
Abstract
Heterogeneity and variability of symptoms due to the type, site, age, sex, and severity of injury make each case of traumatic brain injury (TBI) unique. Considering this, a universal treatment strategy may not be fruitful in managing outcomes after TBI. Most of the pharmacological therapies for TBI aim at modifying a particular pathway or molecular process in the sequelae of secondary injury rather than a holistic approach. On the other hand, non-pharmacological interventions such as hypothermia, hyperbaric oxygen, preconditioning with dietary adaptations, exercise, environmental enrichment, deep brain stimulation, decompressive craniectomy, probiotic use, gene therapy, music therapy, and stem cell therapy can promote healing by modulating multiple neuroprotective mechanisms. In this review, we discussed the major non-pharmacological interventions that are being tested in animal models of TBI as well as in clinical trials. We evaluated the functional outcomes of various interventions with an emphasis on the links between molecular mechanisms and outcomes after TBI.
Keywords
Introduction
Every year, ∼69 million people worldwide sustain some form of TBI. 1 It is estimated that >5 million individuals are currently living with TBI-associated disabilities in the U.S. 2 Depending on the magnitude of the impact (mild/moderate/severe), the area of the injury, and the degree of damage, TBI can have diverse functional outcomes and varied underlying mechanisms. Mild single TBI is usually characterized by headache, dizziness, and confusion, and may not always result in behavioral deficits. However, repeated mild TBI can result in cognitive impairment, short-term memory loss, increased impulsivity and aggression, and susceptibility to chronic traumatic encephalopathy. 3 Moderate and severe TBI result in long-term motor and cognitive disabilities including problems with memory, attention, and the regulation of emotions. 4 In addition, >50% of moderate/severe TBI patients experience major depression and/or sleep disturbances such as insomnia, narcolepsy, and excessive daytime sleepiness. 5
The focused primary injury during the acute phase of TBI comprises subdural/epidural hematomas and contusions, increased intracranial pressure (ICP), disrupted cerebral blood flow (CBF), edema, excitotoxicity, and tissue deformation. 6 Whereas, the diffuse secondary injury during the chronic phase spreads to areas distant from the site of injury and comprises axonal damage, vascular injury, hypoxic-ischemic injury, inflammation, oxidative stress, endoplasmic reticulum (ER) stress, ionic imbalance, and blood-brain barrier (BBB) disruption. 7 The excess release of glutamate into the synaptic cleft combined with a reduced reuptake leads to the overactivation of ionotropic glutamate receptors resulting in increased intracellular Ca2+ levels. 8 This leads to the generation of reactive oxygen species (ROS) that results in oxidative stress and mitochondrial dysfunction. Altered oxidative and glucose metabolism are characteristics of the extensive metabolic dysfunction induced after TBI. 9 Increased glucose utilization (hyperglycolysis) and reduced cerebral metabolic rate of oxygen have been observed in the acute phase of TBI, which is followed by a subsequent rapid metabolic depression and an increased CBF.9,10 This reduced glucose availability results in increased metabolism and uptake of alternative cerebral energy substrates, mainly lactate. Moreover, tissue lactic acidosis is associated with poorer outcomes in TBI patients. 11 All these pathological mechanisms synergistically contribute to neuronal death leading to long-term functional deficits. While the primary injury cannot be controlled, timely interventions can minimize the extent of secondary injury. Hence, an understanding of the underlying mechanisms can help identify potential targets for the therapy and management of TBI.
Since TBI consists of a wide range of pathophysiologic mechanisms that involve multiple pathways, its treatment requires the adoption of a multi-faceted and interdisciplinary approach. Treatment options can vary based on the extent of injury and the particular deficits present in each patient, which can be broadly categorized into either pharmacological or non-pharmacological interventions. Despite extensive testing to mitigate both acute and chronic symptoms of TBI, most pharmacological therapies can only target specific pathophysiologic mechanisms and may even result in the development of new symptoms. 12 In contrast, non-pharmacological therapies often rely on environmental and behavioral approaches for improving functional outcomes. These options encompass a wide range of therapies including hypothermia, hyperbaric oxygen (HBO), dietary modifications like caloric restriction (CR) and intermittent fasting (IF), exercise, environmental enrichment (EE), deep brain stimulation (DBS), decompressive craniectomy (DC), using probiotics and fecal matter transplantation (FMT) to rectify gut dysbiosis, gene therapy, music therapy, and stem cell therapy. This review aims to discuss these non-pharmacological interventions that showed efficacy clinically and/or in animal models of TBI.
Animal models of TBI
Accurately replicating human TBI pathophysiology is challenging due to the immense diversity and complexity of the response to injury. However, many well-established animal models replicate several aspects of human TBI pathology as best as possible. TBI animal models were developed using non-mammals (roundworm, fruit fly, and zebrafish), lissencephalic rodents (mouse and rat), and gyrencephalic mammals like swine, sheep, and non-human primates. In addition, several in vitro TBI models using cultured cells are also in use. Further details on TBI animal models are given in the Supplemental Material.
Pharmacotherapies for TBI
Several experimental drugs have been tested to date using various animal models of TBI. These include molecules that mitigate excitotoxicity, cerebral edema, apoptosis, oxidative stress, ER stress, inflammation, BBB disruption, and mitochondrial dysfunction. 13 Summary of some of the preclinically and clinically tested pharmacotherapies are given in the Supplemental Material.
Nutraceuticals
Nutraceuticals have gained much attention due to the failure of the mono-pharmacological approach and the multimodal pathological nature of brain damage after TBI. The synergism between nutraceuticals such as vitamins, minerals, amino acids, and essential fats and with other drugs was shown to limit secondary brain damage, leading to a faster recovery after TBI in experimental models as well as humans. 14
Zinc (Zn) is an essential mineral that is indispensable for cellular metabolism, growth, and development. Zn supplementation was shown to improve cognitive and neurobehavioral responses in rats subjected to CCI injury. 15 Zn supplementation (12 mg elemental Zn) for 15 days after severe closed-head injury in humans was also shown to reduce mortality and improve motor function between 15 and 28 days after injury. 16 Omega-3 polyunsaturated fatty acids (PUFAs) regulate a wide variety of biological functions, including cell membrane integrity, inflammation, vascular tone, and cell signaling. 17 Treatment with docosahexaenoic acid (DHA; a brain-enriched PUFA) attenuated sensorimotor deficits in rats and reduced anxiety-like behavior in mice, subjected to CCI injury. 18 A non-randomized clinical study showed that supplementation of a mixture of DHA and eicosapentaenoic acid at a ratio of 2:1 improved neurological recovery in patients in coma after severe TBI. 19
The plasma levels of vitamin C (ascorbic acid) were shown to be significantly decreased in head trauma patients compared to healthy subjects on day 1, and inversely correlated with the lesion volume and functional impairment. 20 In a randomized double-blind, placebo-controlled trial, intravenous injections (IV) of a high dose of vitamin C (10 g on the first and fourth days of admission, followed by 4 g/day for 3 more days) in severe head injury patients resulted in decreased perilesional edema. 21 A multicenter, prospective cohort study showed that lower levels of Vitamin E correlate with poor neurological outcomes in TBI patients. 22 A clinical trial showed a lower mortality rate in patients who received vitamin E (400 IU/day intramuscular for 7 days) after severe TBI. 21 A retrospective study showed that the serum levels of vitamin D were low in TBI patients at the time of hospital admission, and supplementation of vitamin D significantly improved long-term performance, better Glasgow Outcome Scale (GOS) score, and ameliorated cognitive dysfunction. 23 In a double-blind randomized trial, a single dose of vitamin D (120,000 IU) within 24 h of hospital admission resulted in a better Glasgow Coma Scale score, decreased duration of mechanical ventilation, and reduced serum levels of inflammatory cytokines such as interleukin (IL)-6, IL-2 and tumor necrosis factor (TNF)-α compared to the placebo. 24
Non-pharmacological interventions for TBI
To minimize the secondary brain damage and improve the quality of life of patients, TBI treatments need more holistic care than pharmacotherapy. Early forms of therapy for TBI were mainly targeted at recovery of motor function through physiotherapy. However, a more holistic treatment including non-pharmacologic interventions in addition to pharmacotherapies might be needed for a better recovery after TBI. Some of those are discussed in the following sections. A summary of study characteristics and major outcomes of preclinical studies included in this review are given in Table 1.
Non-pharmacological intervention for post-TBI neuroprotection and recovery.
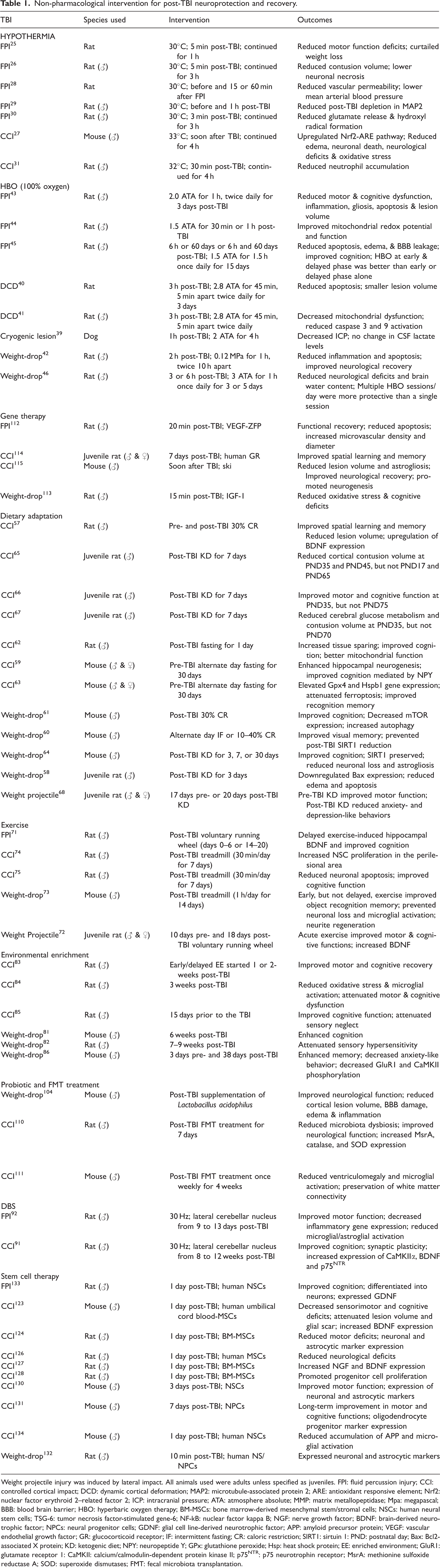
Weight projectile injury was induced by lateral impact. All animals used were adults unless specified as juveniles. FPI: fluid percussion injury; CCI: controlled cortical impact; DCD: dynamic cortical deformation; MAP2: microtubule-associated protein 2; ARE: antioxidant responsive element; Nrf2: nuclear factor erythroid 2–related factor 2; ICP: intracranial pressure; ATA: atmosphere absolute; MMP: matrix metallopeptidase; Mpa: megapascal; BBB: blood brain barrier; HBO: hyperbaric oxygen therapy; BM-MSCs: bone marrow-derived mesenchymal stem/stromal cells; NSCs: human neural stem cells; TSG-6: tumor necrosis factor-stimulated gene-6; NF-kB: nuclear factor kappa B; NGF: nerve growth factor; BDNF: brain-derived neurotrophic factor; NPCs: neural progenitor cells; GDNF: glial cell line-derived neurotrophic factor; APP: amyloid precursor protein; VEGF: vascular endothelial growth factor; GR: glucocorticoid receptor; IF: intermittent fasting; CR: caloric restriction; SIRT1: sirtuin 1: PND: postnatal day; Bax: Bcl2-associated X protein; KD: ketogenic diet; NPY: neuropeptide Y; GPx: glutathione peroxide; Hsp: heat shock protein; EE: enriched environment; GluR1: glutamate receptor 1: CaMKII: calcium/calmodulin-dependent protein kinase II; p75NTR: p75 neurotrophin receptor; MsrA: methionine sulfoxide reductase A; SOD: superoxide dismutases; FMT: fecal microbiota transplantation.
Hypothermia
Many pharmacological cooling agents including antipyretics and non-steroidal anti-inflammatory drugs were tested after TBI. In addition, mechanical cooling provided either by intravascular or surface routes was also widely tested. The efficacy of both mild (∼34°C) and moderate (∼31°C) hypothermia was tested after TBI. 25 In a rat fluid percussion injury (FPI) model, whole body cooling started at 5 min after injury and maintained at 30°C for an hour significantly attenuated motor deficits compared to normothermic control. 25 Furthermore, maintaining the head temperature at 30°C for 3 h post-TBI prevented cortical neuronal necrosis and reduced the lesion volume in rats subjected to FPI. 26 Following weight-drop head injury in mice, lowering the whole-body temperature to 33°C at 30 min after TBI alleviated neurobehavioral dysfunction, and reduced brain edema and neuronal death. 27 Mechanistically, this treatment provided neuroprotection by inducing superoxide dismutases (SOD) and glutathione peroxide (GPx) levels, and upregulating the nuclear factor erythroid 2–related factor 2 (Nrf2)-antioxidant response element pathway, ultimately attenuating oxidative stress. Moreover, rats subjected to moderate hypothermia before and 1 h after FPI exhibited reduced BBB disruption and injury-induced loss of hippocampal microtubule-associated protein 2.28,29 Other benefits of hypothermia observed in rodent models of TBI include reduced excitotoxicity and decreased neutrophil accumulation.30,31
Although the outcomes of preclinical studies that used therapeutic hypothermia were mostly favorable, clinical studies to date were inconclusive. Moderate hypothermia for 24 h was shown to prevent acute derangements of cerebral physiology and metabolism in severe TBI patients without any undesired side effects. 32 Mild hypothermia was also shown to reduce ICP and mortality after DC in humans. 33 Conversely, several meta-analyses and controlled trials reported either no effect or adverse effects of mild hypothermia in TBI patients.34,35 The adverse effects of therapeutic hypothermia such as bleeding during rewarming were thought to outweigh its beneficial effects in clinics. 25 Some of the factors that need optimization in post-TBI hypothermia are the specificity of the cooling device, temperature to be used, time of the start of the intervention, duration, and rewarming rate.
Hyperbaric oxygen
Post-TBI hypoxia promotes oxidative stress, inflammation, and metabolic depression and prevents the regenerative process. HBO therapy (breathing 100% oxygen in a pressurized chamber) is FDA-approved for disorders such as carbon monoxide poisoning, gangrene, radiation and burn injuries, and some types of infections. 36 HBO provides more oxygen to the tissue which promotes wound healing, angiogenesis, and fighting infections. 37
Many preclinical and clinical studies showed the beneficial effects of post-TBI HBO therapy that included sessions lasting one to several hours a day for several days to months at an oxygen pressure between 1.25 to 3 atmosphere absolute (ATA). 38 One of the earliest preclinical studies showed that HBO therapy (100% oxygen at 2 ATA for 4 h) after cryogenic cortical injury in dogs decreased ICP without altering the lactate levels in the CSF. 39 Compared to a 30% and 100% oxygen supply, a 1.5 ATA HBO treatment for 30 min in male rats subjected to FPI elevated the partial pressure of oxygen in brain tissue by ∼3 and ∼6.5 fold, respectively. 40 In a rat dynamic cortical deformation (DCD) model, HBO therapy restored mitochondrial transmembrane potential, prevented caspase-3 and -9 activation and reduced neutrophil infiltration and matrix metalloproteinase-9 expression leading to neuroprotection.40,41 In rats subjected to weight-drop head injury, HBO therapy led to inhibition of TLR4/NF-kB activation and lowered the expression of TNF-α, IL-6, and IL-1β leading to less inflammation. 42 Moreover, improved angiogenesis, neurogenesis, and mitochondrial function were observed in the perilesional cortex of rats treated with HBO following FPI.43,44 Other HBO-mediated benefits following TBI in rodents include upregulation of the anti-inflammatory cytokine IL-10, increased cerebral glucose utilization, axonal sprouting, synapse remodeling, and elevated hippocampal vascular density, reduced apoptosis, lowered brain edema, improved BBB integrity and better recovery of motor and cognitive deficits.38,42 One of the crucial factors for the effectiveness of HBO therapy is timing. A combination of early (started 6 h post-TBI, once a day for 15 days) and delayed (started 60 days post-TBI, once a day for 15 days) HBO conferred better neuroprotection than early or delayed treatment alone, in a rat FPI model. 45 Corroborating this, HBO sessions started at 3 h and 6 h, but not 12 h, 24 h, 48 h, or 72 h, after TBI displayed a significant reduction in brain damage in rats subjected to weight-drop injury. 46
The feasibility of HBO therapy following TBI was also tested in humans. Sessions of 2 ATA HBO therapy for 45 min were observed to reduce ICP by decreasing CBF in 50 patients suffering from acute cerebral edema due to TBI. 47 Furthermore, in a case study involving 6 patients, HBO therapy followed by treatment with drugs such as propranolol, bromocriptine, and gabapentin alleviated paroxysmal sympathetic hyperactivity in patients with severe TBI. 48 When a 54-year-old man with permanent neurological symptoms such as hyperreflexia, impaired cognition and memory, and aphasia from a year-old TBI was subjected to two sessions of HBO one year apart, there was an improvement in sensorimotor functions after the first session that lasted for a year. 49 A second HBO session reinstated sensorimotor improvements in addition to better neuropsychological function. In a retrospective analysis conducted on 154 patients with chronic neurocognitive damage due to TBI subjected to HBO therapy, there was an improvement in all cognitive domains, including memory, attention, and executive function. 50 Neuropsychological gains were also observed in a larger cohort of brain-injured children and adults treated with HBO. 51 HBO therapy was also seen as beneficial in a randomized phase II clinical trial involving 42 severe TBI patients that used a combination of hyperbaric and normobaric oxygen therapy within 24 h of injury. 52 The combined oxygen therapy in this trial improved markers of oxidative metabolism such as blood lactate/pyruvate ratio and glycerol levels and reduced intracranial hypertension and mortality. 52 Post-concussion syndrome (PCS) and post-traumatic stress disorder (PTSD) are prevalent in active military service members and veterans who suffered a blast injury. 53 In a phase I clinical study, military personnel with a history of TBI reported improvement in PTSD/PCS symptoms, cognition, and quality of life following HBO therapy. 53 A clinical trial involving 56 civilian TBI patients with prolonged PCS at a late chronic stage also observed similar benefits. 54
HBO is not an FDA-approved treatment for TBI and/or PTSD. However, HBO is well-tolerated by TBI patients with a minor side effect of mild barotrauma. 55 According to a report by the VA Evidence Synthesis Program, HBO reduced mortality and coma severity in patients with moderate to severe TBI, compared to standard neurosurgical care. 55 On the contrary, no improvement in post-concussion and PTSD symptoms was observed in chronic mild TBI patients subjected to HBO. 55 Moreover, not all the HBO therapy studies were beneficial as contrasting outcomes were observed in service members with PCS. In one such study, no improvement in symptoms was observed in service members when subjected to 40 HBO sessions (1.5 ATA) compared to the control group. 56 Similarly, some clinical studies observed no significant changes in the PCS of service members subjected to either 40 daily HBO sessions using 1.5 or 2 ATA or 30 daily sessions using 2.4 ATA. 54
Similar to hypothermia, the uncertainty in the therapeutic time window and heterogeneity of protocols make HBO therapy a less attractive choice for clinical translation currently.
Dietary adaptations
Studies on animal models have shown that CR and IF, as well as specific diets like the ketogenic diet, can reduce oxidative stress and improve motor and cognitive functions after a brain injury.57–59 CR limits the daily energy intake without promoting malnutrition or vitamin deficiencies, whereas IF limits food intake to a certain period of the day (usually 8 to 12 h) without specific restrictions on calories. Several studies explored the benefits of CR and IF in preclinical TBI models. A 30% CR regimen that spanned 4 months before and 19 days after TBI upregulated brain-derived neurotrophic factor (BDNF) in the perilesional cortical area and hippocampus, improved spatial learning and memory, and attenuated lesion volume in male rats. 57 Similarly, a 10–40% CR post-TBI in adult mice prevented the downregulation of cortical SIRT1 expression which is beneficial for recovery. 60 A 30 days 30% CR in male mice subjected to TBI decreased mTOR and GFAP expression in the hippocampus, increased autophagy, and improved spatial learning and memory. 61 A 24 h, but not 48 h, fast immediately after TBI in adult male rats induced neuroprotection and reduced cognitive deficits that were thought to be mediated by reduced mitochondrial dysfunction and ketosis. 62 Alternate day IF for 30 days before TBI enhanced the hippocampal neuropeptide Y expression, neurogenesis, and cognitive recovery, and these benefits were lost by its knockdown. 59 The above regimen of IF was also shown to attenuate TBI-induced ferroptosis leading to beneficial outcomes. 63 Studies involving dietary adaptations need frequent monitoring of the health conditions of the study participants as there is risk of malnutrition.
In male mice, a post-TBI ketogenic diet (KD) for 30 days that mimics the effect of fasting prevented SIRT1 downregulation, attenuated cognitive deficits, astrogliosis, and cortical and hippocampal neuronal loss. 64 Interestingly, the efficacy of KD following TBI was observed to be age-dependent; KD reduced cortical lesion volume in postnatal day (PND) 35 and 45 rats, but not in PND 17 and 65 rats. 65 Similarly, an age-dependent difference in motor and cognitive function, and cerebral glucose metabolism in juvenile rats given KD was observed following brain injury.66,67 These results suggest an age-dependent utilization and effectiveness of ketones as an alternative fuel in juvenile subjects. 66 Neuroprotective and therapeutic potential of KD before TBI (for 17 days) and a standard diet after TBI, and vice versa were tested in PND 47 rats of both sexes. 68 KD before TBI improved motor function recovery, and KD after TBI reduced anxiety- and depression-like behavior. 68
Exercise
Exercise is known to be beneficial to overall health including cognition and cardiovascular function. Physical activity after TBI has been linked to enhanced levels of neurogenesis, long-term potentiation, and release of growth factors like BDNF. 69 There is some disagreement regarding the therapeutic time window of exercise after TBI based on age. 70 Voluntary exercise at a delayed time point (days 14–20), but not at an acute time point (days 0–6), after TBI led to upregulation of hippocampal BDNF and cognitive improvement in adult male rats. 71 Conversely, improvement in motor and cognitive recovery following physical exercise during the acute phase (days 1–3) after TBI in juvenile rats of both sexes was reported. 72 In male mice subjected to weight-drop injury, an early moderate exercise (days 2 to 7 or 14) prevented memory impairment, neuronal loss, and neuroinflammation. 73 However, a delayed start of moderate exercise (9 days post-TBI) did not affect memory impairment. This indicates the advantage of an early restoration of TBI-induced BDNF levels by exercise. Corroborating this, early exercise elevated the proliferation of neural stem cells (NSCs) and inhibited neuronal apoptosis in the perilesional area of rats subjected to CCI injury.74,75 Some plausible reasons for the discrepancy in the outcomes of these studies might be due to differences in TBI induction and exercise intensity, and the running wheel habituation. 70 Keeping these differences aside, most of the preclinical studies found that exercise enhances motor and cognitive recovery after TBI. 70 Thus, multiple synergistic molecular mechanisms including increased BDNF, neuroplasticity and neurogenesis, reduced apoptosis and neuroinflammation might be responsible for the benefits of post-TBI exercise.
Some of the benefits of post-TBI exercise were translated effectively into clinical studies. 70 Twelve weeks of aerobic exercise training performed 3 times a week for 30 min on a treadmill improved cognitive function in seven individuals with chronic nonpenetrating TBI. 69 A large multicenter cohort study of 2,413 children and adolescents observed a reduced risk of persistent post-concussive symptoms in patients who participated in physical activity within 7 days post-injury compared to the no physical activity cohort. 76 A pilot study with 12 adult participants who suffered a TBI more than 12 months ago observed lesser depression-like symptoms and a higher quality of life following an exercise and self-affirmation intervention compared to the control group. 77
Environmental enrichment
EE with different physical, social, and sensory stimuli has been linked to enhanced neural plasticity and improved functional outcomes in experimental models of TBI. 78 EE given in combination with pharmacotherapies was associated with improved outcomes as compared to pharmacological treatments alone. 78 Furthermore, EE is effective across different age groups and in both sexes. 79 Since there is a dearth of precise therapeutic interventions to manage post-TBI neurologic dysfunction, rehabilitative paradigms like EE are attractive due to their translational potential with minimal or no adverse effects. 80
It was shown that 6 weeks of EE started immediately after weight-drop TBI in mice resulted in a significant improvement in cognitive function and reduced anxiety-like behavior. 81 Another study showed that 6 weeks of EE started at 1 week after weight drop injury in rats resulted in attenuation of injury-associated hyperexcitability in the supragranular sensory cortex and promoted experience-dependent neuronal plasticity. 82 When rats subjected to CCI injury were exposed to EE either continuously for 7, 14, and 21 days or intermittently for 14 days with a gap of 7 days between the two 7-day EE episodes, the continuous and early EE paradigms enhanced motor and cognitive function compared to the intermittent EE and standard housing groups. 83 Furthermore, rats subjected to EE for 14 and 21 days after TBI performed significantly better in the Morris water maze test than the rats that received early and intermittent EE, indicating that a longer period of EE promotes cognitive benefits after TBI. 83 In rats subjected to CCI, EE starting 1 day after TBI and continuing up to 3 weeks curtailed oxidative stress, excitotoxicity, and microglial activation, and promoted motor and cognitive recovery. 84 When adult rats were subjected to EE for 15 days before the induction of CCI injury, social neglect was decreased and cognitive recovery was improved compared to the rats housed in a standard environment. 85 Similarly, EE for 3 days prior and 38 days post-mild repetitive weight-drop TBI (7 injuries in 9 days) in mice significantly enhanced the exploratory activity, cognitive function, and suppressed anxiety-like behavior, compared to the normal housing cohort. 86 At the molecular level, EE inhibited synaptic changes by preventing a TBI-induced increase in phosphorylation of calcium/calmodulin-dependent protein kinase II (CaMKII), and expression of glutamate receptor 1 and calpain-1 in the cerebral cortex compared to the control. 86
EE in preclinical studies is a relative model with a defined arbitrary baseline that compares neurobehavioral changes in animals living in two artificial environments (here we refer to the normal housing of rodents itself as artificial since it’s not their natural habitat). In this scenario, the overall outcomes of EE studies rely on the relative richness of the experience, which is not quantifiable and cannot be extrapolated to humans. The arbitrary baseline of enrichment based on the previous experience of humans under normal conditions is subjective and difficult to define, posing a challenge in adapting EE as a treatment paradigm in the clinic. However, understanding the patient’s perspective of an enriched environment and employing personalized task-specific training as a part of rehabilitation may improve the outcomes after acute brain injuries including TBI.
Deep brain stimulation
Electrical stimulation of specific subcortical areas of the brain showed therapeutic benefits by fine-tuning both the local and distributed neuronal networks in various CNS disorders including TBI. 87 TBI survivors experience defects in executive function resulting in issues with behavior inception, impaired self-control, and mental flexibility. 88 These processes are regulated by monoaminergic transmission predominantly in the frontal cortex, striatum, and hippocampus. 88 In severe TBI patients, DBS for 6 weeks with electrodes implanted bilaterally in the nucleus accumbens and anterior limb of the internal capsule resulted in a significant improvement of cognitive abilities and functional independence after a 2-year follow-up period. 89 Similarly, in an 11-month prospective study with 2 TBI survivors, bilateral stimulation of DBS electrodes targeting the pallido-thalamic region for 5 months increased the mean coma-recovery-revised score and metabolic indices of the cerebral cortex. 90 Stimulation of the lateral cerebellar nucleus (LCN) restores the residual functional capacity of neuronal networks after TBI, partly by regulating the levels of neurotrophic factors such as BDNF, CaMKIIα, and p75 neurotrophin receptor in a rat model of CCI-induced TBI. 91 Moreover, LCN stimulation after FPI in rats improved motor function and mechanistically increased the expression of RNAs related to excitatory and long-term potentiation such as CaMKIIα, postsynaptic density protein 95, and cAMP response element-binding protein. 92 It was also shown that DBS of LCN significantly decreases neuroinflammation and apoptosis, by regulating the levels of IL-1β, inducible nitric oxide synthase, and c-Fos, suggesting its therapeutic potential in regulating multiple pathological events after TBI. 92
Decompressive craniectomy
Intracranial hypertension arising from cerebral edema and hematoma is the leading cause of death and disability following TBI. 93 DC involves inducing one or more small openings in the skull. Following TBI, DC can be used to evacuate the acute subdural hematoma (primary DC) or to relieve refractory intracranial hypertension (secondary DC). 94 However, the unavailability of guidelines to determine the location and size of the DC along with uncertainty in its efficacy and timing makes it still a controversial therapy. 94
In mice subjected to CCI, non-closure or reopening of craniectomy made for the induction of CCI led to reduced edema, and no secondary lesion expansion within the first 24 h of injury compared to the animals in which the craniectomy was closed. 95 However, this protection was lost when DC was performed at 8 h after CCI. 95
In humans, primary DC was not observed to be better than craniotomy in promoting functional recovery after TBI. 96 However, compared to craniotomy, DC was proposed to be beneficial for a subset of patients with severe high-energy injuries. 97 The effects of secondary DC following TBI were evaluated extensively in clinical trials. In 155 adults with severe diffuse TBI, secondary bi-frontotemporoparietal DC reduced the duration and the number of interventions needed for the management of intracranial hypertension and days in the ICU compared to standard care. 98 Conversely, six months after the injury, patients who underwent DC exhibited worse Extended GOS scores. Similarly, medical, or surgical complications such as hydrocephalus and hematoma were higher in the craniectomy group (37%) than in the standard care group (17%). However, 12 months after DC, there was no significant difference in Extended GOS score between craniectomy and standard-care groups. 99 In a randomized clinical trial (408 patients aged between 10–65 years), decreased mortality and increased rate of vegetative state and survival were observed six months after TBI in the DC group compared to the standard care group. 100
Some of the other beneficial effects of DC in TBI patients include improved visibility of mesencephalic cisterns and decreased midline shift of brain tissue. 101 Interestingly, younger (<50 years) TBI patients who had undergone DC showed better GOS score compared to older patients when evaluated 6 months after the injury. 101 Furthermore, an early DC (4.5 ± 3.8 h after TBI) was observed to be more beneficial than a delayed (56.2 ± 57.0 h) DC in TBI patients. 101 The effectiveness of DC also depends on the age of the TBI patient. Patients aged >65 were adversely affected by DC compared to younger patients. 102 However, several clinical studies reported complications such as subdural hygroma, intracranial infection, and bone flap resorption in TBI patients who have undergone DC. 103
Probiotics and fecal microbiota transplantation
As TBI leads to gut microbial dysbiosis, experimental studies explored the effect of rectifying it. Post-TBI supplementation of Lactobacillus acidophilus (LC), which is the most abundant bacteria in the gut, improved neurological function, reduced lesion volume, BBB damage, and brain edema, and attenuated neuroinflammation in adult mice. 104 LC supplementation was also shown to prevent post-TBI disruption of the intestinal wall integrity in mice by regulating the protein kinase C/myosin light chain kinase signaling pathway. 105 In severe TBI patients, administration of probiotics containing Bifidobacterium longum, Lactobacillus bulgaricus, and Enterococcus faecalis reduced the levels of endothelin-1, C-reactive protein, IL-6, IL-10, and TNF-α, and further decreased the hospitalization duration and pulmonary infection rate. 106 Similar benefits of decreased infection rate and shortened hospital stay were observed when brain-injured patients were supplemented with glutamine and probiotics containing Lactobacillus johnsonii. 107 The shift from T-helper cells from type 1 (Th1) to type 2 (Th2) causes immunological dysfunction during TBI leading to increased vulnerability to infections and sepsis. 108 A probiotic supplementation containing Bifidobacterium longum, Lactobacillus bulgaricus, and Streptococcus thermophilus attenuated the Th1 to Th2 shift, increased serum IL-12 and interferon-γ levels, and decreased IL-4 and IL-10 levels in TBI patients. 108 Correspondingly, a decreased incidence of nosocomial infections and shorter ICU stay were observed in patients given probiotics. 108 However, a meta-analysis of randomized controlled trials showed no significant effects of probiotic supplementation on immune response or ICU stay in TBI patients. 109
Experimental studies also observed the benefits of post-TBI FMT. Post-CCI FMT for 7 consecutive days from uninjured donor rats reversed TBI-induced changes in α- and β-bacterial diversity of the gut microbiome. 110 Furthermore, FMT alleviated oxidative stress in the ipsilateral hippocampus and improved neurological recovery compared to vehicle treatment. 110 The molecular mechanisms behind the beneficial effects of FMT after TBI include the elevation of levels of antioxidant enzymes methionine sulfoxide reductase A, catalase, and SOD in the hippocampus. 110 In mice subjected to CCI injury, an FMT regimen that started 2 h after TBI and continued once weekly for 4 weeks resulted in a reduction in ventriculomegaly and lesion volume and preserved the white matter connectivity. 111 FMT in mice also reduced post-TBI inflammatory gene expression and T-cell infiltration into the brain, possibly by regulating the inflammatory signaling between the gut and brain. 111
Gene therapy
Retroviral, lentiviral, adenoviral, and adeno-associated viral vectors as well as non-viral vectors such as lipid- or polymer-based carriers can deliver genetic material to treat a disease condition. Introduction of the vascular endothelial growth factor gene into the rat brain 20 min following TBI improved cortical vasculature, behavioral outcomes, and reduced cell death. 112 Overexpression of insulin-like growth factor 1, which is known to be neuroprotective and anti-inflammatory resulted in reduced oxidative stress in the area surrounding the lesion and prevented cognitive deficits in rats subjected to TBI. 113 Interestingly, overexpression of the human glucocorticoid receptor gene following neonatal injury improved spatial learning and memory during adolescence in rats. 114 Hippocampal delivery of the pro-oncogene ski at 7 days after TBI reduced lesion volume and astrogliosis, and promoted neurogenesis and functional recovery in mice. 115
Music therapy
Music is known to have a calming effect on the brain. Several studies evaluated if singing or listening to music can help in recovery following TBI. When TBI patients chose and sang 3 songs for 15 sessions, there was an enhancement of mood in the longer term. 116 In a cohort of mild TBI patients, playing piano with an instructor (2 sessions per week for 8 weeks, 30 min each time) and then on their own for 15 min a day improved neuropsychological test outcomes. 117
When they heard their preferred music, 14 cognitively impaired severe TBI patients showed reduced agitation compared with a cohort that heard general classical relaxation music. 118 In a randomized controlled clinical trial, 40 patients with moderate or severe TBI showed improved cognitive function when they received neurological music therapy for 3 months. 119 In a cohort of 40 moderate to severe TBI patients, a 3-month neurological music therapy increased grey matter volume in the right inferior frontal gyrus and improved the executive function indicating post-injury rehabilitation by music therapy. 120 In a clinical trial with 25 moderate or severe TBI patients, a 3-month neurological music therapy promoted white matter plasticity and executive function. 121 A recent study showed that in rats subjected to moderate to severe CCI injury, classical music reduced cortical lesion volume and activated microglial number, and induced BDNF expression leading to improved motor, cognitive, and anxiety-like behavior compared to no music cohort. 122 Overall, these studies are still rudimentary but indicate the potential of music therapy in post-TBI rehabilitation.
Stem cell therapy
Injury-induced neurogenesis has been observed in different experimental TBI models. However, due to the scarcity of endogenous NSCs, supplementation of exogenous stem cells is thought to promote functional recovery after TBI. Intracerebroventricular administration of human umbilical cord blood-derived mesenchymal stem/stromal cells (MSCs) 24 h post-TBI decreased sensorimotor and cognitive deficits, attenuated lesion volume and glial scar, and upregulated BDNF expression in adult mice. 123 Similarly, IV injections of rat bone marrow-derived MSCs (BM-MSCs) 24 h post-TBI attenuated motor function deficit and improved neurological severity score (NSS) in rats. 124 Interestingly, transplantation of rat BM-MSCs cultured in a medium containing BDNF and nerve growth factor (NGF) conferred better neuroprotection in rats subjected to CCI compared to the MSCs cultured in a medium deprived of these growth factors. 125 Transplantation of human MSCs in rats subjected to CCI also showed improvement in NSS and motor function that lasted for at least 3 months. 126 Moreover, the transplanted MSCs migrated to the injury site and expressed neuronal and astrocytic markers in the post-TBI rat brain.124,126,127 Migrated BM-MSCs also showed increased expression of NGF and BDNF, and promoted endogenous progenitor cell proliferation in rats subjected to CCI.127,128 The beneficial effects of MSCs are primarily due to the intercellular communication via extracellular vesicles and paracrine signaling rather than their ability to differentiate and substitute injured cells as previously thought. Moreover, MSCs are known to transfer mitochondria, anti-inflammatory cytokines, and RNA to host cells, including immune cells, via extracellular vesicles in a broad range of tissue injury and disease models. Correspondingly, post-CCI IV injection of cell-free exosomes generated from human BM-MSCs attenuated motor deficit and improved modified NSS and spatial learning without altering the lesion volume in rats. 129
NSCs are multipotent but differentiate into neural lineage cells when transplanted. Long-term cultured or cryopreserved human NSCs and induced pluripotent stem cell (iPSC)-derived NSCs were shown to be suitable for successful transplantation. Transplantation of NSCs 3 days post-CCI improved motor function, but not cognitive function in mice. 130 Furthermore, NSCs transplanted into the ipsilateral side expressed both neuronal and astrocytic markers, while those transplanted into the contralateral side expressed only neuronal markers. 130 Conversely, improvement in motor and cognitive functions was observed in mice transplanted with neural progenitor cells (NPCs) 7 days post-CCI. 131 These transplanted NPCs survived in the host brain for at least 14 months and expressed oligodendrocyte progenitor cell markers, but not neuronal or glial markers. 131 Some preclinical studies tested the efficacy of human NSCs in rat models of TBI. One such study observed the proliferation and migration of human NSCs transplanted near a cortical weight-drop injury to other brain regions including the contralateral cortex. 132 Moreover, transplanted human NSCs differentiated into neurons and astrocytes, but not oligodendrocytes. 132 Similarly, grafting of human NSCs one day after FPI improved spatial learning and memory when tested 10 days after grafting. 133 These human NSCs were mainly differentiated into neurons in the injured hippocampus and released glial-cell-line-derived neurotrophic factor. 133 Even though NSCs have the potential to differentiate into any brain cells, in a mouse CCI model, transplantation of human NSCs one day after the injury did not show any significant decrease in lesion volume. 134 Instead, these mice showed a significant decrease in injury-dependent accumulation of amyloid precursor protein. Furthermore, human NSC transplantation reduced TBI-induced microglial activation and promoted the transition of microglia/macrophages towards the reparative inflammatory phenotype. 134
Cell therapy was also tested in TBI patients. In a phase I clinical trial involving 10 children (aged 5–14 years), IV delivery of autologous bone marrow-derived mononuclear cells (BMMNC) within 48 h after TBI preserved brain structure (no significant change in grey matter, white matter, intracranial volume, or CSF volume) and improved adaptive behavior assessment scale, GOS score and the neuropsychological outcomes in a 6-month follow-up. 135 An extension study that compared the above patient data to a control group of age- and severity-matched children found that BMMNC therapy was associated with easier management of ICP measured by Pediatric Intensity Level of Therapy score and reduced severity of organ dysfunction measured using Pediatric Logistic Organ Dysfunction score. 136 Adult patients (aged 18–55 years) who received autologous BMMNC within 48 h after injury also showed structural preservation of brain regions correlated with neuropsychological outcomes such as verbal fluency and memory, and downregulation of major inflammatory cytokines in plasma 96 h post-injury. 137 In another clinical trial, the administration of autologous BM-MSCs directly into the injured area followed by a second dose of IV injection improved neurological function measured using the Barthel index in patients aged 30–55 years. 138 Interestingly, the transplantation of fetal neural cells along with hemopoietic liver cells, and umbilical cord-derived MSCs (UC-MSCs) in TBI patients also improved clinical outcomes evaluated by GOS, Fugl-Meyer Assessments (FMA), and Functional Independence Measures, respectively. 139 Moreover, significant improvement in chronic motor deficits measured by FMA was observed in TBI patients who received intracranial implantation of SB623 (allogeneic modified BM-MSC) cells. 140
Some studies tested stem cells modified genetically to improve their therapeutic potential. BM-MSCs overexpressing SOD2 or human UC-MSCs lacking histone deacetylase 1 transplanted to the injured mouse brain showed improved therapeutic efficacy by protecting transplanted MSCs from oxidative stress-induced apoptosis or promoting their engraftment in the hippocampus, respectively.141,142 Transplantation of UC-MSCs overexpressing BDNF conferred neuroprotection in rat models of TBI. 143 Compared to naïve MSCs, the MSCs overexpressing IL-10 attenuated TBI-induced functional deficits more effectively by reducing inflammation. 144 Similarly, NSCs and NPCs were also genetically modified to elevate their therapeutical potential in experimental TBI; some examples of overexpressed genes in NSCs and NPCs include human multineurotrophin (MNTS1), glial cell line-derived neurotrophic factor (GDNF) and BDNF145–147. Transplantation of NPCs overexpressing MNTS1 one week after FPI in rats exhibited better NPC survival and neuronal differentiation than control NPCs. 145 However, this genetic modification of NPCs did not affect functional and pathological outcomes significantly. Improved cognition along with superior NPC survival and neuronal differentiation were seen in NPCs overexpressing GDNF when grafted into the brains of rats subjected to FPI compared to naïve NPCs. 146 Overexpression of BDNF in NSCs elevated the levels of pre-and post-synaptic proteins (synaptophysin and SHANK2) and growth-associated protein 43 compared to naïve NSCs when delivered to the injured cortex of rats subjected to CCI. 147 Interestingly, NSCs stably transfected with epidermal growth factor receptor variant III showed differential survival rates of 100%, 65%, and 10% when transplanted to the sham, ipsilateral, and contralateral side of rats subjected to TBI, respectively. 148
The efficacy of stem cell therapy increased when combined with other protective biomolecules. Continuous infusion of NGF along with fetal cortical graft transplantation improved post-TBI cognitive and motor function in rats. 149 Supplementation of curcumin-loaded niosome nanoparticles after the transplantation of human NSCs/NPCs was superior in improving motor function and reducing brain edema, astrogliosis, and neuroinflammation in rats subjected to TBI. 150 Furthermore, compared to the control group (standard environment/without iPSC therapy), superior motor and cognitive functions were observed in rats transplanted with iPSCs exposed to an enriched environment following TBI. 151 Another factor that influences the performance of the cell transplant is the extracellular environment. The use of biomaterials that act as extracellular matrices such as fibronectin and collagen scaffolds improved the survival of the transplanted stell cells and promoted functional recovery in rodent models of TBI.152,153 Some of the other parameters that affect the efficacy of stem cell therapy are the route and time of administration, number of cells used and severity of the injury. Similar to pharmacological therapies, these parameters need to be optimized for the clinical success of stem cell therapy.
Pharmacological aspects of non-pharmacological interventions
Even though non-pharmacological interventions, as the name suggests, do not use chemical compounds, many of them act via mechanisms common to pharmacological interventions. For example, hypothermia regulates cerebral metabolism, arterial blood pressure, glutamate release, and ROS formation following TBI.30,32,34 Similarly, hypothermia, HBO, and DC reduce edema and ICP.27,32,38,95 Other molecular mechanisms targeted by non-pharmacological interventions in TBI include ferroptosis (in IF), autophagy via mTOR (in IF and CR), and metabolism via SIRT1 (in IF, CR, and KD).60,61,63,64 Stem cells transplanted at the site of injury secrete factors such as neurotrophic factors, cytokines, and chemokines and regulate the function of surrounding cells.140,154 Furthermore, part of the neuroprotection by probiotics is through the short-chain fatty acids produced by the beneficial microorganisms that colonize the gut. 155 One feature that sets most non-pharmacological interventions apart from pharmacotherapy is its multifunctionality.
Conclusions
Compared to pharmacotherapies, non-pharmacological interventions take a more holistic approach rather than target a particular aspect of TBI pathology. These interventions can confer neuroprotection by modulating some of the TBI-induced processes such as mitochondrial dysfunction, neuroinflammation, oxidative stress, and apoptosis as depicted in Figure 1. Some of the non-pharmacological interventions also promote the post-TBI repair process by inducing neurogenesis and neuroplasticity and by regulating glial scar formation and autophagy. Furthermore, non-pharmacologic interventions have minimal to no side effects. Future studies that explore the benefits of combining non-pharmacological and pharmacological interventions are needed. Also, studies that consider sex and age as biological variables are essential for the better translatability of non-pharmacological interventions. One drawback of some of these interventions like dietary adaptations is that they are more effective as a prophylactic strategy than as a therapy. Moreover, there is a lack of uniform or standard procedures for most of the non-pharmacological interventions.
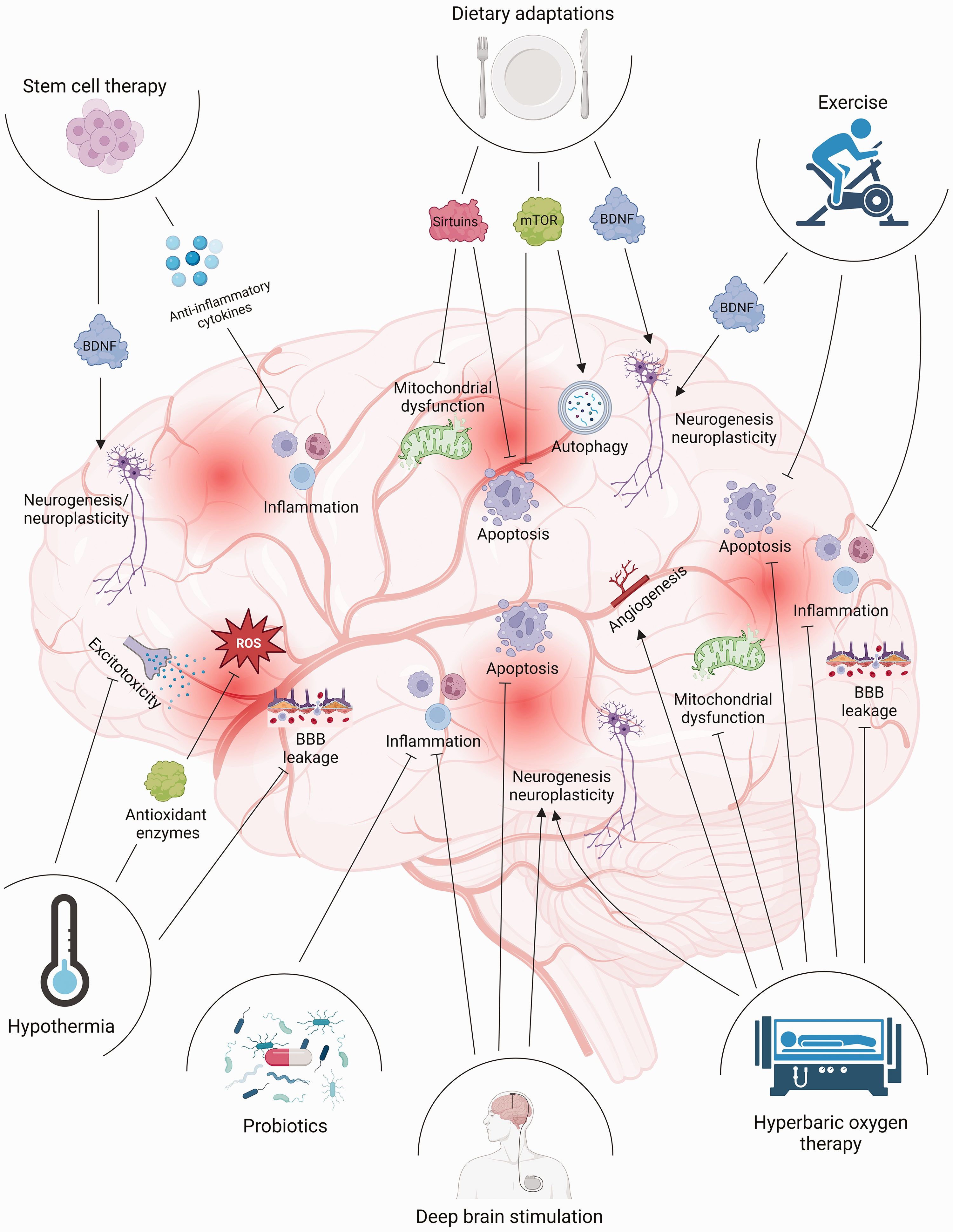
Putative molecular mechanisms involved in the neuroprotection by various non-pharmacological interventions following TBI. Most of the non-pharmacological interventions targeted pathological processes such as inflammation, apoptosis, mitochondrial dysfunction, and BBB leakage. Interventions such as stem cell therapy, dietary adaptations, and exercise upregulated the level of BDNF and promoted neuroplasticity.
Supplemental Material
sj-pdf-1-jcb-10.1177_0271678X241234770 - Supplemental material for Non-pharmacological interventions for traumatic brain injury
Supplemental material, sj-pdf-1-jcb-10.1177_0271678X241234770 for Non-pharmacological interventions for traumatic brain injury by Charles K Davis, Vijay Arruri, Pallavi Joshi and Raghu Vemuganti in Journal of Cerebral Blood Flow & Metabolism
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Partially funded by grants from the US National Institute of Health (R35NS132184, RO1NS130763, and RO1NS109459) and US Department of Veterans Affairs (I01BX004344 and I01BX005127). Dr. Vemuganti is a recipient of a Research Career Scientist Award (IK6BX005690) from the US Department of Veterans Affairs.
Acknowledgements
The authors thank Dr. Y. Sathyamoorthy for some suggestions.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Supplementary material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.