
Abstract
The activation of the bradykinin type 2 receptor is intricately involved in acute post-ischemic inflammatory responses. However, its precise role in different stages of ischemic injury, especially in the chronic phase, remains unclear. Following simultaneous cerebral and retinal ischemia, bradykinin type 2 receptor knockout mice and their controls were longitudinally monitored for 35 days via magnetic resonance imaging, fundus photography, fluorescein angiography, behavioral assessments, vascular permeability measurements, and immunohistochemistry, as well as glycemic status assessments. Without impacting the lesion size, bradykinin type 2 receptor deficiency reduced acute cerebral vascular permeability preventing the loss of pericytes and tight junctions. In the chronic phase of ischemia, however, it resulted in increased astrogliosis and cortical neuronal loss, as well as higher functional deficits. The retinal findings demonstrated a similar pattern. Bradykinin type 2 receptor deficiency delayed, but exacerbated the development of retinal necrosis, increased subacute vascular permeability, and promoted retinal ganglion cell loss in the chronic phase of ischemia. This investigation sheds light on the temporal dynamic of bradykinin type 2 receptor effects in ischemia, pointing to a therapeutic potential in the subacute and chronic phases of ischemic injury.
Introduction
Bradykinin (BK), a nonapeptide of the kallikrein-kinin system (KKS), is among the earliest inflammatory mediators released in response to cerebral and/or retinal ischemia.1 –5 BK exerts its biological effects by activating two types of G-protein coupled receptors: a constitutive bradykinin type 2 receptor (B2R) and an inducible bradykinin type 1 receptor (B1R).6 –10 The role of B2R in the initial stages of cerebral and retinal ischemia has been the subject of extensive research due to its rapid activation and its involvement in acute inflammation and edema induction.3,11 –13 Studies using pharmacological B2R antagonists in rodent models of temporary and permanent middle cerebral artery occlusion (MCAO) have shown a protective effect of B2R inhibition in the acute phase of ischemia.1,14 –16 Moreover, the timing of BK release and B2R expression in the ischemic brain correlates significantly with the progression of cell death, underscoring the contribution of B2R to the initiation of apoptosis and necrosis following ischemia.3,11 BK administration and B2R activation are known to disrupt the blood-brain/retinal barrier (BBB/BRB) through the stimulation of eNOS and PLA2, resulting in increased NO and prostaglandin production, leading to plasma protein extravasation and the formation of vasogenic edema.17 –19 In rats, intravitreal BK administration has been observed to induce changes in vascular integrity and significant retinal thickening. 12 Furthermore, BK plays a chemoattractive role in promoting the migration and infiltration of immune cells, thereby exacerbating the inflammatory response.14,20
Despite the known detrimental consequences of B2R activation, beneficial effects in brain and retinal ischemia have also been shown for BK, as well as for other components of KKS. For example, in a rat MCAO model, it was shown that intracerebroventricular kallikrein gene transfer had a neuroprotective effect after reperfusion. The activation of B2R led to the suppression of oxidative stress and caspase-3 activity, an increase in NO concentration, acute reductions in lesion size, and functional deficits observed in vivo, along with enhanced cell survival, neurogenesis, and angiogenesis in vitro.21,22 Similarly, intravenous administration of tissue kallikrein yielded positive effects in a model of retinal ischemia-reperfusion injury, preventing ganglion cell death, reducing vascular permeability, and improving visual function. 23 B2R activation has also demonstrated its ability to counteract glutamate excitotoxicity in cultured rat brain neurons 24 and retinas in vitro. 25 Additionally, its potential anti-inflammatory effects were highlighted in an in vitro model where BK stimulation of microglia led to a reduction in the secretion of pro-inflammatory cytokines TNF-α and IL-1β. 26
Although past research has generated numerous findings, often with conflicting results, it underscores a possible dual role of B2R activation in ischemia. 27 In this study, we examined the brains and retinas of mice subjected to ischemia with chronic hypoperfusion, using longitudinal magnetic resonance vascular perfusion and morphology imaging, coupled with assessments of functional recovery, to investigate the role of B2R signaling across different stages of ischemic injury.
Materials and methods
Key resources that are essential to reproduce the results are provided in Supplementary material Supplementary Table 1.
Experimental animals
All animal handling and procedures were approved by the Ethics Licensing Committee of the University of Zagreb School of Medicine and the Ethics Committee for the protection of animals used for scientific purposes of the Ministry of Agriculture Republic of Croatia (No. 380-59-10106-18-111/49). Experimental procedures were conducted according to the Croatian Animal Protection Act (NN 102/17, 32/19), Amendments to the Animal Protection Act (NN 37/13), and the Guidelines on the Protection of Animals Used for Scientific Purposes (NN 55/13) in line with the European Directive 2010/63/EU. The reported experiments followed the ARRIVE and STAIR guidelines.28,29 The experiments were carried out on male C57BL/6J (wild type (WT); RRID: IMSR_JAX:000664; n = 32) and B6;129S7-Bdkrb2tm1Jfh/J backcrossed across 11 generations to C57BL/6J (B2R-KO; RRID:IMSR_JAX:002641; n = 32) 3–6 months old mice, purchased from Jackson Laboratories (Bar Harbor, USA) and bred at the animal facility of the Croatian Institute for Brain Research, University of Zagreb School of Medicine. The animals were housed on a 12-hour light-dark cycle in a humidity-controlled environment at 22 ± 2°C. Water and pelleted food were given ad libitum. Experimental group assignments were randomized with a lottery drawing box. All efforts were made to minimize animal suffering and the number of animals used. All procedures and data analyses were performed by investigators blinded to animal genotypes and experimental grouping.
Non-invasive measurement of blood and intraocular pressure
Blood and intraocular pressure measurements in anesthetized mice were assessed one week before ischemia induction. Details are provided in the Supplemental Methods.
Assessment of the glycemic status
Glycemic status assessment was conducted 8 days prior and 34 days after MCAO by tail vein blood sampling. The procedure involved collecting blood for measuring HbA1c levels, and glucose concentration or to perform the intraperitoneal glucose tolerance test (IPGTT; See Supplemental Methods for details).
Induction of cerebral and retinal ischemia
Cerebral and retinal ischemia, followed by chronic hypoperfusion, were induced by a 30-minute left MCAO using the recently redefined Koizumi intraluminal filament method.30,31 Details are provided in the Supplemental Methods.
Magnetic resonance imaging
MRI experiments were conducted 7 days before and 2, 9, and 35 days post-MCAO using a 7T system (Bruker Biospin, Germany) as previously described. 31 The protocols for brain and eye anatomical and angiography imaging, data processing, and analysis are provided in Supplemental Methods.
Fundus photography and fluorescein angiography
Fundus photography and fluorescein angiography were performed 7 days before and 2, 9, and 35 days after MCAO, immediately before the MRI session using an in-house smartphone ophthalmoscopy setup. The protocols for imaging, data processing, and analysis are provided in Supplemental Methods.
Behavioral tests
Animals underwent a battery of motor function tests, including the rotarod, 32 wire hanging, 33 and pole34 tests one day before each MRI session. Before the initial test, mice underwent a four-day training period to adapt and acquire the necessary skills. (see Supplemental Methods).
Neurological deficit scoring and weight measurement
Neurological deficit scoring (NDS) and weight measurements were conducted before each fundus photography and MRI session. NDS involved a series of tests evaluating motor and sensory status, reflex functionality, and appearance, graded on a scale of 0–39,35 –37 with higher scores signifying greater injury severity.
Blood-brain/retinal barrier permeability assessment using Evans blue and sodium fluorescein
The permeability of the BBB and BRB was assessed on days 2 and 9 post-MCAO using Evans blue (EB) and sodium fluoresceine (NAF). Permeability was indirectly assessed by measuring the fluorescence intensity of the cerebral and retinal tissue ex vivo using the IVIS® Spectrum system (Perkin Elmer, USA). See Supplemental Methods for details.
Brain microvasculature isolation
The microvasculature was isolated from the ipsilateral hemisphere on days 2 and 9 post-MCAO, following an adapted isolation procedure. 38 See Supplemental Methods for isolation, immunohistochemical processing, and analysis details.
Histochemistry, immunohistochemistry and data analysis
After the final MRI session 35 days post-MCAO, animals were anesthetized by i.p. injection of 2.5% tribromoethanol (Sigma-Aldrich), transcardially perfused, and fixed. Brains were isolated, cryoprotected, embedded, sectioned into 25-µm-thick coronal brain slices, and immunostained. For astrocyte and neuron fluorescent signal analysis see details in Supplemental Methods and Supplemental Figure 2. showing the precise locations of analysis on representative brain sections. The eyes were enucleated, processed, and analyzed as described previously. 31 See Supplementary Methods for details.
Sample size calculation
Based on our preliminary data, The power analysis (power = 0.8; alpha = 0.05; two-sided Mann-Whitney U-test) performed with G*Power software 39 and based on preliminary data estimated 8 animals per MCAO group would be required to detect differences in acute lesion volume. However, to account for attrition due to an expected mortality rate of up to 40%, the sample size was corrected to 12 animals per MCAO group (Ncorrected = N/(1−[% attrition/100]).
Statistical analysis
The statistical analysis was conducted using GraphPad Prism 9. Animals were excluded from analysis if the MRI conducted on day 2 post-MCAO suggested a failed surgical procedure. To assess survival, we employed the product limit Kaplan-Meier method. The Shapiro-Wilk test was utilized to confirm the normality of distribution. Outliers were assessed using the ROUT method. Differences in systolic, diastolic, and mean arterial pressure, intraocular pressure between genotypes, as well as retinal NeuN and GFAP expression were assessed using a two-tailed Student's t-test. For MCA perfusion, a two-way repeated-measures ANOVA with Šidak's post hoc test was employed. To analyze variations in body weight, basal and fasting glucose, HbA1c, IPGTT, arterial vasculature volume, ischemic lesion volume, normalized hemisphere volumes, tissue loss, neurological deficit, rotarod, wire hanging test, pole test, average retinal thickness, normalized arteriolar and venular diameter, tortuosity and capillary area we used a mixed two-way ANOVA model with time and genotype as variables. Post hoc comparisons between genotypes were performed with Šidak's test, while Tukey's test was applied for comparisons within genotypes at specific time points. For EB and NAF extravasation, pericyte count, ZO-1, cerebral NeuN and GFAP expression, a mixed one-way ANOVA model was used, with false discovery rate-controlled correction for multiple comparisons at an α level of 0.05. Statistical significance was defined as P < 0.05.
Results
In order to investigate the role of B2R deficiency in ischemic brain and retinal injury, a total of 64 male B2R-KO and WT littermates were utilized. Four animals from each cohort were excluded after the initial MRI conducted on day 2 post-MCAO suggested failed surgical procedures, including lacunar lesions outside the area of MCA irrigation (n = 4), hemorrhage from arterial puncture (n = 3), and unsuccessful induction of ischemic injury (n = 1).
B2R deficiency exhibited no discernible impact on blood pressure, intraocular pressure or intraoperative MCA perfusion status
To evaluate the influence of B2R deficiency on blood pressure and intraocular pressure, which could potentially alter the course of ischemic lesion progression, non-invasive measurements were conducted on anesthetized mice pre-MCAO. B2R deficiency did not affect systolic (Supplemental Figure 1B), diastolic (Supplemental Figure 1C), or mean arterial blood pressure (Supplemental Figure 1D), nor intraocular pressure (Supplemental Figure 1E). Moreover, to eliminate the possibility of B2R deficiency altering the intraoperative MCA perfusion status, laser-Doppler flowmetry (LDF) was performed intraoperatively (Supplemental Figure 1F). Both groups exhibited comparable LDF signal values before filament insertion, followed by a substantial decline in perfusion during MCA occlusion (P < 0.0001), and no significant alterations during the 30-minute occlusion period. After filament withdrawal, both groups displayed a similar and substantial increase in LDF values (P < 0.0001), reaching approximately 70% of the initial LDF values.
B2R deficiency does not affect acute ischemic lesion volume despite modulating vascular permeability
Volumetric analysis of ischemic lesions revealed a significant reduction in lesion volume from acute to subacute phase for both groups (P < 0.0001). Notably, there were no differences in ischemic lesion volume between the groups at either time point (Figure 1(a)).
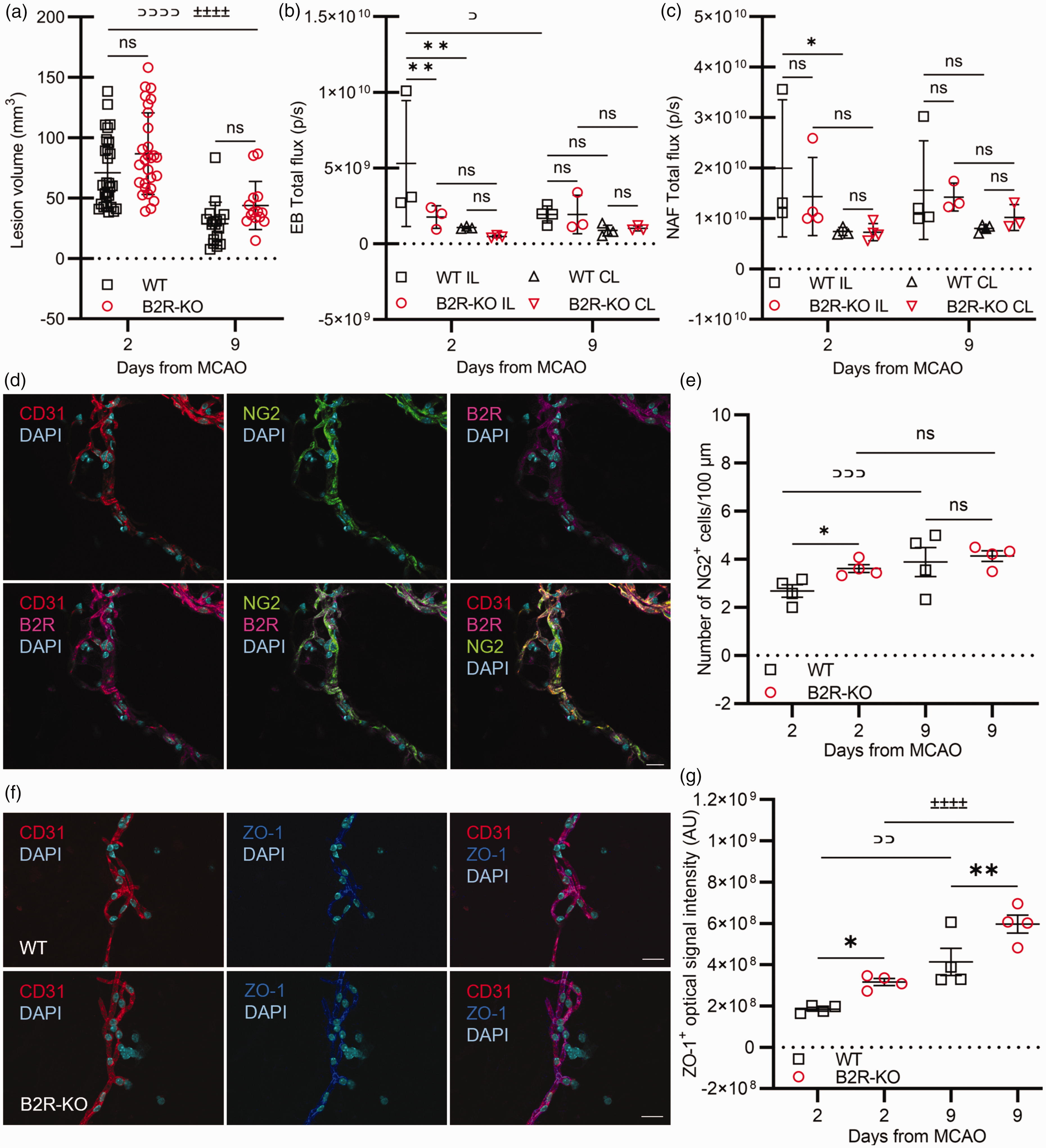
B2R deficiency modulates vascular permeability without affecting the acute lesion volume. (a) B2R deficiency does not have a significant effect on ischemic lesion volume. Repeated measures ANOVA with Šidak's post hoc test within genotype: WT (⊃⊃⊃⊃) P < 0.0001; B2R-KO (±±±±) P < 0.0001. WT day 2 n = 28, day 9 n = 17; B2R-KO day 2 n = 28; day 9 n = 14. (b–c) Analysis of Evans blue (EB) and sodium fluorescein (NAF) extravasation (n = 4 per group/time point). Mixed ANOVA with correction for multiple comparisons controlling for false discovery rate (α = 0.05) among genotypes and between hemispheres (*) P < 0.05; (**) P < 0.01; within genotype: WT (⊃) P < 0.05. (d) Representative images of bradykinin type 2 receptor (B2R; magenta) marker localization on endothelial cells (CD31; red) and pericytes (NG2; green) in triple-stained cerebral microvessels. Cyan – DAPI-positive nuclei. Scale bar: 20 µm. (e) Quantification of NG2+ pericytes and (g) quantification of ZO-1+ proteins in cerebral microvessels at days 2 and 9 post-MCAO (n = 4 mice per group/time point). One-way ANOVA with correction for multiple comparisons controlling for false discovery rate (α = 0.05) among genotypes (*) P < 0.05, (**) P < 0.01; within genotype: WT (⊃⊃) P < 0.01, (⊃⊃⊃) P < 0.001; B2R-KO (±±±±) P < 0.0001. (f) Representative images of tight junction protein ZO-1 (dark blue) marker on endothelial cells (CD31; red) in double-stained cerebral microvessels. Scale bar: 20 µm.
Measurement of EB fluorescence, indicative of albumin extravasation, revealed significantly higher ipsilateral hemisphere (ILH) fluorescence compared to the contralateral hemisphere (CLH) in WT mice on day 2 post-MCAO (P < 0.01; Figure 1(b)). By day 9, ILH fluorescence had significantly decreased compared to day 2 (P < 0.05; Figure 1(b)). In the B2R-KO group, no significant differences were observed in EB fluorescence levels between hemispheres or across days (Figure 1(b)). On day 2 post-MCAO, the B2R-KO group exhibited significantly lower EB fluorescence compared to the control group (P < 0.01; Figure 1(b)). Measurement of NAF fluorescence, representative of small molecule extravasation, also showed significantly higher ILH fluorescence compared to the CLH in WT mice on day 2 (P < 0.05; Figure 1(c)). In the B2R-KO group, no significant differences in NAF fluorescence were observed between hemispheres or across days (Figure 1(c)). Additionally, there were no significant differences in NAF fluorescence between groups (P=ns; Figure 1(c)).
Immunofluorescence analysis confirmed the expression of B2R in endothelial cells and pericytes of isolated cerebral microvessels (Figure 1(d)). On day 2 post-MCAO, WT mice had a significantly lower number of NG2-positive pericytes on microvessels compared to B2R-KO (P < 0.05; Figure 1(e)). This number significantly increased toward day 9 (P < 0.001; Figure 1(e)). Immunolabeling of tight junctions showed significantly lower expression of ZO-1 (Figure 1(f)) in the ILH microvessels isolated from the control group compared to B2R-KO mice on day 2 post-MCAO (P < 0.01; Figure 1(g)). On day 9, both groups exhibited significantly higher ZO-1 expression compared to day 2 post-MCAO (WT P < 0.001; B2R-KO P < 0.0001; Figure 1(g)). However, the expression in WT mice remained significantly lower than in the B2R-KO group (P < 0.0001; Figure 1(g)).
B2R deficiency impacts cerebral arterial perfusion, glycemic status, and tissue loss in the chronic phase of ischemia
Analysis of cerebral arterial perfusion, maximum intensity projections (Figure 2(a)), and arterial blood vessel volumes (Figure 2(b)) indicated that both groups experienced hypoperfusion on day 2 post-MCAO and significantly smaller arterial vasculature volumes of the ILH compared to baseline values (P < 0.0001). On day 9, no significant alterations were observed compared to day 2. In the chronic phase, both groups displayed a substantial perfusion increase (WT P < 0.05; B2R-KO P < 0.0001). However, while WT mice reached baseline values, B2R-KO mice showed a more pronounced rise in arterial perfusion with significantly higher arterial volume compared to baseline and to controls (P < 0.05). The CLH of WT mice did not exhibit significant changes in arterial perfusion (Figure 2(c)), unlike the B2R-KO group, which displayed a significant increase in the acute phase (P < 0.05), a subacute decline, and a notable increase in the chronic phase of ischemia (P < 0.001).
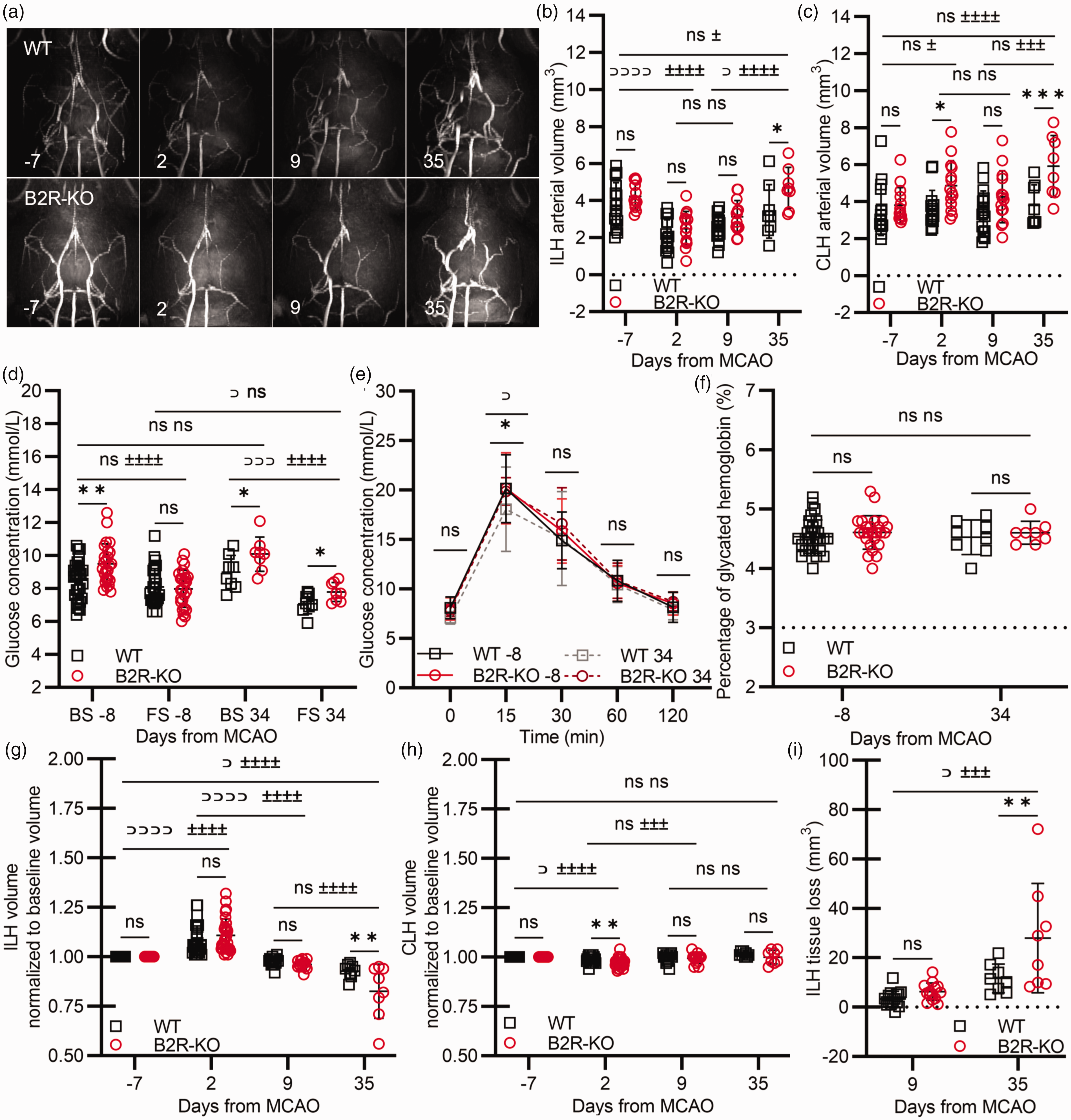
B2R deficiency impacts chronic cerebral arterial perfusion, glycemic status, and tissue loss after cerebral ischemia. (a) Representative maximum intensity projections of MR angiography scans. (b–c) Effects of B2R deficiency on the arterial perfusion status of the ipsilateral (ILH; B) and contralateral (CLH; C) hemispheres pre and post-MCAO. Analysis of (d) blood glucose concentration, (e) intraperitoneal glucose tolerance test, and (f) glycated hemoglobin HbA1c before and 34 days post-MCAO. (g–i) ILH (G) and CLH (H) volume and tissue loss (I). Mixed model ANOVA with Šidak’s post hoc test between genotypes: (***) P < 0.001, (**) P < 0.01, (*) P < 0.05; or Tukey’s post hoc test among time points: WT (⊃⊃⊃⊃) P < 0.0001, (⊃⊃) P < 0.01, (⊃) P < 0.05; B2R-KO (±±±±) P < 0.0001, (±±±) P < 0.001, (±) P < 0.05. Pre-MCAO and day 2 n = 28 per group; day 9 WT n = 17, B2R-KO n = 14; day 35 n = 8 per group.
Since previous research indicates the role of B2R in the regulation of glucose metabolism, 40 glycemic status was evaluated pre and post-MCAO. Basal glucose levels pre-MCAO were significantly elevated in B2R-KO mice compared to controls (P < 0.01; Figure 2(d)). After a 6-hour fasting period, B2R-KO mice exhibited a marked decrease in glucose values, a change not observed in controls (P < 0.0001). In the chronic phase of ischemia, controls experienced a significant drop in fasting glucose levels compared to B2R-KO group (P < 0.05) and to pre-MCAO values (P < 0.01; Figure 2(d)). Moreover, pre-MCAO IPGTT results indicated that blood glucose changes post glucose administration were equal in both groups (Figure 2(e)). However, 34 days post-MCAO, the IPGTT curve exhibited significant flattening in the WT group compared to pre-MCAO values (P < 0.05) and compared to the B2R-KO group (P < 0.05). Interestingly, HbA1c measurements displayed no group differences either pre- or post-MCAO (P = ns; Figure 2(f)).
The evaluation of hemisphere volume indicated a significant increase in ILH volume on day 2 post-MCAO for both groups (P < 0.0001; Figure 2(g)), which then gradually decreased toward day 35 (WT P < 0.05; B2R-KO P < 0.0001). No significant differences in ILH volume were observed between groups in the acute or subacute phases. However, the ILH volume of the WT group was significantly greater in the chronic phase of ischemia compared to the B2R-KO group (P < 0.001), mainly due to more pronounced tissue atrophy in the B2R-KO group (Figure 2(i)). In response to the increased ILH volume, both groups experienced compensatory contralateral hemisphere shrinkage on day 2 post-MCAO (WT P < 0.05; B2R-KO P < 0.0001; Figure 2(h)). Subsequently, the CLH volume returned to baseline values for both groups. Although no significant differences in ILH volumes were observed between groups in the acute phase (Figure 2(g)), the CLH volume of the B2R-KO group was significantly smaller in the acute phase compared to the control (P < 0.01; Figure 2(h)).
B2R deficiency increases acute neurological deficit following ischemia without affecting survival
Most deaths in both groups (n = 7) occurred within the first week post-MCAO. Survival curve analysis revealed no significant difference between groups (Figure 3(a)). Acutely, both groups had a significant decrease in body weight compared to baseline values (P < 0.0001). B2R deficiency did not affect body weight at any measured point post-MCAO (Figure 3(b)).
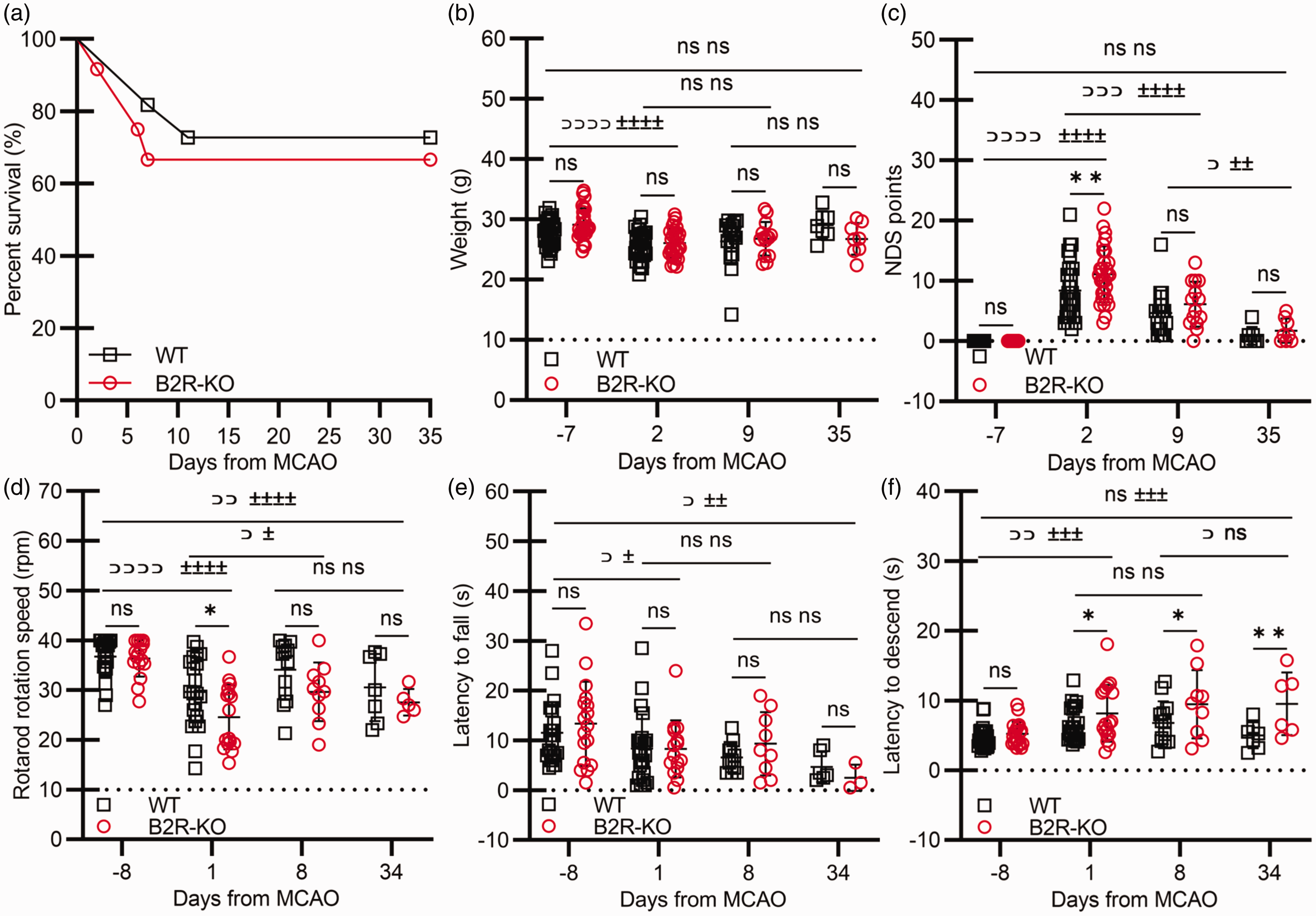
B2R deficiency heightens acute neurological deficit and chronic neurological functions following ischemia without affecting survival. B2R deficiency does not affect (a) survival proportions assessed by the Log-rank (Mantel-Cox) test or (b) animal weight post-MCAO. (c) Neurological status and (d–f) sensorimotor functions assessed using the (d) rotarod test, (e) wire hanging test and (f) pole test. Mixed model ANOVA with Šidak's post hoc test between groups at individual time points: (*) P < 0.05, (**) P < 0.01, or Tukey's post hoc test among time points: WT (⊃⊃⊃⊃) P < 0.0001, (⊃⊃⊃) P < 0.001, (⊃⊃) P < 0.01, (⊃) P < 0.05; B2R-KO (±±±±) P < 0.0001, (±±±) P < 0.01, (±±) P < 0.01. Pre-MCAO and day 2 n = 28 per group; day 9 WT n = 17, B2R-KO n = 14; day 35 n = 8 per group.
On day 2, both groups exhibited significant neurological impairment compared to baseline (P < 0.0001; Figure 3(c)), with the control group displaying better neurological status than the B2R-KO group (P < 0.01; Figure 3(c)). By day 9, there was a noteworthy recovery in neurological function in both groups (WT P < 0.001; B2R-KO P < 0.0001). By day 35, measurements returned to baseline values (P = ns). In the subacute and chronic phases, there were no differences between groups (P = ns).
On day 1 post-MCAO, both groups had shorter rotarod latencies (P < 0.0001; Figure 3(d)). By day 8, these improved (P < 0.05), but did not improve further in the chronic phase or reached baseline values (WT P < 0.01; B2R-KO P < 0.001). In the acute phase, the B2R-KO group showed significantly lower rotational speed at the time of falling compared to the control group (P < 0.05; Figure 3(d)), but there were no differences between groups at other time points.
There was a significant reduction in wire hanging times in the acute phase (P < 0.05; Figure 5(e)), which did not improve by day 34. In the chronic phase, both groups had significantly shorter wire hanging times compared to baseline (WT P < 0.05; B2R-KO P < 0.01), with no differences between groups at any time point (Figure 3(e)).
On day 1 post-MCAO, animals required significantly more time to descend from the pole (WT P < 0.01; B2R-KO P < 0.001; Figure 3(f)). By day 34, the control group regained baseline descent time, while the B2R-KO group still descended significantly slower than at baseline (P < 0.001). The control group exhibited faster descent times than the B2R-KO group in the acute (P < 0.05), subacute (P < 0.05), and chronic phases (P < 0.01; Figure 3(f)).
B2R deficiency is associated with pronounced neuronal loss and increased astrogliosis in the cortical ischemic lesion area
B2R-KO mice displayed pronounced neuronal loss in the cortical ischemic lesion area compared to the control group (P = 0.05; Figure 4(b)). A significant difference was also observed in the density of GFAP+ astrocytes, with WT mice displaying lower density in the cortical ischemic lesion area compared to B2R-KO animals (P = 0.0001; Figure 4(d)). However, no significant differences between the experimental groups were detected in NeuN+ and GFAP+ density in the perilesional cortex (Figure 4(b) and (d)) or the lesional and perilesional striatum (Figure 4(c) and (e)).
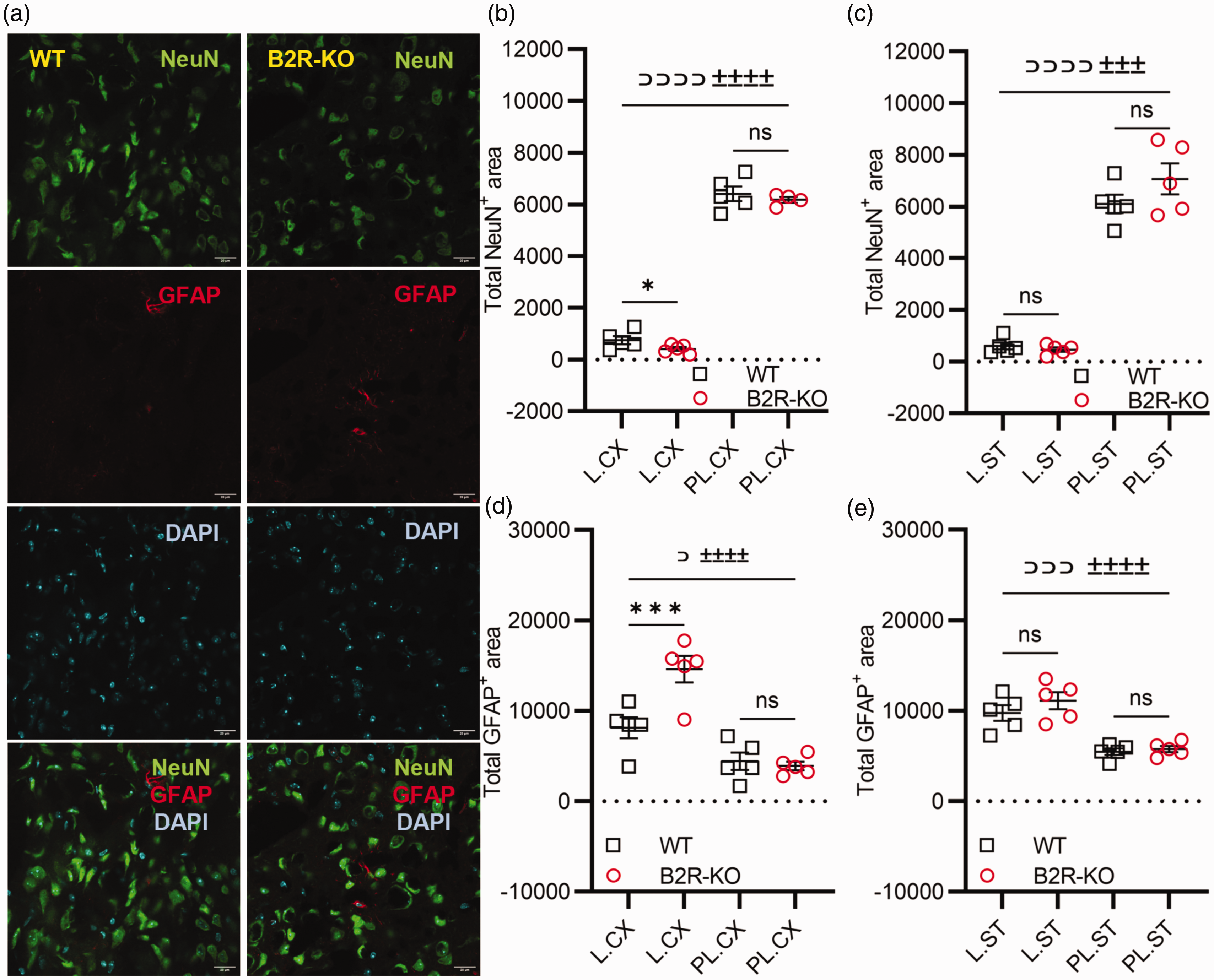
B2R deficiency is associated with pronounced neuronal loss and increased astrogliosis in the ischemic lesion. (a) Representative images of neuronal nuclei (NeuN; green) marker and astrocyte glial fibrillary acidic protein (GFAP; red) marker in the double-stained perilesional area of the cortex 35 days post-MCAO. Cyan – DAPI-positive nuclei. Scale bar: 20 µm. (b) Quantification of NeuN+ neurons and (d) GFAP+ astrocytes in the cortical lesion (L.CX) and perilesional (PL.CX) areas and (c) neurons and (e) astrocytes in the striatal lesion (L.ST) and perilesional (PL.ST) areas 35 days post-MCAO. One-way ANOVA with correction for multiple comparisons controlling for false discovery rate (α = 0.05) between genotypes (*) P < 0.05, (***) P < 0.001; within genotype: WT (⊃) P < 0.05, (⊃⊃⊃) P < 0.001, (⊃⊃⊃⊃) P < 0.0001; B2R-KO (±±±±) P < 0.0001, (±±±) P < 0.01. n = 5 mice per group/time point.
B2R deficiency leads to delayed onset of retinal necrosis
To assess the impact of B2R deficiency on retinal status, fundus photographs were examined pre and post-MCAO (Figure 5(a)). On day 2, two mice in the WT group developed ocular media opacification due to ischemic complications of the anterior segment, hindering image analysis. Among the remaining 26 mice in this group, 84.6% exhibited distinct signs of retinal necrosis (Figure 5(c)). In the B2R-KO group, four mice had ocular media opacification, and of the remaining 24, 70.8% displayed signs of retinal necrosis (Figure 5(a) and (c)). On day 9, three WT mice developed ocular media opacification, while 50.0% of the remaining 14 showed signs of retinal necrosis. In the B2R-KO group, two mice displayed ocular media opacification, and of the remaining 12, 83.3% exhibited signs of retinal necrosis (Figure 5(d)). In the chronic ischemic phase, only one WT mouse had ocular media opacification. In the B2R-KO group, 50% of the mice still displayed clear signs of retinal necrosis (Figure 5(e)).
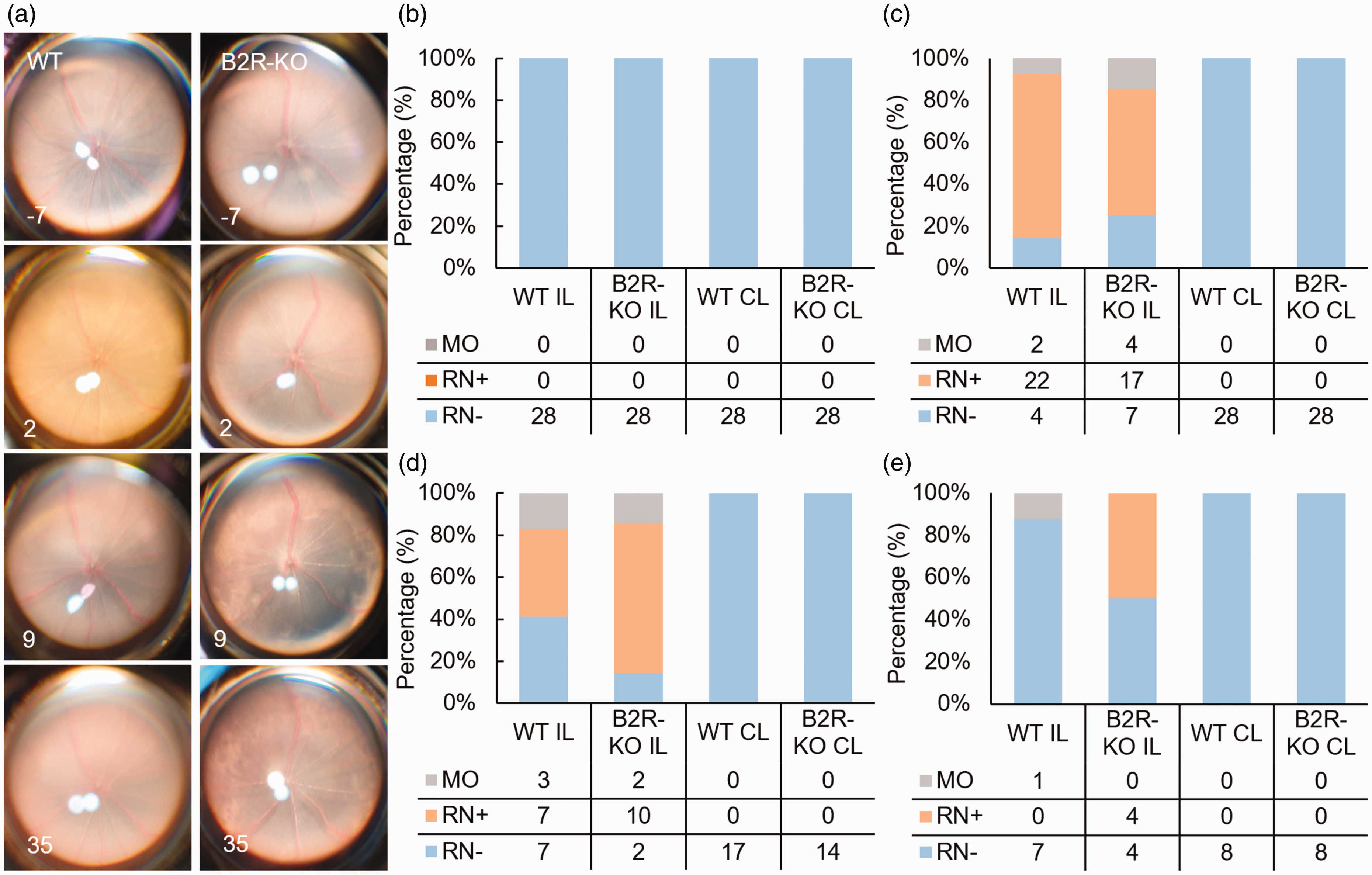
B2R deficiency leads to delayed onset of retinal ischemia. (a) Representative fundus photographs. (b–e) Qualitative fundus analysis over days -7 (B), 2 (C), 9 (D), and 35 (E) shows delayed retinal ischemic necrosis development in B2R-KO animals. MO – media opacification; RN+ – retinal necrosis positive, visible areas of retinal opacification; RN− – retinal necrosis negative, no visible signs of retinal opacification. IL – ipsilateral; CL – contralateral.
Volumetric analysis of the retinal vasculature in post-MCAO B2R-KO animals showed an increase in arteriolar diameter in ipsilateral retinas (ILR; P < 0.01; Figure 6(b)) across all measured time points compared to controls. This was coupled with an enlargement in venular diameter during the acute and subacute phases as well when compared to controls (P < 0.05; Figure 6(e)). There was no significant increase in arteriolar tortuosity post-ischemia (Figure 6(d)). A notable difference in microvascular density was found between the experimental groups, particularly in the acute and subacute phases of ischemia, with B2R-KO animals showing greater capillary area compared to controls (P < 0.05; Figure 6(g)).
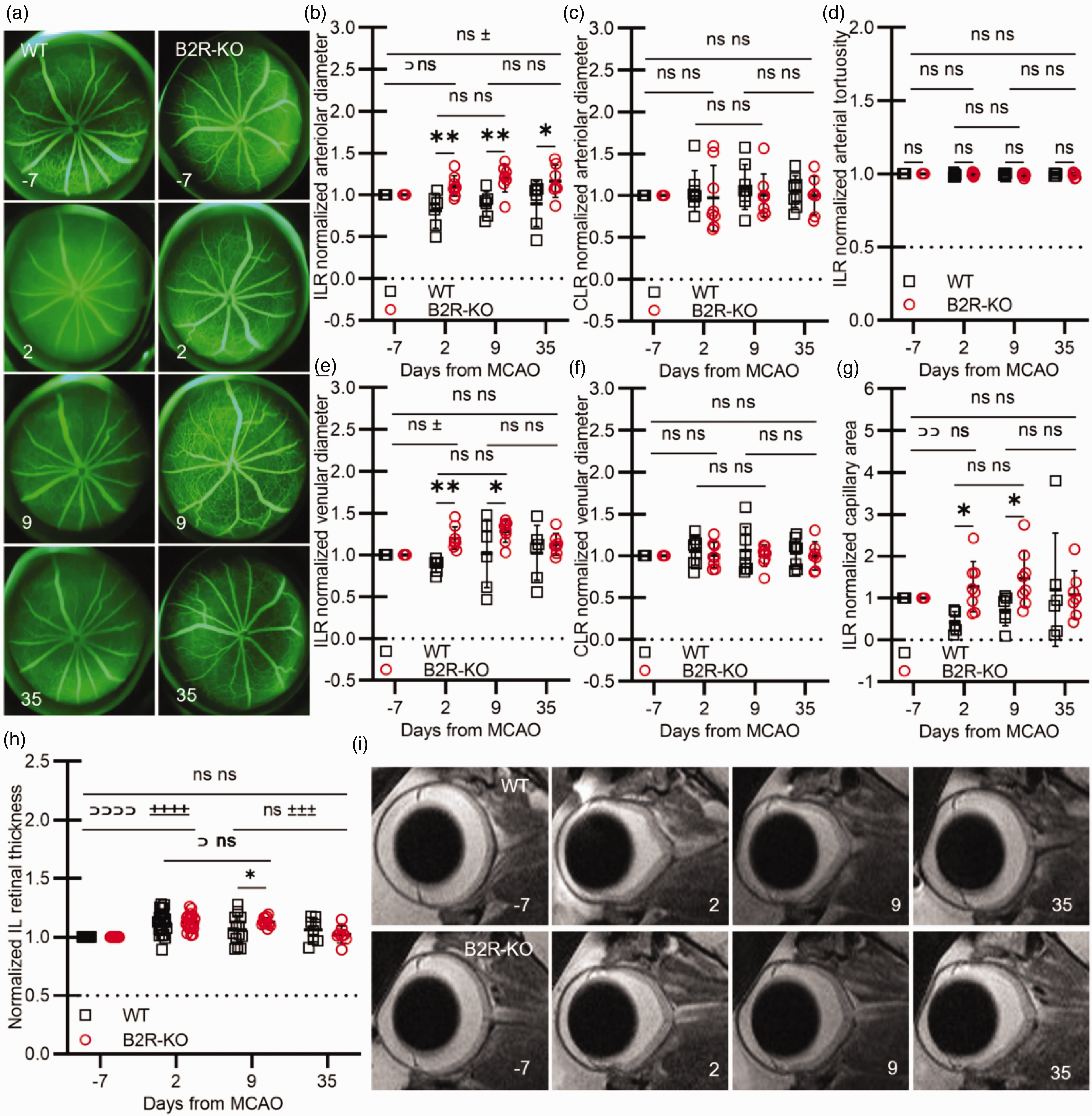
B2R deficiency has discernible effects on retinal vasculature resulting in delayed edema resolution. (a) Representative fundus angiography images of the ipsilateral eyes pre and post-MCAO. (b–g) Analysis of retinal vasculature shows the impact of B2R deficiency on normalized arteriolar diameter (b, c), venular diameter (e, f), capillary area (g), and arteriolar tortuosity (d) pre- and post-MCAO. (h) Normalized retinal thickness measured T2-weighted images pre and post-MCAO. Mixed model ANOVA with Šidak's post hoc test between groups at individual time points: (*) P < 0.05, or Tukey's post hoc test between time points: WT (⊃⊃⊃⊃) P < 0.0001, (⊃) P < 0.05; B2R-KO (±±±±) P < 0.0001, (±±±) P < 0.001. (i) Representative T2 images of the ipsilateral eye pre and post-MCAO. ILR – ipsilateral retina. CLR – contralateral retina.
On day 2, both groups exhibited a substantial increase in ILR thickness (WT P < 0.0001; B2R-KO P < 0.0001; Figure 6(h)). In WT mice, by day 9, retinal edema started resolving, leading to thinning of the retina compared to day 2 (P < 0.05). Throughout the chronic phase, retinal thickness gradually returned to baseline. In the B2R-KO group, there was no resolution of edema by day 9. However, by day 35, the ILR thickness in the B2R-KO group normalized to baseline values (P < 0.001). On day 9 the B2R-KO group had significantly thicker ILR compared to controls (P < 0.05; Figure 6(h)).
B2R deficiency increases ganglion cell loss in chronic retinal ischemia
Assessment of B2R deficiency on BRB permeability changes showed that on day 2, EB fluorescence (Figure 7(a)) in WT mice exhibited a significantly higher level in the ILR compared to the contralateral retina (CLR) (P < 0.05). By day 9, EB fluorescence in the ILR decreased significantly compared to day 2 (P < 0.05), with no notable change in CLR fluorescence for the WT group (P = ns). Conversely, the B2R-KO group displayed no difference in EB fluorescence between retinas on day 2, but exhibited significantly higher ILR EB fluorescence on day 9 compared to day 2 (P < 0.05). On day 2 post-MCAO, the B2R-KO group had significantly lower ILR EB fluorescence compared to the control group (P < 0.05). On day 9, the control group's EB fluorescence was notably lower than that of the B2R-KO group (P < 0.05; Figure 7(a)). As for NAF fluorescence measurements (Figure 7(b)), significant differences were observed between ILR and CLR in both WT and B2R-KO mice on day 2, with ILR showing higher fluorescence (P < 0.05). The WT group displayed a considerable reduction in ILR NAF fluorescence on day 9 compared to day 2 (P < 0.05), while the B2R-KO group exhibited no significant change. Although there were no significant differences between groups on day 2, WT animals' ILR NAF fluorescence was notably lower than that of the B2R-KO group by day 9 (P < 0.05; Figure 7(b)).
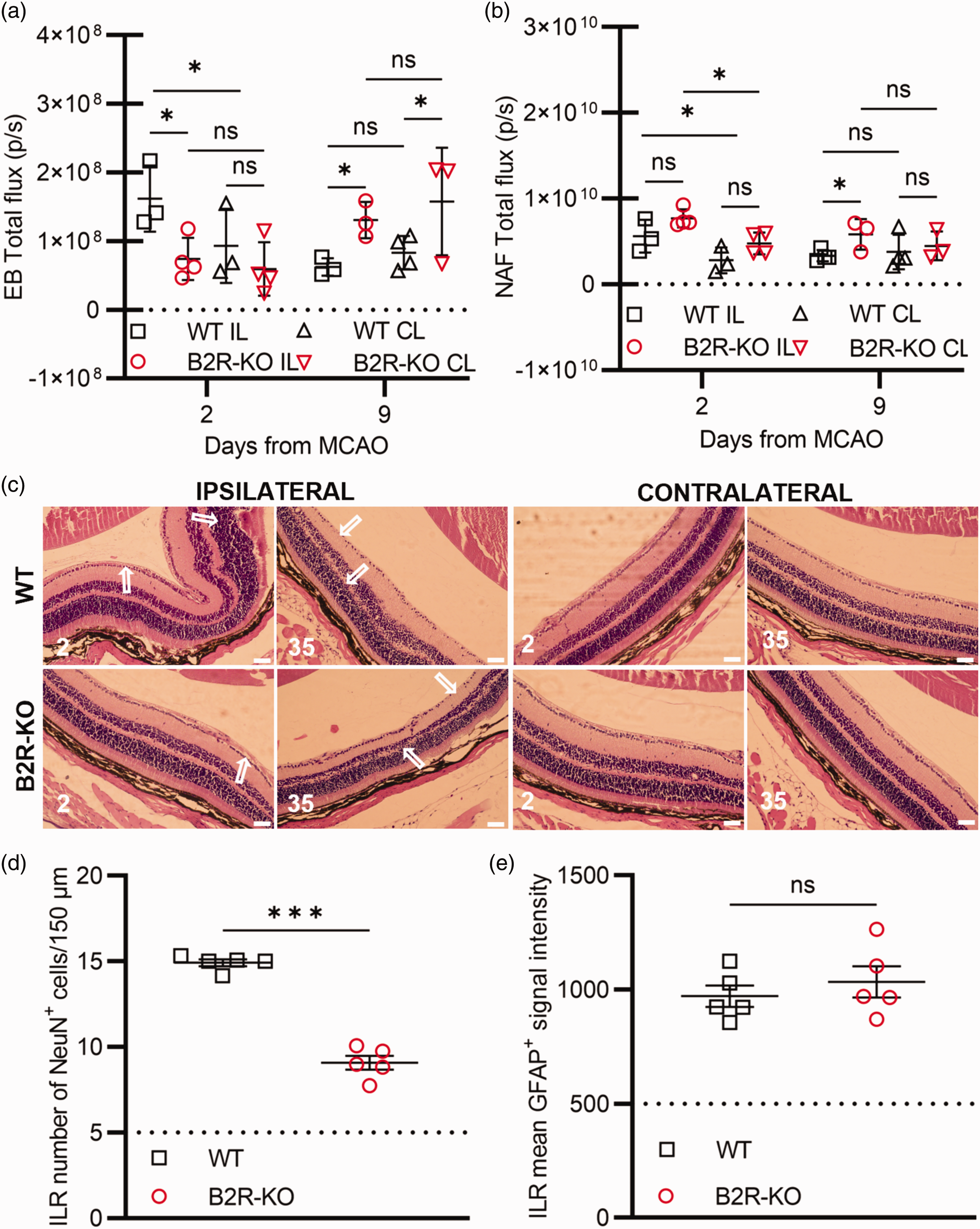
B2R deficiency promotes ganglion cell loss in the chronic phase of retinal ischemia. (a–b) Analysis of Evans blue (EB; A) and sodium fluorescein (NAF; B) extravasation (n = 4 per group/time point). Mixed ANOVA test with correction for multiple comparisons controlling for false discovery rate (α = 0.05): (*) P < 0.05. (c) Representative sections of the ipsilateral and contralateral retinas post-MCAO. White arrows display areas of aberrant morphology. Scale bar: 50 µm. (d) Quantification of NeuN+ ganglion cells and (e) GFAP+ mean signal intensity in the ipsilateral retinas (ILR) 35 days post-MCAO. Student's t-test: (***) P < 0.001 (n = 5 per group).
Histological analysis of retinas isolated on days 2 and 35 for the WT group indicated localized thickening of retinal layers during the acute phase of ischemia, particularly the inner nuclear layer, outer nuclear layer, and ganglion cell layer (Figure 7(c)). Conversely, on day 2, B2R-deficient mice displayed edema affecting primarily the ganglion cell layer with uniform thickening of other layers (Figure 7(c)). In the chronic phase, both groups displayed substantial ganglion cell loss and thinning of the inner nuclear layer, and inner and outer plexiform layers. The ILR NeuN+ cell quantification revealed significant ganglion cell loss on day 35 post-MCAO for the B2R-KO group compared to the control group (P < 0.001; Figure 7(d)). Interestingly, no difference in mean GFAP signal intensity of Muller cells between groups was observed on day 35 post-MCAO (Figure 7(e)).
Discussion
While existing data on ischemic brain injury in B2R-deficient mice are available, our study represents the first longitudinal, in vivo investigation that comprehensively explores all phases of ischemic damage development, with a particular emphasis on vascular perfusion status. Our findings indicate that B2R deficiency does not significantly alter the arterial perfusion status of the ILH during the acute and subacute phases of ischemia. In the chronic phase of ischemia, however, while the control group exhibits a return to the initial perfusion values, B2R deficiency leads to a significant increase in both volume and perfusion of the arterial vasculature. Also, although the volume of vasculature in the CLH remains relatively stable over time, the absence of B2R results in a significant increase in arterial perfusion during both the acute and chronic phases of ischemia. These differences in arterial vasculature perfusion may be attributed to a potential compensatory effect of B1R,41,42 a hypothesis warranting further investigation.
Studies involving pharmacological B2R antagonists in animals and humans have shown that intravascular administration of these antagonists significantly increases blood pressure.43 –45 However, assessing blood pressure in B2R-KO mice has yielded conflicting results. Some studies reported elevated blood pressure in the absence of B2R,46 –49 while others did not find such differences.50 –53 The latter was consistent with our results, showing no difference in blood pressure or intraocular pressure between groups. Although an increase in blood pressure could be expected in the absence of B2R, it has been shown that B1R could have a compensatory role in B2R-KO animals.41,42,54 Considering the vasoactive role of the B2R and the possible compensatory actions of the overexpressed B1R in B2R-KO mice, we performed LDF measurements during MCAO, which ruled out possible differences in MCA perfusion profile between study groups, allowing for an unbiased comparison of postoperative outcomes.
In line with prior research,50,55 our study, which employed a shorter 30-minute MCAO procedure, found that B2R deficiency did not influence the size of the ischemic lesion during the acute and subacute phases of ischemia. Discrepancies with the findings of Xia et al. 56 could be attributed to the use of different MCAO procedures, which have been demonstrated to yield different pathophysiological outcomes. 31 Studies have suggested varying patterns in the extravasation of low and high molecular weight molecules at different stages of BBB damage. 57 In this study, we observed an increase in extravasation of both high and low molecular weight molecules in the acute phase, followed by a gradual decline in high molecular weight permeability during the subacute phase, indicating partial recovery of vascular integrity. However, the B2R-deficient group did not exhibit significant changes in the extravasation of these molecules between hemispheres or across time points. These findings support previous evidence suggesting that B2R activation can have detrimental effects on vascular integrity and increase BBB permeability in the acute stage of ischemic injury.58 –60 While our study did not explore the potential effects of B2R deficiency on transcellular routes across the BBB, it did reveal a significant impact on the disruption of the paracellular route by analyzing isolated cerebral microvessels affected by ischemia. Specifically, the control animals showed a significantly lower number of pericytes and lower expression of the tight junction protein ZO-1 compared to the B2R-deficient animals. Prior research has shown that pericytes can adopt microglia-like properties during ischemia through the activation of G protein-coupled receptors and tyrosine kinase receptors. 61 By activating the Akt/eNOS pathway, PI3K and PLC increase intracellular calcium levels, mediating cytoskeleton reorganization which could induce pericyte migration.62,63 Conversely, when enveloping endothelium, pericytes promote BBB integrity by increasing the number of tight junctions. 64 Our study not only demonstrated the potential effects on pericytes but also on tight junction proteins, consistent with a previous in vitro study that illustrated the role of BK in reducing ZO-1 tight junction protein expression in human endothelial cells through the activation of B2R, resulting in an increased paracellular transport. 65
The MRI analysis of the chronic phase of ischemia unveiled significant differences in the volume of the ipsilateral cerebral hemisphere between the groups. Although both groups experienced considerable atrophy, the loss of cerebral tissue was markedly more pronounced in the absence of B2R. Furthermore, we showed that the absence of B2R led to pronounced neuronal loss and increased astrogliosis in the remaining tissue of the ischemic lesion, suggesting that B2R may play a role in neuroprotection and glial activation post-stroke, contributing to tissue regeneration and repair. In addition to representing a new finding in the literature, this data supports the hypothesis of protective effects of B2R activation in the chronic phase of ischemic brain injury. These could involve reducing inflammation and oxidative damage, and safeguarding against cell damage and death.21,22,24,66,67 That being said, it is worth repeating that, in our study, B2R deficiency resulted in increased arterial vascular perfusion, which could also contribute to more extensive tissue loss, a phenomenon previously observed in reperfusion injury. 68 Additionally, both B2R deficiency 47 and ischemia 69 induce insulin resistance, potentially affecting the process of tissue recovery.
To the best of our knowledge, our study is the first to reveal the chronic effects of ischemic damage on glucose metabolism in mice. Glycemic status plays a pivotal role as a prognostic variable in the acute phase of ischemia, owing to the adverse impact of stress-induced hyperglycemia on functional outcomes. 70 In acute ischemia, hyperglycemia occurs even in non-diabetic patients71,72 due to autonomic system activation, adrenal hormone release, and increased gluconeogenesis.73 –76 In our study, chronic ischemia was found to significantly lower basal blood glucose concentrations and flatten the glucose curve in the control group. However, these changes were not observed in B2R-deficient animals. One explanation for these group differences could be that, apart from its role in regulating the vasomotor response, the B2R signaling pathway also affects glucose metabolism, 40 with the increase in B2R expression following ischemia 3 leading to improved insulin sensitivity or glucose uptake. 47
Survival was not impacted by B2R deficiency, which aligns with previous studies that also did not report a survival effect in B2R-KO mice 55 or following pharmacological inhibition of B2R during the acute phase of ischemic damage.1,15,77 Neurological deficit scoring confirmed the deleterious impact of ischemic damage, followed by gradual recovery from the subacute to the chronic phase of ischemia. Intriguingly, in the acute phase of ischemia, B2R deficiency had a negative effect on neurological status, despite no differences in the volume of the ischemic lesion or body mass loss. This finding contradicts the results of previous research showing reduced ischemic lesions and neurological deficit in the absence of B2R, 3 or no differences between groups. 50 These discrepancies could be attributed to variations in methods used for assessing the volume of the ischemic lesion and neurological deficit. However, two of the additional three motor function tests corroborated our findings. The rotarod test showed a significantly shorter time of successful maintenance on the rod in B2R deficient animals in the acute phase of ischemia. The possible influence of B2R deficiency on the endurance of mice during aerobic physical activity 78 should also be considered, as it has the potential to introduce bias to our results. The wire-hanging test did not reveal significant differences between groups. Given that the groups did not differ in ischemic lesion volume, it is possible that paw function and strength were equally impaired. Finally, the pole test revealed significant differences between groups at all post-MCAO time points, with B2R deficiency leading to significantly delayed descent times. This extended descent time during the chronic phase of ischemia corresponds to the increased tissue loss observed in the absence of B2R, providing additional evidence for a protective role of B2R during the chronic phase of cerebral ischemia.
As our previous study suggested that most retinal responses to ischemia overlapped with those in the brain when the Koizumi MCAO procedure was performed, 31 we also monitored the evolution of concomitant retinal ischemic injury. Interestingly, using fundus photography a fundamental method in both clinical and preclinical settings for detecting and monitoring retinal ischemia-related changes79,80 we recorded a subacute increase in the frequency of retinal opacification phenomena in the B2R-deficient mice, as opposed to the decrease seen in the control group. These findings align with previous studies indicating protective mechanisms in ischemic retinal injury associated with B2R activation.23,25 Both groups also exhibited ocular media opacification during the acute and subacute phases of ischemia, despite ruling out factors like low ambient temperatures, hypothermia, prolonged anesthesia, and corneal dryness.81,82 The observed opacification of the cornea and lens could also be attributed to reduced perfusion in the ophthalmic artery. 83
The analysis of fluorescein angiograms highlighted the dysregulation of arteriolar caliber in control mice post-ischemia, characterized by constriction. Remarkably, this constriction was absent in B2R-deficient mice, possibly due to a compensatory vasodilatory mechanism involving overexpressed B1R.4,84 Venular dilation was also observed in B2R-deficient animals, a response not seen in control mice. Moreover, B2R deficiency prevented the acute reduction in microvascular density, contrasting the reduction seen in the control group, suggesting an acutely protective effect on microvascular integrity. While both venular dilation and microvascular density preservation appear physiologically consistent with the observed absence of arteriolar constriction, the exact mechanism for these changes warrants additional investigation.
In cases of acute retinal ischemia, clinical observations typically reveal retinal swelling due to edema development, followed by thinning of retinal layers caused by cell loss and axonal atrophy.85,86 Our MRI analysis confirmed substantial retinal thickening during the acute phase, supported by histological sections of ipsilateral retinas on day 2 post-MCAO, displaying morphological damage, edema, and layer thickening. Although there was no difference in retinal thickness between groups during the acute phase, the B2R-deficient group exhibited significant thickening during the subacute period, accompanied by a higher incidence of retinal necrosis in fundus photographs. To further explore ischemia-induced morphological changes indicating delayed edema resolution in the absence of B2R, we analyzed the vascular permeability of isolated retinas. The quantification revealed a significant increase in extravasation of EB and NAF in the B2R-deficient group from the acute to subacute phase, suggesting a subacute deterioration in BRB vascular integrity. The deleterious effect of B2R deficiency on retinal ischemic damage and delayed edema resolution might be attributed to a compensatory mechanism among bradykinin receptors, given prior evidence of the role of B1R in promoting retinal vascular permeability.4,84,87
While retinal thinning was not apparent in the chronic phase of ischemia based on T2 measurements, it should be noted that the sensitivity of the retinal thickness measurement method on MR images was limited by low image resolution. 88 However, histological analysis unveiled significant morphological changes, layer thinning, and cell loss, indicating long-term degenerative alterations following ischemic injury. Although literature extensively reports the degeneration of retinal layers following ischemic injury,31,89,90 there is limited information on the chronic effects of ischemia on retinal cells in the absence of B2R. Previous data demonstrated no protective effect of the B2R antagonist icatibant against retinal ischemic damage in the first week post-ischemia, 91 which is in agreement with our in vivo data indicating delayed deterioration in B2R deficiency. Moreover, immunohistochemical analysis of retinal ganglion cell counts in the chronic phase revealed a detrimental effect of B2R deficiency on ganglion cell viability.
Our study sheds light on the multifaceted role of B2R in different phases of ischemic damage. While B2R deficiency did not significantly affect blood pressure or acute ischemic lesion size, it influenced BBB integrity, cerebral arterial perfusion, functional motor impairment, glycemic status, and promoted loss of cortical neurons and astrogliosis in the chronic phase. In retinal ischemia, B2R deficiency also affected retinal permeability and BRB integrity, and demonstrated a progressive detrimental effect, leading to a notable reduction in the number of retinal ganglion cells. These findings collectively illuminate the contrasting effects of B2R in different phases of cerebral and retinal ischemic injury, revealing their temporal dynamic and their therapeutic potential in subacute and chronic phases of post-ischemic tissue recovery and neuroprotection. Ultimately, as the dichotomous role of B2R activation indicates that both agonists and antagonists of B2R could be evaluated as potential therapeutics in different stages of ischemic injury development, the optimal therapeutic window for both should, ideally, be further investigated in studies employing pharmacological modulation. This should also include evaluating their effectiveness in the presence of common stroke comorbidities, such as hypertension and diabetes mellitus, to assess the risk of negative side effects in different patient sub-populations.
Supplemental Material
sj-pdf-1-jcb-10.1177_0271678X241270241 - Supplemental material for The temporal dynamic of bradykinin type 2 receptor effects reveals its neuroprotective role in the chronic phase of cerebral and retinal ischemic injury
Supplemental material, sj-pdf-1-jcb-10.1177_0271678X241270241 for The temporal dynamic of bradykinin type 2 receptor effects reveals its neuroprotective role in the chronic phase of cerebral and retinal ischemic injury by Helena Justić, Anja Barić, Martina Ratko, Iva Šimunić, Marin Radmilović, Marta Pongrac, Siniša Škokić and Marina Dobrivojević Radmilović in Journal of Cerebral Blood Flow & Metabolism
Footnotes
Ethics approval
All animal handling and procedures were approved by the Ethics Licensing Committee of the University of Zagreb School of Medicine and the Ethics Committee for the protection of animals used for scientific purposes of the Ministry of Agriculture Republic of Croatia (No. 380-59-10106-18-111/49).
Consent for publication
All authors give their consent and approval for publication of this manuscript.
Availability of data and materials
Key resources that are essential to reproduce the results are provided in the Supplementary Information (Supplemental Table 1). The raw data used and/or analyzed in the current study are available from the corresponding authors upon request. The angiography processing macro is available from GitHub (![]() ).
).
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded by the Croatian Science Foundation project BRADISCHEMIA (UIP-2017-05-8082), Scientific Centre of Excellence for Basic, Clinical and Translational Neuroscience project GA KK01.1.1.01.0007 “Experimental and clinical research of hypoxic-ischemic damage in perinatal and adult brain” and project “Sinergy of molecular markers and multimodal in vivo imaging during preclinical assessment of the consequences of the ischemic stroke (SineMozak-KK.01.1.1.07.0071)” funded by the European Union through the European Regional Development Fund. The work of doctoral student Marta Pongrac has been fully supported by the “Young researchers' career development project – training of doctoral students” of the Croatian Science Foundation funded by the European Union from the European Social Fund.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
Helena Justić assessed the glycemic status, fundus imaging, functional tests, histochemistry, extravasation imaging and analysis, magnetic resonance data analysis, and wrote the manuscript; Anja Barić. performed blood and intraocular pressure measurements, magnetic resonance imaging, neurological scoring, and analysis; Martina Ratko performed immunohistochemistry and analysis; Iva Šimunić performed magnetic resonance angiography analysis, fluorescein angiography image postprocessing; Marin Radmilović designed research, fundus and fluorescein angiography analysis, contributed to writing and editing of the manuscript; Marta Pongrac performed isolation, immunohistochemical processing and analysis of microvasculature; Siniša Škokić. designed and performed magnetic resonance imaging and postprocessing; Marina Dobrivojević Radmilović designed research, performed MCAO, statistical analysis, and revised the manuscript. All authors reviewed and critically edited the manuscript.
Supplementary material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.