modeled ischemia.20,30,31 Greater expression of glutamate receptor, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor (AMPA-R) was found in the piriform cortex, hippocampus, and ventral striatum. 32 The N-methyl-D-aspartate receptor (NMDA-R) subunit (NR2A) was found predominantly in the cortex, hippocampus, and striatum. 33 Glutamate transporters, Glutamate-Aspartate Transporter (GLAST) and the Excitatory Amino Acid transporter (EAAT) subtype, EAAT4, are found predominantly in the Purkinje neuronal subtype found in the cerebellum34,35 and are linked with increased tolerance to complete global ischemia35,36 Following stroke, neurons increase the release of glutamate compared to GABA, 16 resulting in excitotoxicity37 –40 the overexcitation of glutamate receptors triggering an aberrant influx of calcium (Ca2+). 38 A high concentration of cytosolic Ca2+ results in cell death in vitro 41 and in vivo 42 through the activation of pro-apoptotic pathways and increased generation of free radicals.43,44
Free radical generation due to differential cellular activity
The generation and accumulation of free radicals during I/R injury varies amongst the regions in the brain like the pattern observed in selective vulnerability. Under physiological conditions, free radicals are generated from aerobic metabolism in the mitochondria, 45 which are manageable by antioxidant defenses. Following reperfusion of the brain, the reintroduction of oxygen can exacerbate mitochondrial dysfunction 46 and increase ROS generation 47 resulting in increased tissue damage. 48 The hippocampus generates more ROS after I/R injury than the cortex 49 due to the higher energy requirements to support the higher levels of neuron activity 50 needed for memory formation and learning functions of the region.51,52 The linkage between energy production and ROS generation is further illustrated by the reduction of ROS when aerobic respiration is inhibited under ischemic conditions. 49 Within the hippocampus, there are higher levels of oxidative activity in the CA1 compared to the CA3 53 and resulting cell injury in vitro 54 and in vivo. 53
Definition: Differential vulnerability
As defined above, selective vulnerability describes the susceptibility of tissue to injury based on the function of the target region. Recently, we introduced the term differential vulnerability to describe the intrinsic properties of cells that are cell type-specific and dictate their biological response to stressors, such as nutrient depletion. 10 In monocellular cultures, neurons are the most vulnerable to modeled cerebral ischemia using oxygen glucose deprivation (OGD). In our model, an OGD duration of two hours results in 80% cell death of neurons. Pericytes and endothelial cells follow, reaching similar cell death levels of 80% after 6 hours. Astrocytes are the most tolerant, with cell viability remaining high even after 12 hours of OGD 10 (Figure 2).
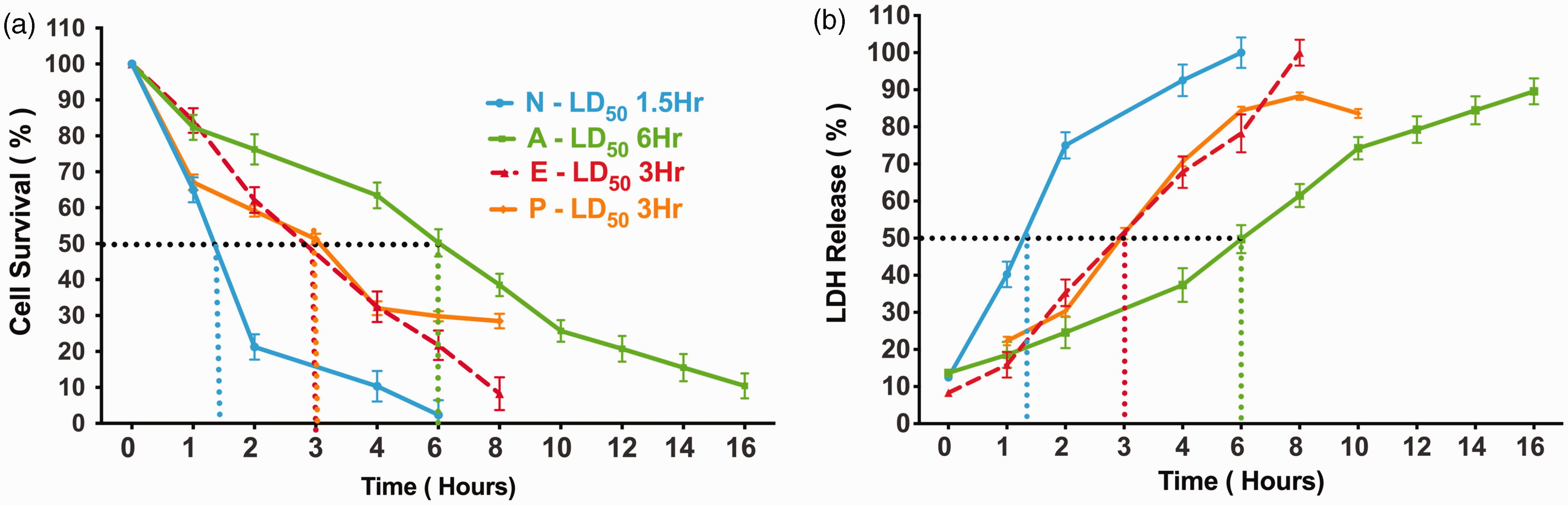
Estimated cell survival and cell death over durations of substrate deprivation. Percentage relative to control cultures of cell survival (a) and cell death (b) was estimated 24 hours following increasing durations of oxygen-glucose deprivation (OGD) for primary isolated cultures of neurons (N), astrocytes (A), endothelial cells (E), and pericytes (P). Neurons were observed as the most vulnerable cell type to OGD reaching 50% of cell death/survival (LD50) at 1.5 hours. Astrocytes were the most tolerant cell type to OGD reaching LD50 after 6 hours of OGD. Endothelial cells and pericytes reached LD50 at 3 hours of OGD. Furthermore, each component of the NVU has different tolerances to modeled cerebral ischemia. While this figure has not been previously published, the levels of cell viability and cell death after OGD for neurons, astrocytes, and endothelial cells was previously published. 12
We are not the first to observe this differential vulnerability of cells. In prior literature, the term ‘differential vulnerability’ has been used to describe the regional differences in cellular subtypes, for example the vulnerability differences between neuronal subtypes in the brain54 – 56 We seek to limit the term ‘differential vulnerability' to focusing on variations among all elements of the NVU beyond regional differences. Other studies have investigated the cell-type-specific response through a similar lens as used in this review without explicitly defining the observation. 57
Potential mechanisms underlying differential vulnerability
An explanation for cell type-specific responses to substrate deprivation has yet to be determined. We will now focus on several potential and non-mutually exclusive mechanisms that could underlie differential vulnerability in the NVU.
Energy metabolism
Under physiological conditions, each cell type exhibits a unique metabolic profile, and in response to ischemic injury, the metabolic characteristics of each cell type undergo distinct changes. The intricate link between metabolism and cell response to injury suggests variations in metabolic pathways as potential explanations for differential vulnerability.
Neurons produce a majority of adenosine triphosphate (ATP) through aerobic metabolism 58 due to the ATP production efficiency to support neuronal energy-demanding functions 59 making neurons more dependent on oxygen compared to other cell types. Oxygen is the final electron acceptor in the electron transport chain (ETC), so when oxygen is limited, the ETC is disrupted and aerobic metabolism fails. As mentioned previously, this exacerbates mitochondria dysfunction 46 and increases ROS generation. 47 In addition, neurons are immensely reliant on access to energy substrates as demonstrated by the close coupling between neuron firing and glucose utilization 60 and localization. 61 In contrast, astrocytes produce a majority of their ATP through glycolysis and lactate synthesis, which allows them to participate in the Astrocyte-Neuron Lactate Shuttle (ANLS). The ANLS postulates astrocyte neuronal support using the conversion of glucose into lactate, instead of Acetyl CoA, for transport to neurons via monocarboxylate transporters.62 –64 Glycolysis and lactate production produces less ATP per mole of glucose compared to aerobic metabolism, but it is oxygen-independent and is not associated with mitochondria dysfunction during hypoxia. Similar to astrocytes, endothelial cells produce a majority of their ATP through glycolysis instead of aerobic metabolism. 65
Another key metabolic difference between the cellular elements of the NVU is glycogen metabolism. Glycogen metabolism plays an important role in brain function: it plays a key role during memory consolidation and learning, 66 is upregulated during increases in neural activity, 67 and glycogen dysmetabolism is implicated in neurodegeneration. 68 Studies have demonstrated the localization of glycogen stores68,69 and essential glycogen synthesis enzymes (glycogen synthase) and glycogen degradation enzymes (glycogen phosphorylase) is specific to astrocytes68,70 and supports ANLS. 71 The enhancement of glycogen mobilization during reperfusion resulted in a decrease in apoptotic astrocytes.72,73 Different access to glycogen among NVU cell types may provide a potential explanation for differential vulnerability.
Neuroinflammation
Neuroinflammation is a secondary source of damage induced by cerebral ischemia and is a target for novel therapeutic development 74 with little success.75 –77 The progression of pro-inflammatory immune response is important to cerebral ischemia pathophysiology but is outside the scope of this review. This review will focus on the cell-type-specific responses to pro-inflammatory mediators and how this would influence treatment development.
Microglia are the resident immune cells in the brain that have a dual response to ischemic injury. Activated M1-like phenotype (pro-inflammatory) microglia release pro-inflammatory cytokines, chemokines, and free radicals. 78 ROS 79 and other injury response mediators act as ligands for inflammasome activation and trigger pro-inflammatory cytokine release through pathways like NF-κB80,81 Studies have demonstrated neuronal elevation of NF-κB in response to focal cerebral ischemia results in increased cell death. 82 When NF-κB function is impaired, microglia polarization shifts to a M2-like phenotype (anti-inflammatory) resulting in reduced neuronal death and IR injury. 83 Franke, et. al demonstrated an increased expression of the inflammasome, NLRP3, during reperfusion after transient MCAo in neurons, which was associated with higher levels of apoptosis. 84 While data indicate endothelial cells respond to the pro-inflammatory mediators by upregulating adhesion molecules, disruption of tight junctions, and overall increase of BBB permeability, 85 there is little information about how the viability of endothelial cells is impacted.
As mentioned throughout this review, the generation of ROS and resulting oxidative stress is a hallmark of cerebral ischemia and reperfusion injury. As pro-inflammatory cytokines induce the production of ROS in cells, neurons, being more vulnerable to oxidative stress compared to astrocytes, 86 may be more susceptible to neuroinflammatory-mediated injury. Neurons have a lower antioxidant defense than astrocytes. 87 Astrocytes generate glutathione (GSH), a powerful antioxidant, through the NRF2 pathway. 88 Studies have demonstrated astrocyte endogenous GSH is critical for astrocyte tolerance to ischemia. 89 Data has also connected astrocyte production of GSH to the astrocyte-mediated protection of neurons. 88 A potential therapeutic, citicoline, resulted in elevated levels of GSH90,91 and reduced IR injury. 91 To date, treatments targeting GSH haven’t been successful. 6 Endothelial cells have direct contact with the periphery immune response and suffer the initial insult of periphery ROS-generating mediators as demonstrated by the attenuation of cell death and vasogenic edema with improved antioxidant treatment. 92
Excitotoxicity
Increased glutamate concentration is a hallmark of cerebral ischemia that triggers a complex cascade resulting in excitotoxicity and cell death, as mentioned previously, making the cell-type-specific management of this toxic ligand of interest.
EAATs (GLT-1) transport excess glutamate from the synapse and are almost exclusively expressed in astrocytes to control a majority of the glutamate reuptake. 93 The expression of GLT-1 is downregulated during cerebral ischemia 94 and its downregulation is associated with worsened outcomes. 95 The activation of GLT-1 in animals that underwent focal photothrombosis successfully decreased lesion size in only male animals, 96 suggesting a greater but not exclusive role of GLT-1 in stroke treatment. The role of GLT-1 during ischemia becomes even more complex when the response of individual components of the NVU is evaluated. While GLT-1 may protect neurons during the initial onset of ischemia, after longer durations of ischemia, GLT-1 reverses and releases glutamate, inducing neuronal excitotoxicity. 97 The inhibition of GLT-1 in neuronal cultures improved neuron viability after OGD, while astrocyte cultures with enhanced GLT-1 expression improved astrocyte viability. 98 This cell-type-specific response to glutamate uptake suggests astrocytes may benefit from the increased intracellular concentrations. In astrocytes, glutamate is converted into glutamine by glutamine synthase and transported to neurons via phosphate-activated glutamase.99,100 Glutamate is also converted to alpha-ketoglutarate by glutamate dehydrogenase before being used in ATP production in astrocytes101,102 potentially acting as an astrocytic fuel source during ischemia.
The expression and activity of NMDA-R in neurons under physiological and pathophysiological conditions are well-studied and are a target for potential therapeutics. NMDA-R activity is unique to each cell type, making it a possible explanation for differential vulnerability. Conventionally, NMDA-R expression is accepted as neuron-specific, but recent investigation demonstrated NMDA-R localization in astrocytes.103,104 A previous study demonstrated that when treated with glutamate and H2O2, neurons upregulated the expression/activation NMDA-R, but astrocyte NMDA-R expression/activation stayed constant. 105 Activation of NMDA-R is associated with increased Ca2+ influx and cell death, 37 so neurons presenting a higher expression of receptors and activity than the other cell types in the NVU may explain neuronal vulnerability to excitotoxicity.
Endothelial cells also express AMPA-R and NMDA-R which impact the viability and function of the cells during stroke. 106 The activation of NMDA-R under ischemic conditions resulted in increased blood-brain barrier (BBB) permeability in vitro. 107 After glutamate treatment, occludin, a tight junction (TJ) protein linked with maintaining BBB integrity, is downregulated in endothelial cells. The activation of NMDA-R and AMPA-R disrupted TJ assembly through occludin post-translational modifications. 107 Decreased expression of claudin-5, a transmembrane TJ protein, and disruption of TJ assembly was observed after in vitro 108 and in vivo 109 ischemia. These glutamate-induced alterations resulted in increased BBB permeability, but the impact on endothelial cell viability has not been demonstrated.
Clinical implications
Historically, stroke therapeutics have been developed with the unrecognized assumption that the elements of the NVU respond as a unit. The majority of tested neuroprotectants were developed in models of neuron ischemia. 110 In some cases, astrocyte ischemia was also used to characterize putative therapies. 111 In contradistinction, while evaluating the efficacy of drug treatments that work to limit thrombin cytotoxicity, we observed cell type-specific responses to treatments. 10 Monocultures for each of the cell types were treated with cytoprotective agents known to act on thrombin and its receptor, protease-activated receptor-1 (PAR1), and subjected to OGD. Treatment outcomes varied, highlighting how differential cell responses affect therapeutic efficacy. We then confirmed in vivo that different elements of the NVU respond quite differently to therapies.10,11 In fact, we showed that at certain doses of treatment candidates, while one cell type was protected, other cell types were harmed. 10 In another example, we demonstrated that short duration treatment with therapeutic hypothermia benefited neurons, but long duration hypothermia harmed astrocytes and neurons. 12 Therapeutic hypothermia, despite powerful preclinical evidence of benefit, has not proven successful in human clinical trials.112,113 These data show that without considering differential vulnerability and response to treatment, clinical trials are at risk of failure for using dosing schemes that harm one or another element in the NVU.
Throughout this review we have addressed the complexity of stroke pathophysiology and related it to the failures of potential therapeutics. However, it is important to note that there are other possible and non-exclusive reasons a proposed drug treatment fails to alleviate the negative effects of cerebral ischemia. These include time to treatment, 114 patient variability,115,116 and penetration of the blood brain barrier. 117 A drug’s ability to penetrate the BBB has been a long-standing obstacle when developing treatments for any disease that affects the brain. The BBB is a network that has a complex response to the onset of stroke that affects its permeability to cerebroprotective agents. Mandeville, et al. discusses these complexities of the BBB and how researchers should consider them when developing potential therapeutics. When discussing cell-type-specific responses to treatment, it is important to consider target engagement. 118 Webster, et al. describes an antibody therapy that was designed to both penetrate the BBB and specifically target neuron expressed metabotropic glutamate receptor type 1. 119 As the technology to specialize target engagement improves it will be important to consider what cells are being affected and their differential vulnerability to ischemia.
Conclusion
Here we demonstrate the innate heterogeneity of response to injury among the cellular components of the NVU. The postulate of selective vulnerability improves our understanding of how regional topography influences cerebral responses to both global and focal ischemia injury, while differential vulnerability describes the inherent biological mechanisms that influence the cell type-specific responses to insults and intended cerebroprotective treatment. We illustrate a non-exhaustive list of the potential mechanisms that regulate the cellular responses to cerebral ischemia. The different cell-types of the NVU show marked differences in energy metabolism and glutamate maintenance that influence the overall response to cerebral ischemia of each cell-type differently. Astrocytes have a greater tolerance to ischemia, which could be explained by the differential expressions and activities of various candidate players involved in the well-known mechanisms of I/R injury. The astrocyte cellular mechanisms that are meant to support NVU homeostasis allow for tolerance to ischemia-induced insults. Neuronal receptors and transmembrane proteins that are essential for neuron communication of electrochemical signals may increase their vulnerability to nutrient deprivation. Further research on these candidates will offer insight into the failures of stroke therapeutics and will allow researchers to develop treatment options that preserve the viability and cell function of all cell types in the NVU.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Supported by the National Institute of Neurological Disorders and Stroke R01 NS075930 (PL), by U24 NS113452 (PL) and by American Heart Association – Predoctoral Fellowship GR1065747 (AB).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.