
Abstract
The major pathophysiological process of vascular cognitive impairment (VCI) is chronic cerebral ischemia, which causes disintegration of the blood–brain barrier (BBB), neuronal death, and white matter injury. This study aims to test whether sulforaphane (Sfn), a natural activator of nuclear factor erythroid 2-related factor 2 (Nrf2), reduces the chronic ischemic injury and cognitive dysfunction after VCI. Experimental VCI was induced in rats by permanent occlusion of both common carotid arteries for six weeks. This procedure caused notable neuronal death in the cortex and hippocampal CA1, myelin loss in the corpus callosum and hippocampal fimbria, accumulation of myelin debris in the corpus callosum, and remarkable cognitive impairment. Sfn treatment alleviated these ischemic injuries and the cognitive dysfunction. Sfn-mediated neuroprotection was associated with enhanced activation of Nrf2 and upregulation of heme oxygenase 1. Sfn also reduced neuronal and endothelial death and maintained the integrity of BBB after oxygen-glucose deprivation in vitro in an Nrf2 dependent manner. Furthermore, Nrf2 knockdown in endothelial cells decreased claudin-5 protein expression with downregulated claudin-5 promoter activity, suggesting that claudin-5 might be a target gene of Nrf2. Our results demonstrate that Sfn provides robust neuroprotection against chronic brain ischemic injury and may be a promising agent for VCI treatment.
Introduction
As human lifespan increases, the aged population expands rapidly.1,2 Some of the major challenges that elderly people face are progressive memory loss, cognitive impairment, and dementia. The estimated prevalence of dementia in the USA in 2010 was 14.7% among the elderly people (>70 years), with the annual cost of care estimated at $159–$215 billion.3,4 Increasing cases of dementia pose a huge burden to the society;2,5 therefore, we are in urgent need to find effective drugs that could alleviate cognitive impairment. Only after Alzheimer’s disease, vascular cognitive impairment (VCI) is the second most common type of dementia.5–7 Vascular dysfunction and abnormality not only cause VCI itself, but also accelerate other types of dementia.6–11
Following vascular dysfunction and abnormality, the integrity of the blood–brain barrier (BBB) is compromised, which is considered as an important pathological factor, even at the starting point of the brain injury in VCI.12–14 Though the causes of the brain injury are not clear, increasing evidence suggests an ischemic origin, which contributes to neuronal loss and white matter (WM) injury, leading to cognitive impairment and dementia.5,9,15–19 Neurons are the electrically functional cells of the neurovascular units in the brain. They require large amount of energy to maintain membrane potentials and therefore demand a continuous supply of glucose and oxygen. This leads to their vulnerability to ischemic injuries. On the other hand, WM conducts the electric signals. Intact myelin allows axons to conduct at least 50 times faster than unmyelinated axons, and damage or loss of myelin affects the function of the axons which may even lead to axonal injury.20,21 Recent studies have shown that axonal abnormalities and myelin loss are the main manifestations of WM injury,14,22 and the WM injury underlies profound cognitive impairment, especially in VCI.7,23–25
Nuclear factor erythroid 2-related factor 2 (Nrf2) is the master transcription factor that controls the intrinsic antioxidative system. 26 Under unstressed conditions, Nrf2 is not transactive; following stress, such as oxidative and nitrosative ones, Nrf2 translocates to the nucleus, binds to the antioxidative response elements (AREs) on its target genes and triggers the transcription of these genes. Previous studies have elucidated the neuroprotective effects of Nrf2 against oxidative stress and ischemic injury in the central nervous system.26–30 Among Nrf2 targets, heme oxygenases-1 (HO-1) is an anti-oxidative enzyme with neuroprotective effects.26,31–33 It is not expressed under physiological condition, but robustly upregulated following Nrf2 activation. As a result, HO-1 can also function as a marker of Nrf2 activation.
Sulforaphane (Sfn) is a potent natural Nrf2 activator with BBB permeability and neuroprotective effects against acute stroke, traumatic brain injury and Parkinson’s disease.30,34–37 However, it is not clear whether Sfn can also protect the brain against chronic ischemia and VCI via activating the Nrf2/HO-1 pathway. The purpose of this study is to test our hypothesis that Sfn alleviates neuronal damage, WM injury, BBB disruption and cognitive impairment induced by chronic cerebral hypoperfusion through activating the Nrf2 pathway.
Materials and methods
Rats, surgery, and treatments
Sprague-Dawley rats (male, six to seven months) were purchased from the Experimental Animal Center of Lukang Pharmaceuticals (Shandong, China). All animal experiments in the study were approved by the institutional review board of Taishan Medical University and conducted in accordance with the Guide for the Care and Use of Laboratory Animals (NIH) and ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines. All the rats were housed at constant humidity and temperature with a 12:12-h light/dark cycle.
Rat VCI was induced by permanent occlusion of bilateral common carotid arteries (2VO) as previously described.38,39 This is an established VCI model40,41 that mimics large vessel diseases with cognitive impairment.16,42,43 The rats were randomly assigned to three groups: sham-operation (Sham), 2VO, and Sfn-treated 2VO (2VO+Sfn) groups. In brief, rats were anesthetized with 1.5% isoflurane in a mixture of 30% oxygen and 70% nitrous oxide. Under a surgical microscope, both common carotid arteries were gently exposed after a midline cervical incision and then ligated with a 5–0 silk suture. The Sham group went through similar procedure without ligation of the carotid arteries. The wounds were closed, and rats were returned to their cages.
Sfn was purchased from LKT Lab (St Paul, MN), and the working solution was prepared by diluting the stock solution with phosphate-buffered saline (PBS). Sfn (10 mg/kg) was injected intraperitoneally after the surgery and, subsequently, twice a week for the 2VO+Sfn group, while PBS was administered for other groups as vehicle.
Behavioral tests
Behavioral tests were performed in a blind manner to evaluate the sensorimotor and cognitive function of rats. The rats were pre-trained for three days before surgery. At the indicated time-points (Figure 1(a)), rotarod and corner tests were performed to evaluate sensorimotor function, while Morris water maze (MWM) test was performed to evaluate the cognitive function, according to previous reports.44,45
Sfn improves sensorimotor and cognitive functions after 2VO in rats. (a) Diagram showing the time-points for outcome evaluation. (b) Rotarod test showed that rats in 2VO+Sfn group stayed longer on the rods than 2VO group. (c) Corner test did not reveal turning preference in any group. (d) Representative images of the swim paths of rats during learning phase (the platform present) and memory phase (the platform removed) in the MWM test. Time to find the hidden platform (escape latency) was measured for spatial learning ability, and time spent in target quadrant was measured to evaluate the memory capacity. Sfn improved both learning and memory deficiency after 2VO. n = 6, *p < 0.05, ***p < 0.001 vs. Sham; ##p < 0.01, ###p < 0.001 vs. 2VO.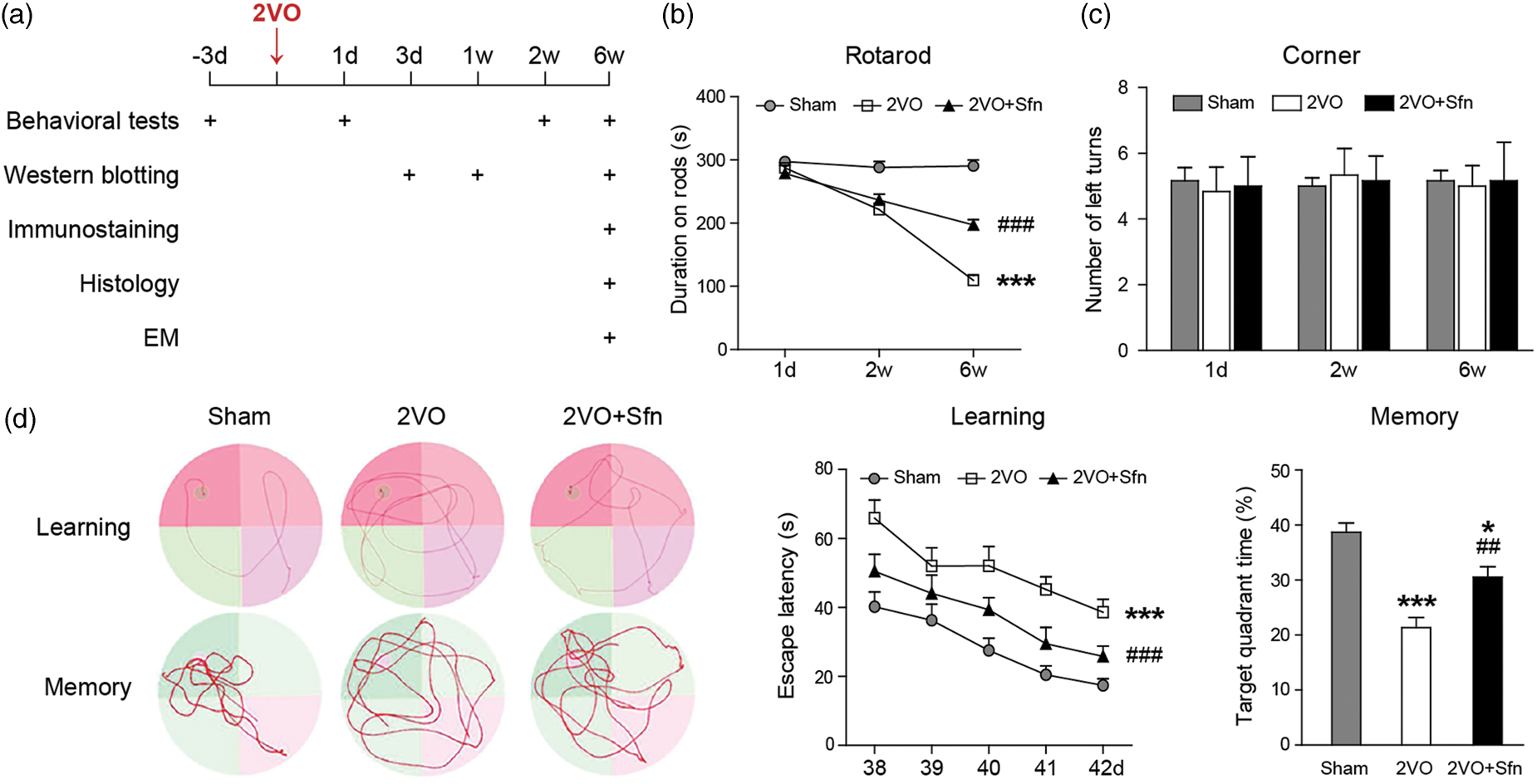
Rotarod (IITC Life Science Inc., Woodland Hills, CA) test evaluates sensorimotor coordination and balance. After the rats were placed on the rods, the rods began to rotate and accelerated to 10 r/min within 30 s. The rods rotated steadily at 10 r/min for the remainder of the trial (300 s in total), and the duration that the rats remained on the rods were recorded. If the rats could remain on the rod for 300 s without falling, the rotation would gradually stop, and the duration on rods was recorded as 300 s. In the corner test, the rats were placed between two boards angled at 30° facing the corner. When the rats entered the corner, both sides of their vibrissae would be stimulated simultaneously. Normal rats typically turned back randomly from either side. Animals with focalized sensorimotor dysfunction tended to turn preferentially toward the non-impaired side. The number of left turns was recorded from 10 trials for each test.
The MWM test was performed in the sixth week from day 39 through day 42 after 2VO. In brief, the water maze pool was filled with opaque water and an 11-cm diameter platform was submerged under the water surface. Each rat received three trials per day with 90 s per trial to locate the platform from one of four quadrants. The rats were supposed to perform a search for the platform and gradually remember the location of the platform per external spatial cues. The latency to find the hidden platform was recorded (learning phase). On day 42, an additional probe trial was performed with the platform removed. The rat was placed into the most distal quadrant from the previous platform. Each rat was allotted 60 s for the trial. Time spent in the goal quadrant and the swim speed were recorded (memory phase).
Western blot
At indicated time-points after surgery, the rats were subjected to transcardiac perfusion with cold normal saline. The brain tissues from the cortex, the corpus callosum (CC)/external capsule (EC), and the striatum were collected. Cytosolic and nuclear fractions of the samples were extracted using a nuclear and cytoplasmic protein extraction kit (Beyotime, Shanghai, China) and subjected to Western blotting analysis using standard methods.44,45 The blots were probed with antibodies recognizing Nrf2 (1:1000, Enzo Life Science, Farmingdale, NY), HO-1 (1:1000, Enzo Life Science), 200 kDa neurofilament heavy polypeptide (NF-200, 1:10000, Abcam, Cambridge, MA), myelin basic protein (MBP, 1 µg/ml, Abcam), non-phosphorylated neurofilament H (SMI-32, 1:1000, Calbiochem, San Diego, CA), claudin-5 (CLD5, 1:1000, Invitrogen, Coraopolis, PA), occludin (1:1000, Invitrogen), β-actin (1:3000, Sigma-Aldrich, St. Louis, MO), and histone H3 (1:1000, Abcam). The blotted bands were obtained by the ChemiDoc MP imaging system (Bio-Rad, Hercules, CA) and then analyzed with NIH ImageJ software.
Immunohistochemistry
The rats were perfused at indicated time-points after surgery with cold normal saline and fixed by 4% paraformaldehyde. The brains were embedded in paraffin and then cut into 5 -µm sections. The sections at the specific levels were selected for immunohistochemical stains. Paraffin sections were sequentially immersed through the following solutions: xylene, 100% alcohol, 90% alcohol, 70% alcohol, and 50% alcohol. After being blocked with 5% donkey serum for 1 h, the sections were stained with the following primary antibodies at 4℃ overnight: Nrf2 (1:1000), HO-1 (1:400), MBP (1:1000), SMI-32 (1:1000), NeuN (1:500, EMD Millipore, Burlington, MA), glial fibrillary acidic protein (GFAP, 1:1000, Sigma-Aldrich), Iba1 (1:2000, Wako, Richmond, VA), adenomatous polyposis coli (APC, 1:400, EMD Millipore), von Willebrand factor (vWF, 1:1000, Abcam), and CLD5 (1:200). After washing, the sections were incubated for 1 h at room temperature with secondary antibodies conjugated with DyLight 488 or Cy3 (1:1000, Jackson ImmunoResearch Laboratories, West Grove, PA). Fluorescence images were captured using a fluorescent microscope (Olympus BX51, Japan).
Luxol fast blue-Cresyl violet staining
Sections were stained with a Luxol fast blue (LFB, staining myelin)-Cresyl violet (CV, staining neuronal cell bodies and processes) staining kit (American MasterTech, Lodi, CA) according to the manufacturer’s instructions. In brief, paraffin sections were incubated with LFB solution at 60℃ for 1 h. The sections were differentiated by dipping them several times in 0.05% lithium carbonate, and this differentiation was continued by dipping the sections in 70% reagent alcohol (65% absolute alcohol, 5% isopropyl alcohol, and 30% water) until the gray matter and WM was clearly distinguished. The sections were then stained with CV solution for 10 min. The microimages of the brain sections were photographed (Olympus BX51, Japan).
Electron microscopy
The ultrastructure of the CC/EC at day 42 after surgery was visualized by a transmission electron microscope (EM) as described previously. 46 In brief, the CC/EC tissues were fixed in 2.5% glutaraldehyde for 12 h, followed by 1% osmium tetroxide for 1 h. The tissues were embedded in resin after the dehydration with serial treatment of increasing ethanol concentrations. Ultra-thin tissue sections were prepared with a Reichert ultra-microtome, and the images were captured with a Philips CM120 electron microscope (F.E.I., Hillsboro, OR). G-ratio and abnormal axons defined as axolysis and/or dense axoplasms were analyzed according to the previous reports.47,48
Primary neuronal cultures and treatment
The primary cortical neurons of the rats were dissected from E17 fetal rats and maintained in the Neurobasal-A medium supplemented with B27 (Invitrogen), as previously described.32,44 Experiments were performed 10 days after seeding. Oxygen-glucose deprivation (OGD) was performed with standard procedures as previously published,32,44 with or without 1.5 µM Sfn pretreatment for 24 h.49,50 Twenty-four hours after reoxygenation, the cell death was quantitatively evaluated.
Primary brain endothelial cultures, treatment, and the in vitro BBB model
Primary mouse brain microvascular endothelial cells (MBMECs, Cell Biologics, Chicago, IL) were maintained in growth culture media on gelatin-coated dishes. Passages 3–8 of the cultures were used for experiments. To mimic ischemia in vitro, cultured MBMECs were exposed to OGD.32,44,45 The durations of OGD were 16 h for normal cells and 10 h for lentiviral-transfected cells, respectively. For the Sfn treatment groups, Sfn was added to culture media at a concentration of 1.5 µM for 16 h before experiments.
In vitro BBB model was established as previously described. 45 In brief, transwell inserts (pore size 0.4 µm, Falcon, Corning, NY) were used to separate the wells into a luminal (top) chamber and an abluminal (bottom) chamber. MBMECs were seeded into the luminal chambers and let grow till 100% confluence. Fluorescein-dextran (4 kDa, Sigma-Aldrich) was added into the luminal chamber at 0.05 mg/ml for 6 h, and fluorescence intensity in an aliquot of 50 µL media from the abluminal chamber was measured to represent the BBB permeability.
In vitro Nrf2 knockdown
To knock down Nrf2 in neurons and MBMECs, lentiviral particles carrying shRNA targeting Nrf2 (sh-Nrf2) or a scramble sequence (sh-Sc) were added to cells together with 8 µg/ml polybrene (all from Santa Cruz, Dallas, TX) for 16 h. The virus-containing culture media were then replaced with fresh growth media. Experiments were performed 72–96 h after the transduction.44,45
Viability and death assays for primary cells
The live/dead cell viability/cytotoxicity assay was performed according to the manufacturer’s instructions (Molecular Probes, Eugene, OR). 44 In this assay, red dots (fluorescent ethidium homodimer-1) represent dead cells with compromised membranes, while green stains (fluorescent membrane-permeant calcein AM) represent live cells. Both dyes were added to cells and incubated at room temperature for 30 min before the representative images were captured. For Hoechst 33258 (Invitrogen) nuclear staining, the percentage of cells showing nuclear condensation was quantified. For cell counting, three random fields were captured per well by a blinded observer. Four to six wells per condition were selected for every experiment, which were repeated on three independent occasions.
Extracellular lactate dehydrogenase (LDH) released from damaged cells was measured by an LDH detection kit (Pointe Scientific Inc, Canton, MI). The LDH release from a total cell lysate in the control group was set as 100%, and all the data were expressed as a relative percentage compared with it.
Luciferase assay
Mouse CLD5 promoter expression plasmids (Chromosome X+: 76262902-76264591) with both Gaussia luciferase (GLuc) and secreted alkaline phosphatase (SEAP) reporters were packaged with a third-generation lentiviral system (GeneCopoeia, Rockville, MD). 51 Briefly, human embryonic kidney cells 293 T were grown to 70–80% confluent, and CLD5 promoter expression plasmids together with lentiviral HIV-packaging plasmids were co-transfected. Forty-eight hours after transfection, supernatants containing lentiviral particles were collected for subsequent experiments. To assess the role of Nrf2 on MBMEC CLD5 promoter activity, MBMECs were infected by CLD5 promoter expression lentivirus with or without sh-Sc/sh-Nrf2 lentivirus. A secrete-pair dual luminescence kit (GeneCopoeia) was used to measure GLuc and SEAP luminescence intensities in the supernatants 72 h after the transduction. SEAP was set as an internal control, and the GLuc/SEAP ratio was calculated for each sample.
Data analysis
All the data were presented as mean ± SEM. Data were analyzed with the appropriate analysis of variance (ANOVA), followed by the Bonferroni post hoc test. A p value of less than 0.05 was considered statistically significant.
Results
Sfn treatment improves cognitive functions after chronic cerebral hypoperfusion
Animal experiments were scheduled as shown in Figure 1(a). We first investigated the protective role of Sfn in rats after 2VO. As shown in the rotarod test, 2VO led to a gruadual decrease in the durations that rats remained on the rods, indicating that rats developed chronic deficits in their sensorimotor coordination and balance function (Figure 1(b)). On the other hand, 2VO did not induce significant turning preference in the corner test (Figure 1(c)), indicating that this model did not cause focal or asymmetric brain injury. Sfn treatment improved the rats’ performance in the rotarod test (Figure 1(b)).
The cognitive function was assessed six weeks after surgery. As shown in Figure 1(d), the rats in the 2VO group demonstrated severe cognitive impairment, as indicated by learning and memory deficiencies in MWM test. Sfn treatment improved their performance. There was no difference in the swim speed among groups (data not shown), suggesting that the measurements in learning and memory abilities were not biased by differences in gross motor functions. Taken together, these findings suggest a beneficial role of Sfn in improving long-term coordination and balance functions as well as spatial cognitive functions following VCI.
Sfn treatment reduces neuronal death in vivo and in vitro
To analyze the structural basis of the observed behavioral deficits, we sacrificed the rats six weeks after the surgery and stained the brain sections with LFB-CV, and neurons with normal-like morphology were counted. As shown in Figure 2(a) to (c), 2VO reduced normal-like neurons in the cortex and hippocampal CA1 region, which was partially reversed by Sfn treatment. Next, we tried to determine whether Nrf2 is necessary for Sfn to protect neurons. Since there are neither specific Nrf2 inhibitors nor Nrf2 knockout (KO) rats, we performed Nrf2 knockdown in the primary neuronal cultures using sh-Nrf2. We found that Sfn reduced neuronal death after OGD (Figure 2(d) and (e)). While sh-Sc could not abolish the protective effects of Sfn (data not shown), sh-Nrf2 largely abolished the protection of Sfn (Figure 2(d) and (e)), indicating the key role of Nrf2 in Sfn-mediated neuroprotection against neuronal death.
Sfn reduces neuronal death in vivo and in vitro. Six weeks after 2VO, brain sections at dorsal hippocampus were collected and stained with LFB-CV. (a) Representative images of the parietal cortex (upper panels, scale bar = 100 µm) and hippocampal CA1 (lower panels, scale bar = 200 µm). (b, c) Neurons with normal-like morphology were counted and data were expressed as number per mm2. 2VO reduced normal-like neurons in both cortex and CA1, which was partially reversed by Sfn. n = 6, *p < 0.05, **p < 0.01, ***p < 0.001 vs. Sham; ##p < 0.01, ###p < 0.001 vs. 2VO. Nrf2 knockdown in rat primary neurons was induced by sh-Nrf2 transfection, and OGD was performed with or without Sfn pretreatment. (d) Hoechst 33258 staining with cell counting and (e) LDH assay showed that Sfn ameliorated OGD-induced cell death, which was abolished by Nrf2 knockdown. *p < 0.05, **p < 0.01, ***p < 0.001 vs. Ctrl; #p < 0.05, ##p < 0.01 vs. OGD; &p<0.05 vs. OGD+Sfn.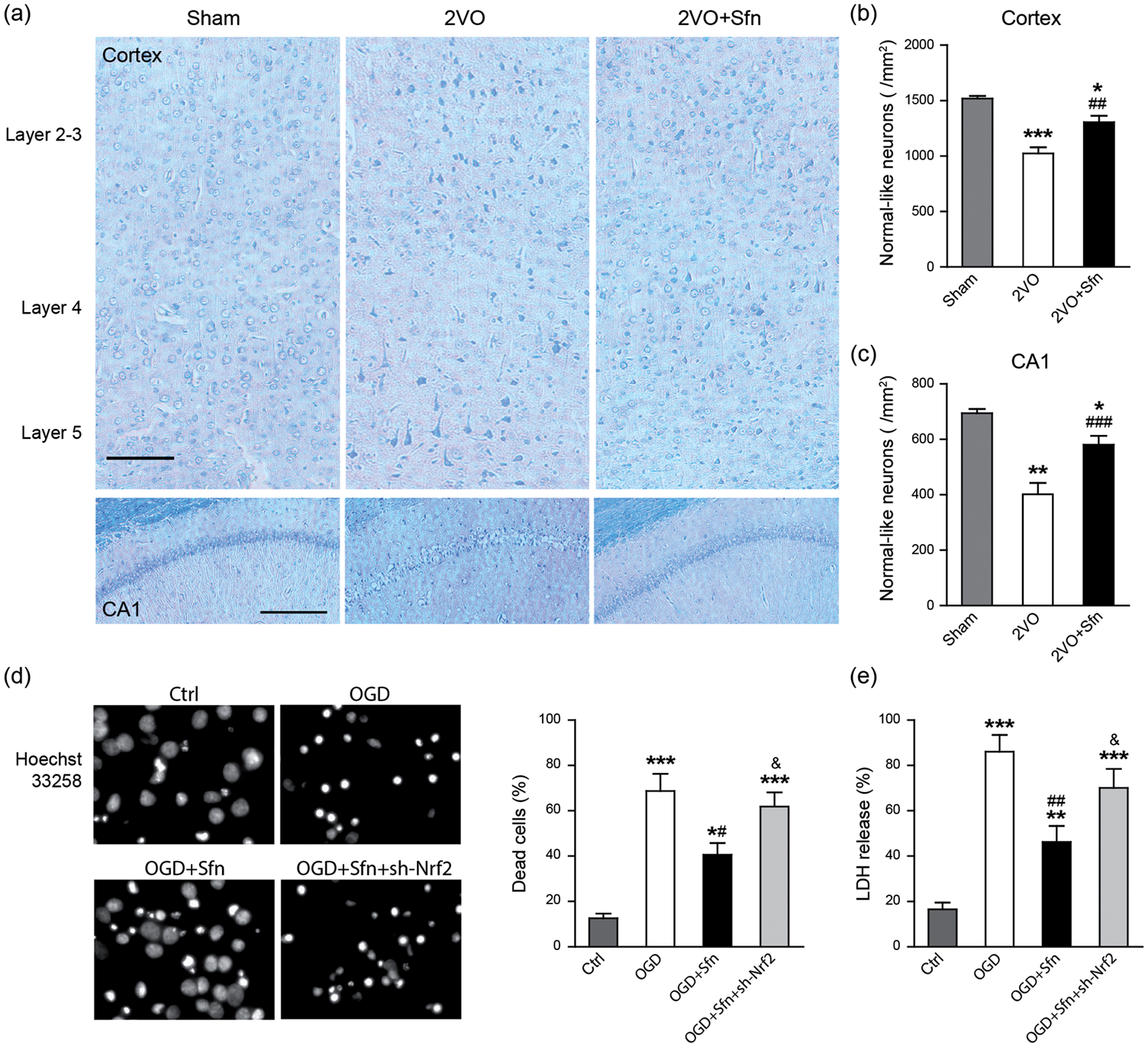
Sfn treatment attenuates myelin loss, myelin debris accumulation, and axonal injury in the WM after 2VO
Since WM injury has been considered as a characteristic feature of VCI pathology, we next investigated whether Sfn improves WM integrity after 2VO, including both myelin and axonal components. LFB staining showed that 2VO caused significant myelin loss in the CC/EC (Figure 3(a)) and the fimbria of hippocampus (Figure 3(b)) at day 42 after surgery, and Sfn treatment reduced myelin loss in these areas (Figure 3(a) and (b)). EM images showed that Sfn reduced abnormal axons (defined as axolysis and/or dense axoplasms) and myelin loss (Figure 3(c) to (e)). Compared to the Sham group (Figure 3(c)), 2VO caused the accumulation of myelin debris in the CC/EC, which is also alleviated by Sfn treatment (Figure 3(f)).
Sfn reduces myelin loss, myelin debris accumulation and axonal damage after 2VO. Paraffin sections at dorsal hippocampus were collected six weeks after 2VO and stained with LFB-CV. (a, b) Representative images of LFB staining in CC/EC (scale bar = 100 µm) and hippocampal fimbria (scale bar = 200 µm), with semi-quantitative analysis of LFB optic density (OD). Sfn reduced myelin loss in both regions after 2VO. n = 6, **p < 0.01, ***p < 0.001 vs. Sham; #p < 0.05, ##p < 0.01 vs. 2VO. Tissues from the CC/EC were processed for EM six weeks after 2VO. (c) Representative EM images of the CC/EC (scale bar = 2 µm), showing that Sfn reduced myelin loss (arrows) and abnormal axons (stars) defined as axolysis and/or dense axoplasms. (d) Analyses of abnormal axons and (e) assessment of G-ratio in the CC/EC confirmed Sfn-mediated axonal protection. n = 300 axons, *p < 0.05, ***p < 0.001 vs. Sham; #p < 0.05 vs. 2VO. (f) Representative EM images (scale bar = 1 µm) revealed accumulated myelin debris (arrowheads) in the CC/EC after 2VO, which was decreased by Sfn.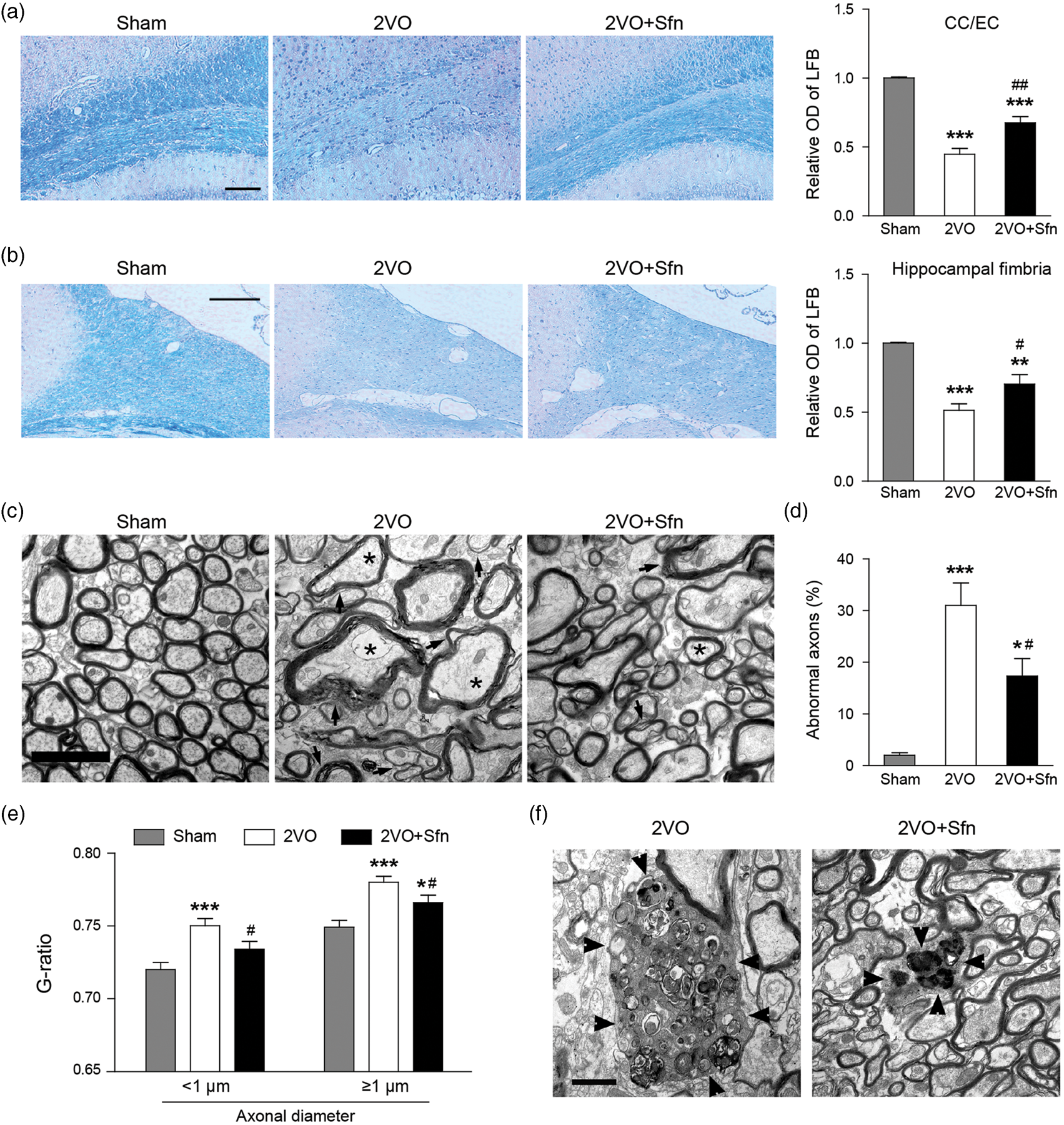
Then we tried to confirm the above findings by using specific WM markers, including MBP for myelin contents, SMI-32 for non-phosphorylated neurofilament H which represents injured axons, and NF-200 for total axonal neurofilaments.
52
Immunostaining showed that 2VO caused severe myelin loss and axonal injury in the CC/EC and striatum (Figure 4(a) to (e)) as indicated by the decreased levels of MBP and concomitant increased levels of SMI-32. Sfn treatment significantly attenuated the decreases in MBP and the increases in SMI-32 (Figure 4(a) to (e)). Western blots validated the immunostaining data (Figure 4(f) and (g)). The levels of NF-200 were also decreased in the CC/EC and striatum in 2VO, and Sfn treatment evidently attenuated loss of neurofilaments after 2VO in both CC/EC and striatum (Figure 4(h) and (i)). Collectively, these data indicated that Sfn reduced WM injury after 2VO, which may underlie the improved cognitive functions.
Sfn preserves myelin integrity after 2VO in rats. Six weeks after 2VO, the brain sections or tissues were collected. (a) Representative immunostaining images of MBP (green) and SMI-32 (red) in the CC/EC and striatum with (b–e) semi-quantifications of the respective fluorescence intensities. Sfn preserved myelin component and protected against axonal damage after 2VO. Scale bar = 50 µm. (f, g) Representative Western blots and semi-quantifications of MBP and SMI-32 were consistent with the immunostaining results. (h, i) Representative Western blots and semi-quantification of NF-200 in the CC/EC and striatum, showing that Sfn preserved axonal neurofilaments. n = 3–5. *p < 0.05, **p < 0.01, ***p < 0.001 vs. Sham; #p < 0.05, ##p < 0.01, ###p < 0.001 vs. 2VO.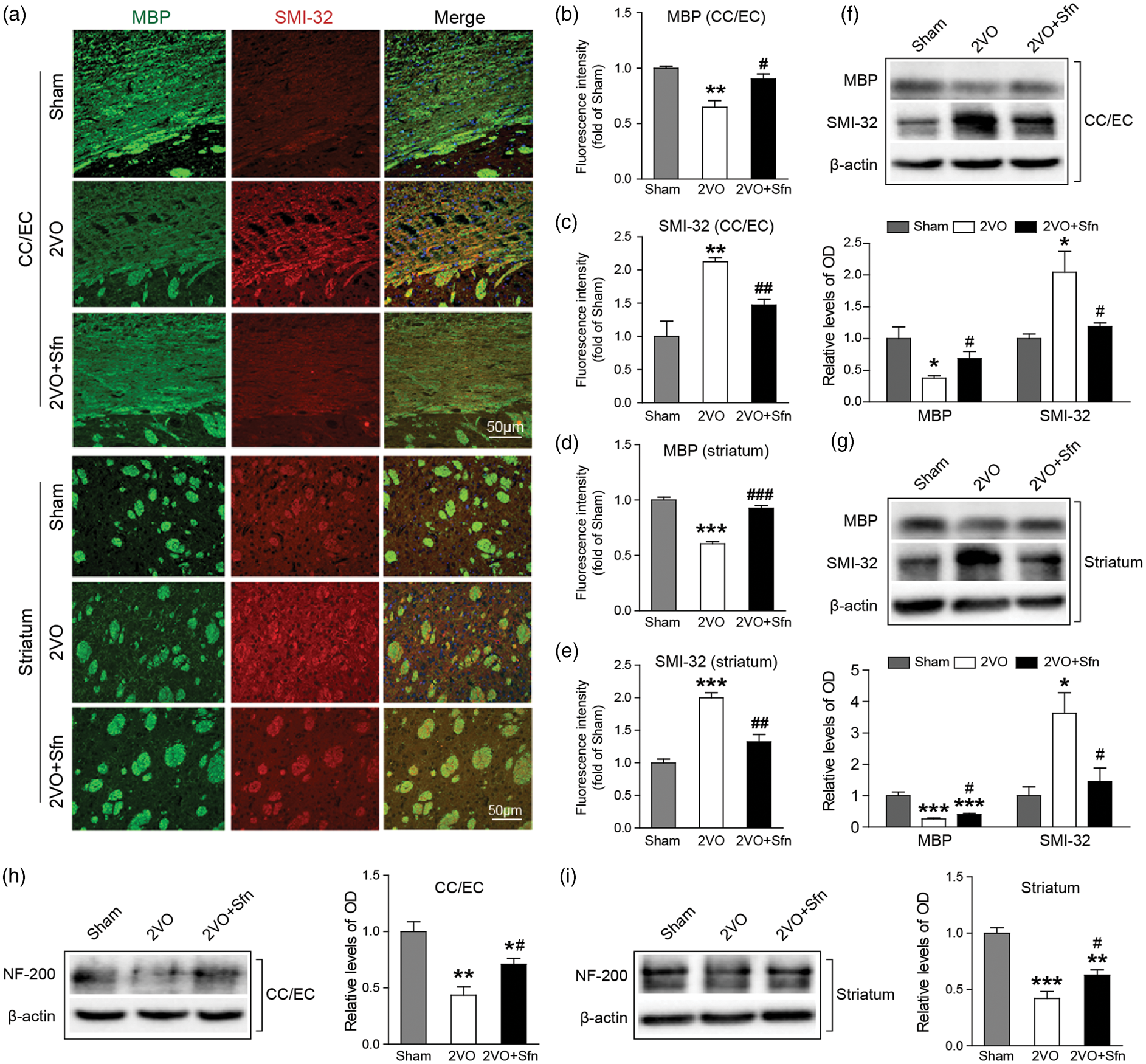
Sfn activates the Nrf2/HO-1 pathway in the brain after 2VO
We next tried to investigate the neuroprotective mechanism of Sfn. It has been reported that Sfn is a potent activator of Nrf2,
53
which may be the underlying mechanism of Sfn-mediated neuroprotection. Western blotting showed that the Nrf2 nuclear translocation and HO-1 expression were increased at day 3 and day 7 after 2VO, and Sfn treatment further enhanced the activation of Nrf2 (Figure 5(a)). Similarly, Sfn increased the activation of Nrf2 and expression of HO-1 up to six weeks after 2VO (Figure 5(b)), indicating that Nrf2 activation contributes to the long-term neuroprotective effects of Sfn.
Sfn activates Nrf2 and upregulates HO-1 expression after 2VO in rats. Cortical tissues were harvested at indicated time-points, and nuclear and cytosolic proteins were extracted. (a) Representative Western blots and semi-quantitative analysis within one week after 2VO, showing that 2VO-induced Nrf2 activation was enhanced by Sfn administration. (b) Representative Western blots and semi-quantitative at six weeks after 2VO, showing long-lasting activation of Nrf2 pathway by Sfn. *p < 0.05, **p < 0.01, ***p < 0.001 vs. Sham; #p < 0.05, ##p < 0.01 vs. 2VO. (c) Representative images of double-labeling of HO-1 with cellular markers (NeuN for neurons, GFAP for astrocytes and microvessels, Iba-1 for microglia/macrophages, and APC for mature oligodendrocytes) revealed wide-spread HO-1 expression after 2VO. Scale bar = 50 µm.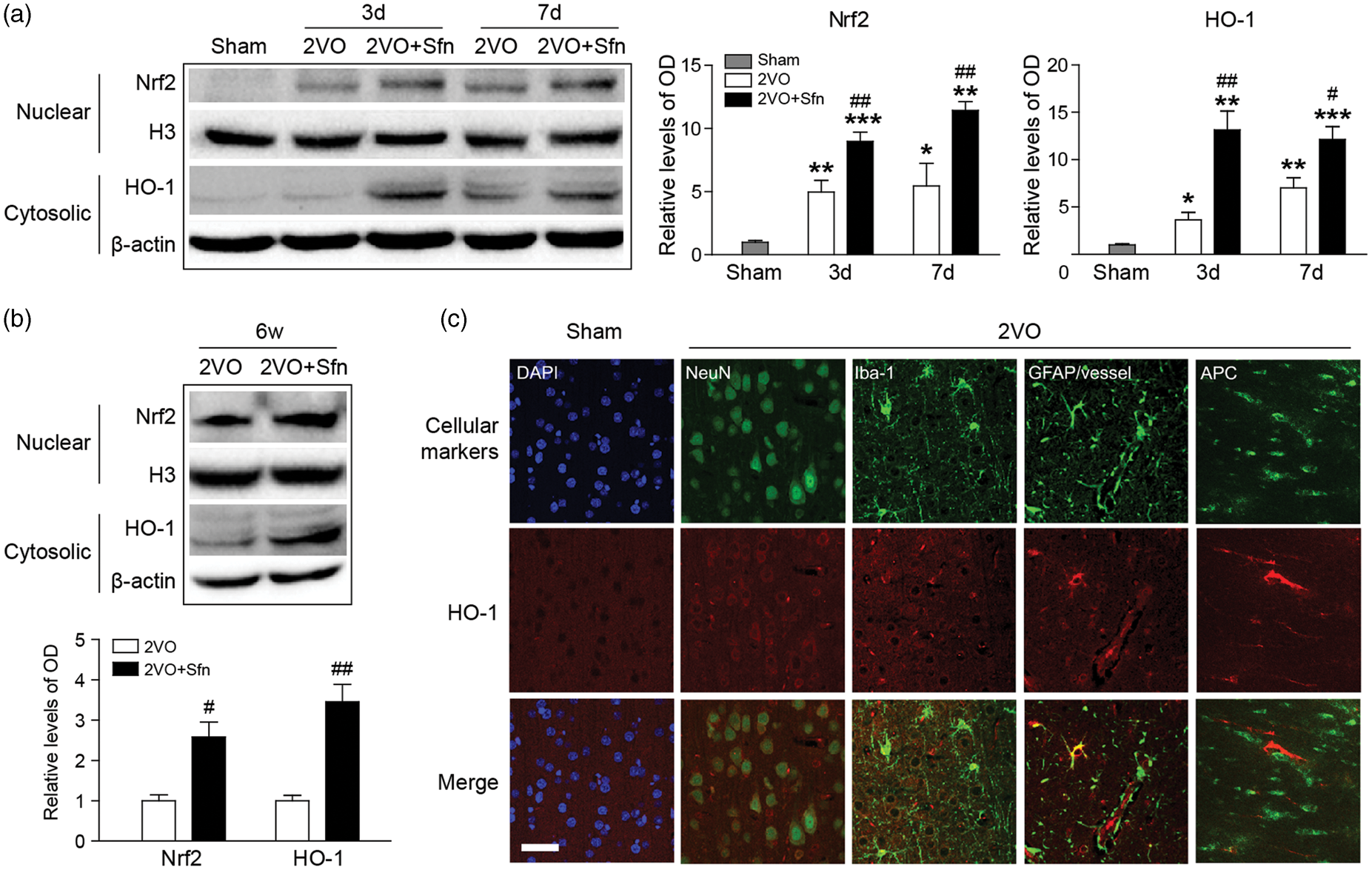
The cellular distribution of HO-1 was also examined in the cortex after 2VO (Figure 5(c)). Double-labeling studies showed that most of the NeuN-positive neurons expressed HO-1, though at a relatively low level. The GFAP-positive astrocytes near the CC/EC and microvessels expressed robust HO-1, while the astrocytes in other regions seldom expressed it. Microvessels also expressed strong HO-1. APC-positive mature oligodendrocytes expressed HO-1, too, especially for those at the boundary areas of the WM and gray matter. Iba-1-positive microglia/microphages expressed HO-1 in selected areas where tissue damages occurred. These results suggest that the Nrf2 pathway is activated in multiple cell types in the brain after 2VO in rats.
Sfn treatment preserves BBB integrity in vitro and in vivo
BBB disruption has been reported following VCI.13,14,54 To determine if Sfn protects the BBB, we first performed OGD in primary MBMECs. As shown in Figure 6(a) and (b), Sfn treatment significantly reduced MBMEC death induced by OGD, indicating that Sfn protects the MBMECs against ischemic injury. To detect if Sfn reduces BBB leakage, we then established an in vitro BBB model using MBMECs and transwells and subjected them to OGD. As shown in Figure 6(c), OGD severely increased BBB permeability and Sfn treatment partially but significantly ameliorated the BBB leakage, confirming its protective role against BBB disintegrity.
Sfn protects the BBB via Nrf2 activation and TJ preservation. MBMECs were subjected to OGD with or without Sfn pretreatment. (a) Live/dead staining (scale bar = 50 µm) with cell counting and (b) LDH assay showed that Sfn protected against OGD-induced cell death. (c) In an in vitro BBB model, Sfn partially reversed OGD-induced BBB leakage. ***p < 0.001 vs. Ctrl; #p < 0.05, ##p < 0.01, ###p < 0.001 vs. OGD. Nrf2 was knocked down by sh-Nrf2 transfection. (d) Live/dead staining (scale bar = 50 µm) with cell counting and (e) LDH assay revealed that Sfn-provided protection was abolished by Nrf2 knockdown. n.s.: not significant, **p < 0.01. (f) In vitro BBB permeability assessment without OGD showed that Nrf2 knockdown disrupted the BBB integrity. *p < 0.05, **p < 0.01, ***p < 0.001 vs. sh-Sc. (g) Dual-luciferase assay revealed that CLD5 promoter activity was decreased by Nrf2 knockdown. n.s.: not significant, **p < 0.01, ***p < 0.001. (h) Six weeks after 2VO, the brain sections were collected and immunohistochemistry showed that Sfn reduced CLD5 loss in the cortex (scale bar = 20 µm). (i–k) Representative Western blots and semi-quantitative analysis showed that Sfn reduced loss of CLD5 and occludin after 2VO. *p < 0.05 vs. Sham; #p < 0.05, ##p < 0.01 vs. 2VO.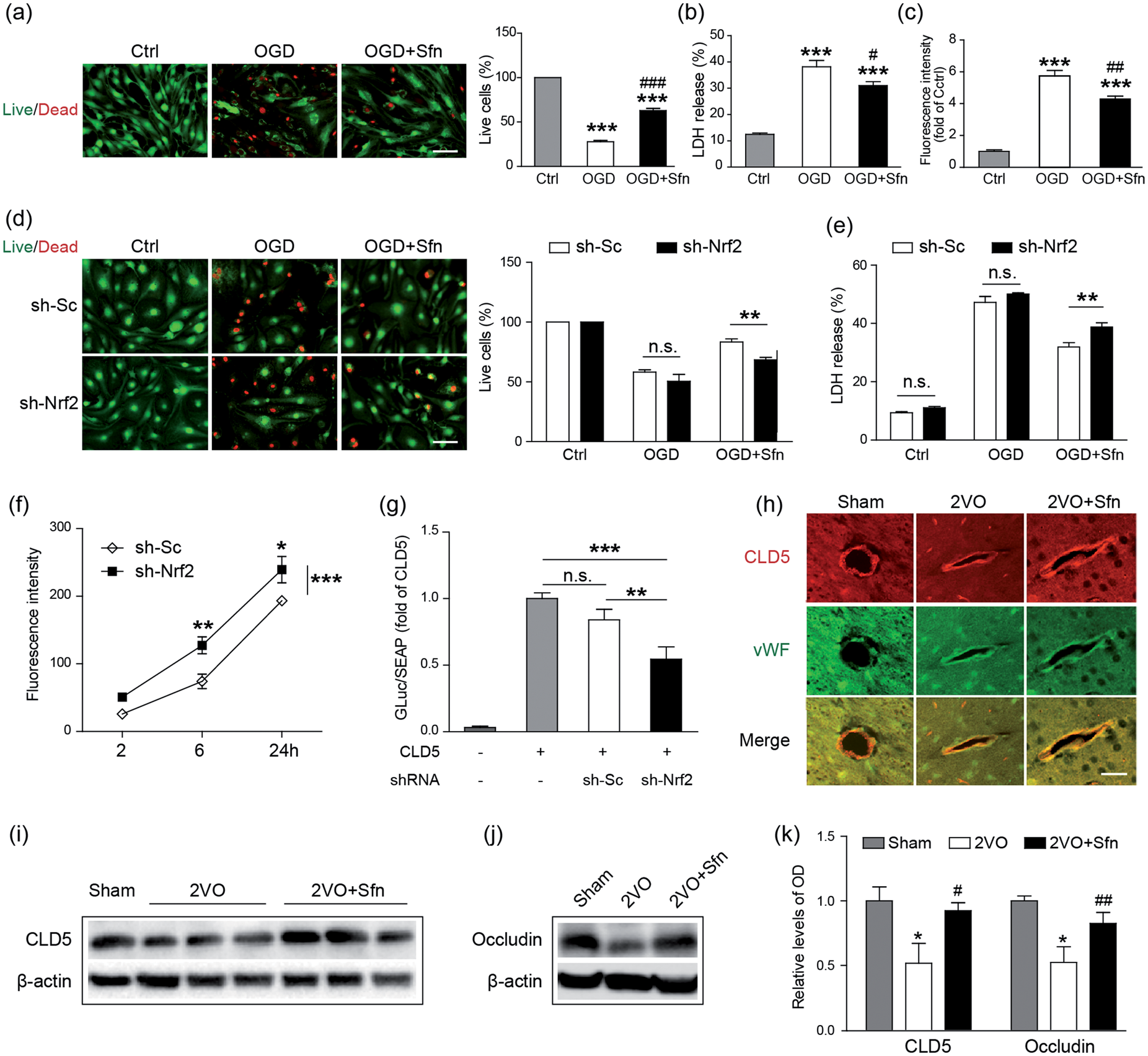
To detect the necessity of Nrf2 in Sfn-mediated MBMEC protection, we transfected the cells with lentiviral particles carrying sh-Sc or sh-Nrf2. While sh-Sc had no effects on the endothelial cell death, Nrf2 knockdown significantly blunted the protective role of Sfn against OGD (Figure 6(d) and (e)), indicating that Sfn protects endothelial cells in an Nrf2-dependent pattern.
On investigating the role of Nrf2 in maintaining BBB integrity, we found that Nrf2 knockdown in MBMECs led to increased BBB leakage in a time-dependent manner even without OGD (Figure 6(f)), indicating the critical role of Nrf2 in BBB preservation. Tight junctions (TJs) are the key structures of BBB. To investigate the mechanism for Nrf2 to preserve BBB function, we performed the promotor analysis of the three essential TJ proteins: CLD5, occludin, and zonula occludens-1 (ZO-1). Although we did not detect any binding sequences of Nrf2 on the promotors of occludin and ZO-1, we did identify a potential binding candidate of Nrf2 on CLD5 promotor. The DNA sequence for Nrf2 binding is known as ARE, and its consensus sequence is 5′-nTGACnnnGC-3′, in which TGA and GC are the core motifs and n can be any nucleotide.55–57 In the position of −1252 to −1243 of the CLD5 promotor lies a sequence of ATGACCTGGC that matches with ARE.58,59 To detect the effect of Nrf2 on CLD5 transcription, we co-transfected the MBMECs with plasmids carrying sh-Nrf2 and CLD5 promoter conjugated with luciferase reporters. As shown in the Figure 6(g), sh-Nrf2 significantly reduced the CLD5 promoter activity compared with sh-Sc, indicating that CLD5 may be a target of Nrf2, and that Nrf2 preserves BBB integrity through maintaining TJ protein CLD5.
To confirm the above findings, we detected the levels of the TJ proteins in the rat cortices six weeks after 2VO. The immunostaining showed that Sfn significantly attenuated CLD5 loss in the microvessels compared with the 2VO group (Figure 6(h)). Western blots confirmed these findings (Figure 6(i) and (k)). In addition, Sfn also reduced the loss of occludin (Figure 6(j) and (k)), probably via an Nrf2-independent mechanism. Taken together, these results suggest that Sfn treatment protects the BBB integrity via Nrf2.
Discussion
In this study, we showed that Sfn treatment significantly reduced neuronal death in the cortex and the hippocampal CA1, ameliorated the WM injury in the CC/EC, striatum, and hippocampal fimbria, and preserved the integrity of BBB, and whereby improved cognitive function after 2VO-induced VCI. We also demonstrated that the Sfn protective effects depend on the Nrf2 pathway. Moreover, we discovered that VCI is associated with the accumulated myelin debris in the CC/EC and that Nrf2 might directly upregulate CLD5. To our knowledge, these findings have not been reported previously.
Several potent Nrf2 activators are available, including Sfn, 2-cyano-3,12 dioxooleana-1,9 dien-28-oyl imidazoline (CDDO-Im) and 15-deoxy-Δ12,14-prostaglandin J2 (15-d-Δ12,14-PGJ2),26 etc. Although Sfn is not as strong as the latter two in activating Nrf2, we have decided to use Sfn based on its following advantages. First, Sfn is safe to use as it is a dietary component derived from cruciferous vegetables such as cauliflower, broccoli, and broccoli sprouts.34,60 Second, Sfn has BBB permeability. For example, Sfn and its metabolites can be detected in the animal brains shortly after admistration by gastric gavage. 61 Third, Sfn is more Nrf2 specific than CDDO-Im and 15-d-Δ12,14-PGJ2. The latter two have been reported to activate PPARγ,62,63 while Sfn has not been reported to do so.
As an electrophile, Sfn readily reacts with the thiol groups within the proteins to form thionoacyl adducts and changes the conformation of the proteins. This is considered the way in which Sfn inhibits Kelch-like ECH-associated protein 1 (Keap1) and activates the Nrf2 pathway.37,64 Consistent with the notion, we found that Sfn treatment induced Nrf2 nuclear translocation and HO-1 upregulation in the brain following 2VO, and the expression of HO-1 was widespread, including in neurons, astrocytes, oligodendrocytes, microglia/macrophages, and vascular endothelial cells. We had previously reported that HO-1 upregulation and Nrf2 activation induced by CDDO-Im protected the brain against acute ischemic injury, 32 and the data presented here moved forward to indicate that Sfn-mediated Nrf2 activation could also protect the brain against mild but chronic ischemic brain injury. However, we could not exclude the possibility that Sfn might protect via additional mechanisms, as it may react with the thiol groups in proteins other than Keap1. Nevertheless, our data suggest that Nrf2 contributes to the protective effects of Sfn.
WM injury is common in VCI and can exhibit as axonal injury, rarefaction of the WM tracts, enlarged perivascular spaces, and neuroinflammation.39,65 These pathological mechanisms ultimately lead to cognitive impairment.66,67 In the present study, we reported apparent axonal injury and myelin loss in both CC/EC and striatum following 2VO, and Sfn reduced these WM pathologies. Myelin sheath is a static structure and might maintain its assembly for a while even after the oligodendrocytes dies. Therefore, myelin debris tends to accumulate after WM injury, especially in the WM bundles or tracks. However, few studies have previously reported definite myelin debris in the WM, especially the accumulated debris. By using Oil-Red-O staining and conventional microscopy, Weinger et al. 68 showed myelin debris in the model of autoimmune encephalomyelitis. By using EM, the Johnson group marked the scattered myelin debris in sciatic nerve seven days after injury. 69 In the current study, we noticed a few clustered cylinder-like structures in the CC/EC that matched the myelin structure70,71 and which were therefore considered to be accumulated myelin debris.
The accumulation of myelin debris in the WM could serve as an indicator for the severity of WM injury for the following reasons. First, the amount of myelin debris represents the subtract between the total detached myelin and the portion that has been scavenged. And second, the myelin debris itself is neurotoxic. For instance, direct injection of myelin debris into the CC or cortex induced profound and comparable inflammation in mice with multiple sclerosis. 72 In rats, the myelin debris also inhibits remyelination in the spinal cord after a lysolecithin-induced demyelination. 73 Our results showed that Sfn reduced the accumulation of myelin debris in CC/EC, indicating that Nrf2 activation protects the WM after 2VO.
It is critical to clear the myelin debris to boost neural recovery. Microglial Nrf2 might play a role in the clearance of the myelin debris. For example, Nrf2 KO impaired the clearance of myelin debris and increased myelin debris in sciatic nerve seven days after the injury. 69 Two mechanisms might contribute to Nrf2-mediated myelin debris clearance. First, the activation of Nrf2 pathway could polarize microglia/macrophages to the M2-phenotype. One major function of the M2 microglia/macrophages is to clear cellular debris through phagocytosis.74–76 Second, Nrf2 has been implicated in the upregulation of the autophagy cargo protein sequestosome 1 (also known as fluorimeter p62). The Nrf2 activation creates a feed-forward loop for enhanced p62 expression, which strengthens both autophagy and phagocytosis.74,77–79
The BBB breakdown could be an upstream detrimental factor for WM injury and cognitive impairment in VCI, which occurs as early as the third day after the ischemic surgery in rodents.14,53,80 In addition to causing chronic ischemia and oxidative stress, BBB disruption leads to the accumulation of neurotoxic materials in the brain parenchyma and the perivascular spaces. For example, immunoglobin and albumin could induce brain edema and weaken the capillary blood flow, and thrombin could lead to neuronal injury and cognitive impairments. 81 IgG leakage in the CC/EC could affect the spatial memory performance. 82 In our study, we found that Sfn treatment effectively attenuated endothelial cell death and loss of TJ proteins, which contributes to its BBB preservation function.
One interesting finding of this study is that CLD5, a key component of the TJ, might be a new target gene of Nrf2. Several previous reports foreshadowed our findings. For instance, Sfn protects against autoimmune encephalomyelitis associated with an improved expression and distribution of CLD5. 83 The Nrf2 activation by Barleria lupulina induced increased expression of CLD5 mRNA in human endothelial cultures 84 and silencing Nrf2 abrogated CLD5 expression in human cerebral microvascular endothelial cultures. 85 Based on these and our own findings, we speculate that CLD5 might be a target gene of Nrf2. TRANSFAC-based promoter analysis shows that there is an ARE motif on the CLD5 promoter and the matrix score or binding strength of Nrf2 to the CLD5 ARE is as high as 0.825. Our results in Figure 6 suggest that CLD5 is a target gene of Nrf2.
The age of the subject animals is an important factor to model VCI as VCI is an elderly disorder. The rats we used in this study were six to seven months old, which is considered to be middle-aged to old. One reason why we have chosen these rats is that the pathophysiological process of VCI might start early in the middle-age or early old-age. Another reason is that the mid-aged rats have been used in numerous VCI studies, including those earliest ones,38,86 which makes our study comparable with previous ones. Even so, the age of the rats could be considered a limitation of the current study, and it should be cautious to interpret or apply our findings to the more elderly population. Further study with aged animals will help to determine the protective effects and mechanisms of Nrf2 activation.
In conclusion, our data reveal that Sfn treatment suppresses the neuronal loss and WM injury against VCI and improves cognitive functions in the rats, including their learning and memorizing abilities. We also suggested that the neuroprotection of Sfn might be related to the suppression of BBB breakdown, especially the maintenance of the TJ proteins. Further investigation should target to clarify the precise mechanisms that Sfn induces the ischemic tolerance to the brain. In summary, our study suggests that Sfn might be a promising therapeutic agent for the treatment of VCI.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the National Institutes of Health (NS092810 to F.Z.) and the Natural Science Foundation of China (81271276 to F.Z.). We thank Dr. Ken Arai of Harvard University for his academic suggestion, Jessica Zhang for her editorial support, and Patricia Strickler, Angela Tassari, and Michelle Jarvis for their administrative support.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
FZ and BS designed and supervised the project. LM and TY performed the in vitro experiments. LM, XLi, XLei, WZ, YS, YZ, and FZ performed the in vivo experiments. LM, TY, and FZ wrote and edited the manuscript, and YG, BS, and FZ read and edited the manuscript.