
Abstract
Aneurysmal subarachnoid hemorrhage (SAH) carries significant mortality and morbidity, with nearly half of SAH survivors having major cognitive dysfunction that impairs their functional status, emotional health, and quality of life. Apart from the initial hemorrhage severity, secondary brain injury due to early brain injury and delayed cerebral ischemia plays a leading role in patient outcome after SAH. While many strategies to combat secondary brain injury have been developed in preclinical studies and tested in late phase clinical trials, only one (nimodipine) has proven efficacious for improving long-term functional outcome. The causes of these failures are likely multitude, but include use of therapies targeting only one element of what has proven to be multifactorial brain injury process. Conditioning is a therapeutic strategy that leverages endogenous protective mechanisms to exert powerful and remarkably pleiotropic protective effects against injury to all major cell types of the CNS. The aim of this article is to review the current body of evidence for the use of conditioning agents in SAH, summarize the underlying neuroprotective mechanisms, and identify gaps in the current literature to guide future investigation with the long-term goal of identifying a conditioning-based therapeutic that significantly improves functional and cognitive outcomes for SAH patients.
Keywords
Introduction
Aneurysmal subarachnoid hemorrhage (SAH) affects ∼30,000 people in the United States per year. 1 Although, SAH accounts for only 6–8% of all strokes, it is responsible for ∼25% of all cerebrovascular-related deaths and years lost due to stroke,2 –4 due to its early occurrence (median age 53 years) and high mortality (45% at 30 days) and morbidity (50% of survivors have long term neurological deficits).2 –5 While improved neurocritical care and the advent of endovascular therapy for vasospasm have likely improved SAH outcomes to some degree, no pharmacologic agent has been effective in preventing or attenuating secondary brain injury after SAH since nimodipine was introduced in the early 1980s. Nonetheless, modern era morbidity and mortality remain unacceptably high, and new methods of treating SAH-induced brain injury are desperately needed.
The pathophysiological drivers of secondary brain injury after SAH are commonly divided between those leading to early brain injury (EBI) versus those contributing to delayed cerebral ischemia (DCI), although these mechanisms are not mutually exclusive (Figure 1).5 –15 EBI refers to secondary brain injury occurring within the first three days after SAH,6 –9 with BBB breakdown, cerebral edema, neuroinflammation, increased intracranial pressure, and neuronal cell death identified as key contributors to this form of secondary brain injury.6 –9,13 –15 DCI is a phenomenon in which new neurological deficits develop well after ictus, typically 4–12 days, with a peak incidence of 7–10 days post-SAH.9 –15 Cerebral vasospasm was once thought to be the sole contributor to, and was used synonymously with, DCI; however, recent evidence implicates that microvascular autoregulatory dysfunction,16,17 microthrombosis,18,19 and cortical spreading depression20 –22 also play significant pathophysiological roles.
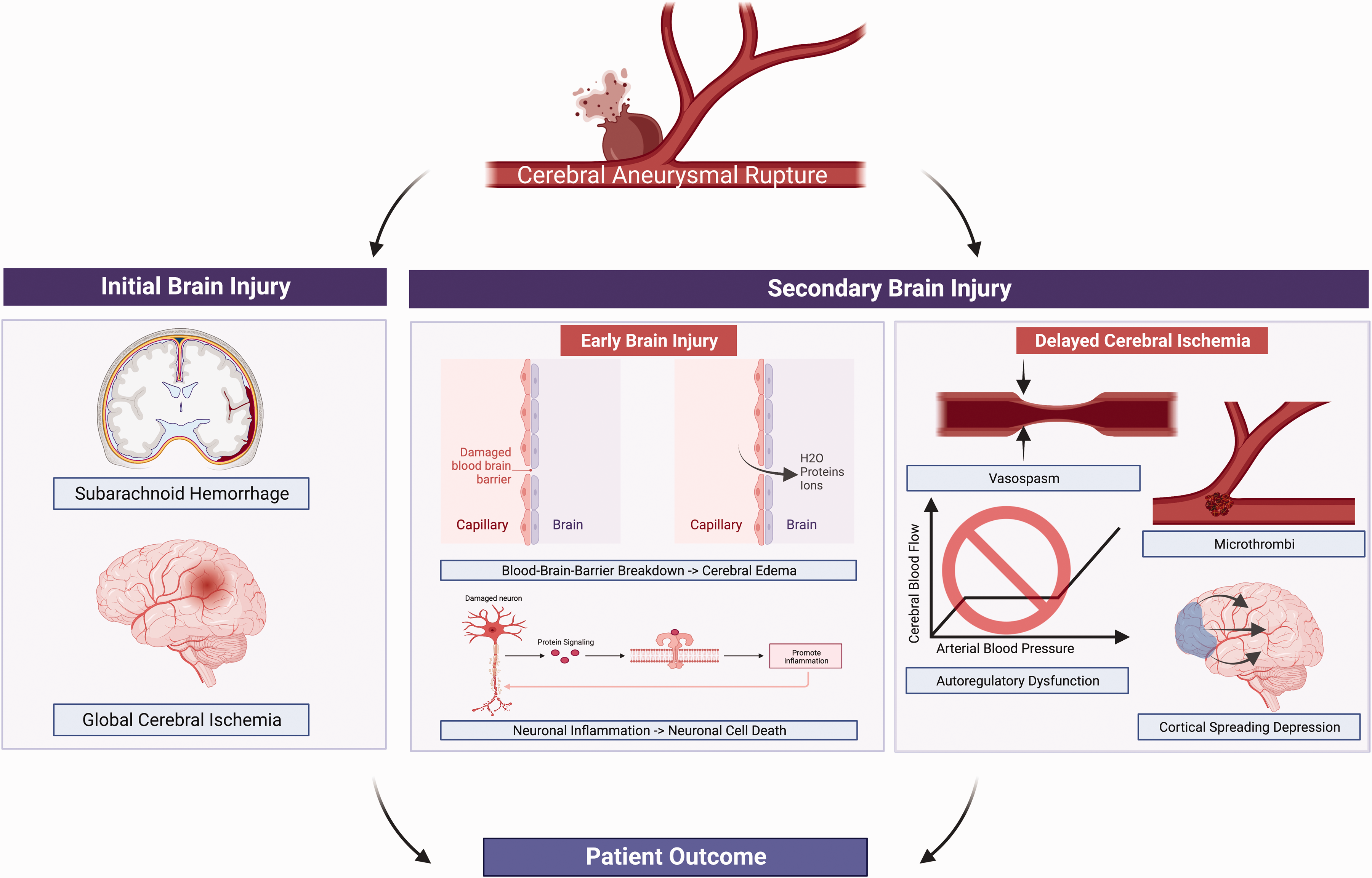
Primary drivers of patient outcome after aneurysm rupture.
Given that EBI and DCI are driven by multiple molecular cascades affecting multiple CNS cell types (vascular cells, neurons, and glia), and that several therapies targeting single elements (e.g., large-artery vasospasm) 23 or single factors (e.g., endothelin 1, tirilizad, statins)24 –26 implicated in these brain injury pathways have failed to translate in late phase clinical trials, new approaches towards preventing or reducing EBI and DCI are needed. Likely, this will require therapeutic strategies that target multiple molecules and/or cell types ultimately to effectively reduce SAH-induced secondary brain injury and improve long-term functional outcome. Conditioning is one such therapeutic strategy that leverages endogenous protective mechanisms to exert powerful and remarkably pleiotropic protective effects against injury to all major cell types of the CNS including neurons, glia, and vascular cells. 27
In the present review, we review the existing literature on the diversity of conditioning agents studied to date and the underlying mechanisms by which they exert their powerful protective effects against brain injury. Next, we assess and synthesize the growing body of preclinical and clinical data that suggests conditioning-based therapy for SAH is a promising new therapeutic approach. Finally, we explore the necessary next steps towards potential translation of a conditioning-based therapy to SAH patients.
Conditioning-based therapy for acute brain injury
Conditioning is a neuroprotective strategy whereby the brain’s inherent resistance to injury can be enhanced by exposure to a sub-lethal injurious stimulus – much of which stems from the resulting epigenetic response. The adaptive responses induced by conditioning involve molecular sensors and transducers, transcription factors, genes, and effectors that ultimately produce a cerebroprotective phenotype.27 –29 In particular, epigenetic reprogramming of the brain plays a key role in producing this protective phenotype. 29 Experimental studies have shown that conditioning-based therapeutics provide powerful protection in variety of acute brain injury conditions including ischemic stroke, traumatic brain injury, intracerebral hemorrhage, and most recently SAH.30 –35 A number of molecular pathways have been implicated in the protective phenotype provided by conditioning. Important for the topic of this review, many of these molecules have been implicated in SAH pathophysiology including endothelial nitric oxide synthase (eNOS), Hypoxia inducible factor- 1 alpha (HIF-1α), Matrix Metallopeptidase 9 (MMP-9), Sirtuin 1 (SIRT1) and others.36 –42
Conditioning-based therapy for SAH
A wide therapeutic window of opportunity in SAH patients (EBI develops 1–3 days and DCI develops 4–12 days after ictus) makes conditioning-based therapeutic strategies particularly attractive for this devastating condition. Conditioning is traditionally categorized based on timing of intervention, with preconditioning referring to stimulus application prior to SAH and postconditioning referring to stimulus application following SAH. Both forms of conditioning have been examined in the setting of SAH. Herein, we critically review and synthesize the preclinical and clinical evidence for conditioning-based therapeutics as a novel means to reduce EBI and DCI and improve neurologic outcome after SAH.
Experimental studies examining early brain injury (EBI)
Anesthetic conditioning
Volatile anesthetics, which are routinely used in a variety of patients requiring general anesthesia, have been shown in preclinical studies to provide robust neuroprotection against EBI after SAH.43 –48 The earliest studies exploring this topic came from Altay and colleagues who were the first to demonstrate that isoflurane postconditioning could reduce or delay EBI following SAH.43 –45 First, they noted that a brief exposure of 2% isoflurane for 1 hour (initiated 1 hour after induction of SAH) in a mouse endovascular perforation model provided strong protection against several components of EBI including cerebral edema, blood brain barrier disruption, neuroinflammation, and neuronal apoptosis leading to improved neurobehavioral outcomes at 24 hours. 43 This protection against neurobehavioral outcomes, however, was lost at 72 hours. In a follow up study, these authors examined a dose effect, showing that 2% and not 1% isoflurane postconditioning provided EBI protection in SAH. 44 They also explored the mechanistic underpinnings for observed EBI protection, showing that isoflurane-induced EBI protection was blocked by a sphingosine kinase antagonist (N, N dimethyl sphingosine) and a sphingosine-1-phosphate receptor-1/3 receptor antagonist (VPC23019) – pharmacologic data indicating isoflurane’s neuroprotective effect against EBI is mediated by the sphingosine pathway.43 –45 Subsequent studies by the same authors examined the impact of a commonly used volatile anesthetic, sevoflurane, on EBI and compared its effect to that of isoflurane in an endovascular perforation mouse model. They found that administration of 1.5% sevoflurane for 60 minutes and 3% sevoflurane for 30- and 60-minutes (both initiated 1-hour post SAH) significantly attenuated EBI and improved neurological outcomes 24 hours after SAH. Other doses and durations of sevoflurane (e.g. 1.5% for 30 and 90 minutes, 3% for 90 minutes, and 4.5% for 30, 60, and 90 minutes) had no protective effect, indicating sevoflurane’s EBI protection is dose dependent. 46 Another study directly compared the EBI protection afforded by isoflurane vs. sevoflurane, finding that 2% isoflurane and 3% sevoflurane had similar levels of EBI protection. 47
Other groups have examined the protective effect of anesthetic conditioning in SAH. For example, Beck-Schimmer and colleagues compared the neuroprotective effects of differing classes of anesthetics in SAH. They found that while sevoflurane postconditioning provided robust protection against EBI in a rat endovascular perforation model of SAH, postconditioning with propofol (a commonly used intravenous anesthetic agent) had no impact on EBI. They also showed that sevoflurane attenuated the downregulation of cytosolic Beta-catenin and speculated that sevoflurane’s effect on EBI may be due to preserving adherens junctions and stabilizing membrane integrity through Beta-catenin. 48 In contradistinction, other studies have demonstrated a neuroprotective effect of propofol on EBI. The first showed that intravenous administration of propofol (10 or 50 mg/kg) provided significant and dose-dependent protection against cerebral edema, blood brain barrier disruption and improved neurological outcomes in a rat endovascular perforation model. 49 A subsequent study by the same authors indicated that intraperitoneal administration of propofol (50 mg/kg) also provided EBI protection. They also showed that this propofol-induced EBI protection was lost with co-administration of LY294002, a specific inhibitor of Phosphatidylinositol 3-kinase/Akt (PI3K/Akt) signaling pathway, suggesting involvement of the PI3K/Akt pathway in the observed EBI protection. 50 It is important to note that the pharmacokinetics of a drug varies with the route of administration and the intraperitoneal administration does not mimic the route of propofol administration in patients.
Interestingly, inert or noble gases (very low anesthetic activity at standard temperature and pressure) have also been shown to provide EBI protection in rodent models of SAH. Postconditioning with Argon (mixture of 50 vol% argon/50 vol% oxygen x 1 hour) in a rat endovascular perforation model was associated with reduced mortality, although no difference in cerebral edema or neuroscore was noted. 51 Improved survival was attributed due to the increased expression of HIF-1α, and heme oxygenase 1. Other studies showed that postconditioning with Xenon (50 vol% O2/50 vol% Xe x 1-hour, echogenic liposomes-600 μL per animal, 10 mg lipid/mL) in a rat endovascular perforation model reduced microglia activation, neuronal damage, improved neurological function, and decreased mortality.52,53 Mechanisms for the EBI protection afforded by Xenon was not explored.
Lipopolysaccharide (LPS) conditioning
Preconditioning with low dose of LPS, a component of gram-negative bacteria and a regulator of immune system has been shown to provide EBI protection in preclinical studies. For example, intraperitoneal administration of LPS (200 mg/kg, Escherichia coli serotype 055; B5) 24 hours before SAH in a rat prechiasmatic injection model provided significant protection against cerebral edema, neuronal apoptosis, blood brain barrier disruption and improved neurological dysfunction at 24 hours post SAH.54,55 The expression of MMP-9, and caspase 3 were upregulated after SAH which was attenuated by LPS preconditioning, suggesting the neuroprotective mechanism of LPS is possibly through downregulating MMP-9 and caspase 3.
Remote limb ischemic conditioning
Remote limb ischemic conditioning (RIC) is a simple, noninvasive, easily accessible, conditioning strategy that involves repetitive inflations followed by deflations in the upper or lower extremities. Hu et al., utilizing a murine endovascular perforation SAH model, demonstrated that repeated RIC (3 cycles of 10 min occlusion/10 min release of the bilateral femoral artery per day for 3 days) provided robust protection against cerebral edema, neuronal apoptosis, short-term and long-term neurobehavioral deficits (1 month) induced by SAH. 56 Though the underlying mechanism for this EBI protection was not fully evaluated, the authors did show that repeated RIC markedly upregulated expressions of Beclin-1 and LC3 (biomarkers of autophagy), suggesting that the observed EBI protection could be mediated via autophagy activation. Interestingly, a protective effect of repeated RIC was not found against cerebral vasospasm. 56
Exercise conditioning
A preconditioning exercise protocol (exercised for 5 days/week for 3 weeks; running speed, 25 m/min; running duration, 30 min/day) has been shown to mitigate EBI and provide protection against neurological deficits in a rat endovascular perforation model. 57 After SAH induction, animals in the exercise group demonstrated significantly reduced neurologic deficits and improved sensorimotor function compared to rats in the non-exercise group. Authors also showed that rats in the exercise group had elevated levels of anti-oxidative stress markers (Nrf2, HO-1, 14-3-3γ), and reduced levels of apoptotic and inflammatory markers (4HNE, NT, Iba1, TNF-α, IL-6, IL-1β, Bax, caspase-3, and TUNEL-positive cells) suggesting that the protective mechanism of exercise preconditioning may be mediated via reducing oxidative stress, inflammation and apoptosis. 57
Clinical studies examining early brain injury (EBI)
A preliminary prospective study comparing the effects of different anesthetics (isoflurane, sevoflurane, desflurane, and propofol) on the plasma and serum caspase- 3 levels in SAH patients undergoing aneurysm clipping/coiling, showed that all four anesthetics significantly reduced intraoperative CSF caspase- 3 levels, suggesting that these anesthetics may have a potential neuroprotective role in SAH patients. 58 More extensive and rigorous studies examining the impact of volatile and intravenous anesthetics on SAH-induced EBI in patients are needed.
Experimental studies examining delayed cerebral ischemia (DCI)
No general consensus exists for the definition of DCI in the animal models. Majority of the experimental studies demonstrate the occurrence of cerebral vasospasm on day 2 and neurological deficits between 0–5 days, post SAH induction. 59 Hence, DCI is defined as the presence of vasospasm and neurological deficits on day 2 or after in animal SAH models.
Hypoxic conditioning
The initial application of a conditioning-based strategy in SAH was focused on the vasospasm protection afforded by hypoxic preconditioning in a rodent model of SAH. 36 Our group demonstrated that hypoxic preconditioning (8% O2/92% N2 for 4 hours, administered 24 hours before SAH) prevented the onset of cerebral vasospasm and significantly reduced neurological deficits in mouse endovascular-perforation SAH. 36 We also implicated endothelial nitric oxide synthase (eNOS) as a key mediator of the observed DCI protection. Specifically, we showed that 1) hypoxic preconditioning mitigated the negative effect of SAH on nitric oxide bioavailability; 2) hypoxic preconditioning increased eNOS activity; and 3) genetic deletion of eNOS blocked the DCI protection afforded by hypoxic preconditioniong. 36 A subsequent study by our group elucidated the important role of Sirtuin 1 (SIRT1), an upstream inducer of eNOS, in the DCI protection afforded by hypoxic preconditioning. Specifically, we showed that 1) hypoxic preconditioning upregulates SIRT1 expression; 2) the SIRT1 activator, resveratrol, produced robust protection against vasospasm and neurological dysfunction after SAH; and 3) the SIRT1-specific inhibitor, EX-527, abolished the DCI protection afforded by hypoxic preconditioning. 39
In a follow up study, we demonstrated the translational potential of conditioning-based therapy against DCI when we showed that hypoxic conditioning initiated 3 hr after SAH (hypoxic postconditioning) provides robust protection against large artery vasospasm, microvessel thrombosis, and neurological deficits. 40 We again implicated SIRT1 in the observed DCI protection, showing 1) Genetic overexpression of SIRT1 or pharmacological activation of SIRT1 via resveratrol mimicked the DCI protection afforded by hypoxic postconditioning; and 2) Genetic deletion of SIRT1 or pharmacological inhibition of SIRT1 via EX-527 abolished the DCI protection afforded by hypoxic postconditioning. 40 More recently, we showed that hypoxic postconditioning also provides strong protection against SAH-induced deficits in functional connectivity, as measured by optical intrinsic signal imaging. We also showed that this protection against functional connectivity deficits was SIRT1-mediated, given that this neurovascular protection was blocked by the SIRT1-specific inhibitor, EX-527, and mimicked by the SIRT1 activator, resveratrol. 41
Anesthetic conditioning
Our lab demonstrated that a structurally distinct conditioning agent – the volatile anesthetic, isoflurane – initiated at clinically relevant time points after SAH (15 min, 1 hr, and 3 hr after SAH) provides robust protection against multiple components of DCI including large artery vasospasm, microvessel thrombosis, and autoregulatory dysfunction in a mouse endovascular perforation SAH model. 37 We also showed that this anesthetic conditioning-induced DCI protection was mediated by HIF-1α, as we found that the afforded DCI protection was lost in the presence of 2ME2, a HIF-1α inhibitor, and in endothelial cell specific HIF-1α null mice. 37 Finally, as opposed to the earlier study by Altay and colleagues, our results indicated that anesthetic postconditioning can lead to sustained neurologic protection after SAH (i.e. beyond post-SAH day 1). 37 Subsequently, studies from an independent laboratory demonstrated that isoflurane postconditioning (2% isoflurane for 1 hour) combined with inotropic support improved cerebral perfusion, reduced DCI related infarcts, and improved neurological outcome in an endovascular perforation mouse model of SAH.60,61 These effects were abolished by administration of HIF-1α inhibitor, 2ME2, implicating a role for HIF-1α in isoflurane-induced DCI protection in SAH.60,61
More recently, through pharmacologic and genetic interventions, we causally linked eNOS to the DCI protection afforded by isoflurane postconditioning, 38 but we did not find a contribution from SIRT1. 62 The latter suggested that the molecular underpinnings of anesthetic vs. hypoxic conditioning for SAH-induced DCI are not entirely overlapping. To explore this possibility, additional studies were conducted to elucidate the potential role of other candidate molecules in the DCI protection afforded by anesthetic conditioning. In one study we implicated NF-kB (a transcriptional factor and a key regulator of inflammation) as a key upstream inducer, noting that 1) Isoflurane postconditioning attenuates NF-kB activation after SAH; 2) Administration of the NF-kB-specific inhibitor, PDTC, mimics the protection afforded by isoflurane conditioning; and 3) Coadministration of PDTC with isoflurane did not provide any additive DCI protection. 63 In another study, we also implicated iNOS – a key downstream mediator of NF-kB. Specifically, we showed that 1) iNOS upregulated after SAH and that was attenuated by isoflurane conditioning; 2) Administration of the iNOS-specific inhibitor, 1400 W, mimics the protection afforded by isoflurane conditioning; 3) Genetic deletion of iNOS mimics the protection afforded by isoflurane conditioning; and 4) Neither pharmacologic inhibition or genetic deletion of iNOS provided any additive DCI protection to isoflurane conditioning. 64
Because the dose of isoflurane (2%) used in all above-mentioned studies is not commonly employed for the patients during surgical procedures (approximately 1.2% or 1 MAC is generally applied for achieving anesthesia in surgical patients; 2% or 1.8 MAC would therefore be supratherapeutic for most patients; MAC = minimum alveolar concentration of anesthetic required for 50% of patients to not respond to a surgical stimulus), we performed a dose exploration for isoflurane conditioning in SAH. We found that not only does a supratherapeutic dose of isoflurane (2%) provide robust DCI protection, but so did an anesthetic dose (1%) and even a subanesthetic dose (0.5%). 65 The neuroprotective effects of other commonly utilized volatile anesthetics have also been investigated in SAH. We have shown that exposure to standard anesthetic concentrations (i.e. 1 MAC) of sevoflurane (2%) and desflurane (6%) also provided significant DCI protection in an endovascular perforation mouse model of SAH. 66 Finally, we recently examined the impact of intravenous anesthetic conditioning in SAH to begin to examine whether different anesthetic classes have differing effects on DCI. In contradistinction to various volatile inhalational anesthetics, intravenous administration of an anesthetic dose of propofol (2 mg/kg/min × 1 hr) did not afford any protection against vasospasm or the neurologic deficits in an endovascular perforation mouse model of SAH. 67
Lipopolysaccharide (LPS) conditioning
Two studies have examined the impact of LPS conditioning on SAH-induced DCI. In the first study, a single low dose of LPS (0.6 mg/kg) administered intraperitoneally 24 hours before SAH (vein transection model) was shown to protect against cerebral vasospasm and neurobehavioral deficits in mice. 68 In another study conducted by the same authors, administration of a single high dose of LPS (200 mg/kg) administered intraperitoneally 24 hours before SAH was found to exacerbate cerebral vasospasm and worsen neurological deficits in mice. 69 In combination, these data show LPS conditioning attenuates DCI in a dose-dependent manner and this effect was attributed to the increase or decrease in chemokines such as keratinocyte derived chemokine68,69 and Chemokine (C-C motif) ligand 5. 69
Remote limb ischemic conditioning
One recent study examined the impact of remote limb ischemic conditioning (RIC) on SAH-induced DCI. They found that remote limb ischemic conditioning (10 min hind limb ischemia × 3 sessions per day × 3 days) initiated 24 hours after induction of SAH (cisterna magna injection model) provided significant protection against cerebral vasospasm but not against neurobehavioral deficits in rats. 70 Underlying mechanism for this DCI protection was not explored.
Clinical studies examining delayed cerebral ischemia (DCI)
Anesthetic conditioning
Supplementing the very promising preclinical findings on anesthetic conditioning in SAH, several clinical studies have suggested a protective role of inhalational anesthetics against DCI in SAH patients. In a preliminary study examining plasma concentrations of endothelin-1(ET-1) and calcitonin gene related peptide (CGRP) during desflurane anesthesia in SAH patients undergoing aneurysm clipping, Wang et al. 71 showed that desflurane anesthesia significantly reduced ET-1 levels, but had no impact on CGRP levels, during the perioperative period. In contradistinction, it was subsequently shown that intravenous propofol anesthesia significantly reduced CGRP levels, but had no effect on ET-1 levels. 72 Given the well-known vasoconstrictive vs. vasodilatory effects of ET-1 vs CGRP, it was posited that desflurane might reduce cerebral vasospasm in SAH patients and that propofol could have the opposite effect. Supporting this intriguing hypothesis, Lee et al. later found that SAH patients exposed to desflurane anesthesia during aneurysm clipping experienced lower incidence of transcranial doppler evident cerebral vasospasm, as compared to the SAH patients exposed to intravenous propofol anethesia. 73 Importantly, however, no difference in angiographic vasospasm, cerebral infarction, or the neurological deficits were noted between the two anesthetic groups.
More recently, our group has completed a series of clinical studies that strongly suggest volatile anesthetics have a substantial protective effect against cerebral vasospasm and DCI in SAH patients as compared to intravenous anesthetic, propofol. In the first study, we demonstrated that SAH patients exposed to volatile anesthetics (sevoflurane or desflurane) during their aneurysm repair (clipping/coiling) had lower incidence of angiographic vasospasm, compared to patients who received combined anesthetics (sevoflurane or desflurane and intravenous anesthetic propofol). 74 In a follow up study with a larger patient cohort from the same institution, we provided evidence that volatile anesthetics provide not only protection against angiographic vasospasm but also DCI. 75 Most recently, we directly compared SAH patients who received volatile anesthetics during aneurysm repair from one institution vs. SAH patients who received intravenous anesthetics during aneurysm repair from another institution. We found that volatile anesthetic use (sevoflurane or desflurane) was associated with significantly lower incidence of both angiographic vasospasm and DCI as compared to those who received the intravenous anesthetic, propofol. 76 No difference in functional status at time of patient discharge, however, was noted between anesthetic groups.74 –76 We posited that dose, duration, or timing of volatile anesthetic exposure had not been optimized to provide maximal neuroprotection and that further rigorous study was warranted.
Remote limb ischemic conditioning
Other investigators have also applied conditioning-based therapeutic strategies to SAH patients. Koch, Gonzalez, and colleagues have conducted a series of early phase clinical trials examining the feasibility and potential efficacy of remote limb ischemic conditioning (RIC) in SAH. First, they showed that a strategy of repeated RIC is feasible and can be performed safely in critically ill SAH patients.77,78 Second, they showed that repeated RIC led to a subtle but significant increase in PT and INR, raising the intriguing possibility that this conditioning-based treatment could decrease microvessel thrombosis (an important component of DCI). 79 Third, they showed that RIC in SAH patients caused transient cerebral vasodilation (with return to baseline immediately after the completion of the RIC protocol) and transient improvement in cerebral metabolism (as indicated by reduced glycerol levels and decreased lactate-to-pyruvate ratio). 80 Fourth, they showed that RIC alters genetic expression and methylation patterns in blood of SAH patients, particularly in genes regulating cell cycle and inflammation. 81 Finally, they showed in a single institution matched cohort study (21 SAH patients treated with RIC vs. 61 matched historical controls) that RIC treatment was associated with good functional outcome (as determined by modified Rankin scale 0–2 at discharge) as well as a non-significant trend towards lower rates of ischemic stroke and death. 82 Most recently, two single institution randomized controlled trials conducted at independent medical centers found that RIC treatment in SAH patients appeared to reduce the incidence of angiographic vasospasm, shortened ICU and overall hospital length of stay, and improved patient outcome as measured by Glasgow outcome scale extended (GOSE) at 6 months.83,84
Ischemic conditioning
To examine if the protective effects of preconditioning in animal models also exists in patients, a multicenter retrospective study was designed to examine if SAH patients with a preexisting steno-occlusive cerebrovascular disease (CVD) and or cerebral infarct had a protective effect on radiographic vasospasm, symptomatic vasospasm, vasospasm-related DCI and the functional outcome as measured by modified Rankin Scale score at discharge. 85 Intriguingly, authors noted that SAH patients with a preexisting CVD were less likely to develop radiographic vasospasm, but no differences were noted in other measured end points. Although this study does not establish causation, it provides correlative support for the notion that SAH patients may benefit from conditioning agents targeting the cerebrovasculature in similar ways as seen in animal studies. 85
Exercise conditioning
A retrospective study investigating the impact of exercise in SAH patients found that mild exercise with ambulation initiated after aneurysm repair prior to post-SAH day 4, significantly reduced symptomatic vasospasm compared to the non-exercised SAH patients. 86 In combination, authors have also demonstrated that implementing mild exercise in a rat SAH model did not increase iNOS, (a marker of neuronal injury), indicating the safeness of this conditioning strategy. 86
Experimental and clinical studies examining long-term functional outcome for SAH
Very few studies have thus far examined the impact of conditioning strategies on long-term functional outcomes after SAH. The aforementioned preclinical study that showed repeated RIC lead to significant reduction in EBI in a rat endovascular perforation model also demonstrated a significant protective effect against SAH-induced neurobehavioral deficits (up to 1 month). 56 Clinically, a small prospective randomized pilot study applying remote limb ischemic conditioning (RIC) in SAH patients demonstrated that RIC protected against both cerebral vasospasm and long-term functional outcomes (up to 6 months) as measured by GOSE score. 83 Though encouraging, these results are preliminary and more rigorous studies examining the impact of conditioning strategies on long-term functional end points are required.
Discussion
A growing body of preclinical and clinical evidence shows that several conditioning-based therapeutic strategies provide strong multifaceted protective effects against two of the most important drivers of SAH outcome – EBI and DCI (Figure 2, Tables 1 and 2). To date, the most rigorously studied conditioning-based therapies include brief exposures to volatile anesthetics, hypoxia, and RIC, which are the focus of the following discussion.
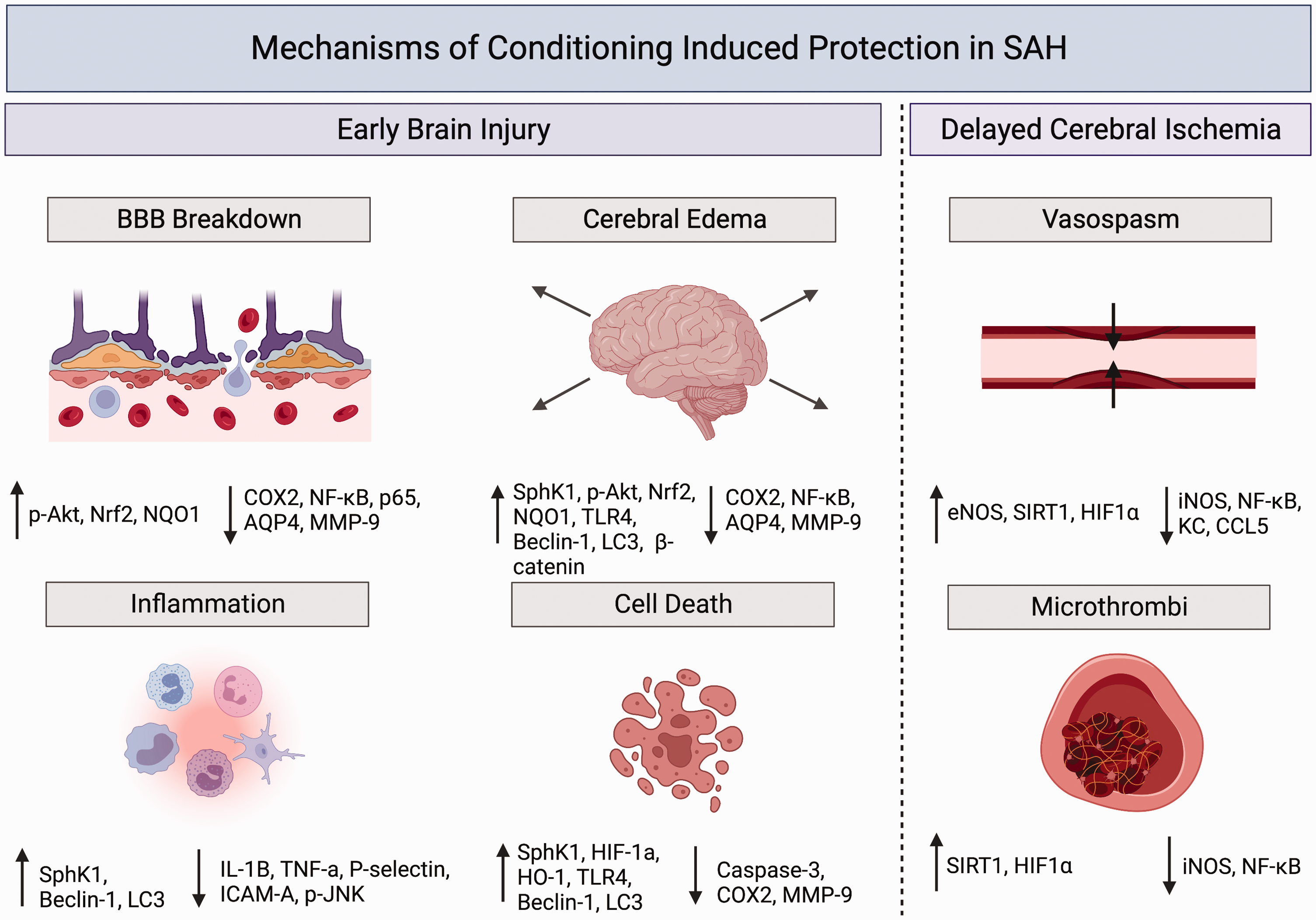
Summary of early brain injury and delayed cerebral ischemia mechanisms altered by cerebral conditioning for subarachnoid hemorrhage. ↑ indicates an increase in markers in response to protective conditioning in SAH. ↓ indicates a decrease in markers in response to protective conditioning in SAH. p-Akt: phospho-protein kinase B; Nrf2: nuclear factor erythroid 2-related factor 2; NQO1: NAD(P)H quinone oxidoreductase 1; COX2: cyclooxygenase-2; NF-kB: Nuclear factor kappa B; AQP4: Aquaporin 4; MMP-9: Matrix metalloproteinase-9; SphK1: Sphingosine kinase 1; TLR4: toll-like receptor 4; LC3: Light chain 3B; IL-1β: Interleukin-1β; TNF-α: Tumor necrosis factor- alpha; ICAM-A: Intercellular Adhesion Molecule-A; p-JNK: p-Jun N-terminal kinase; HIF-1α: Hypoxia inducible factor-alpha; SIRT1: Sirtuin 1; iNOS: inducible nitric oxide synthase; KC: keratinocyte derived chemokine; CCL5: Chemokine (C-C motif) ligand 5.
Conditioning agents and their effects on EBI in SAH.
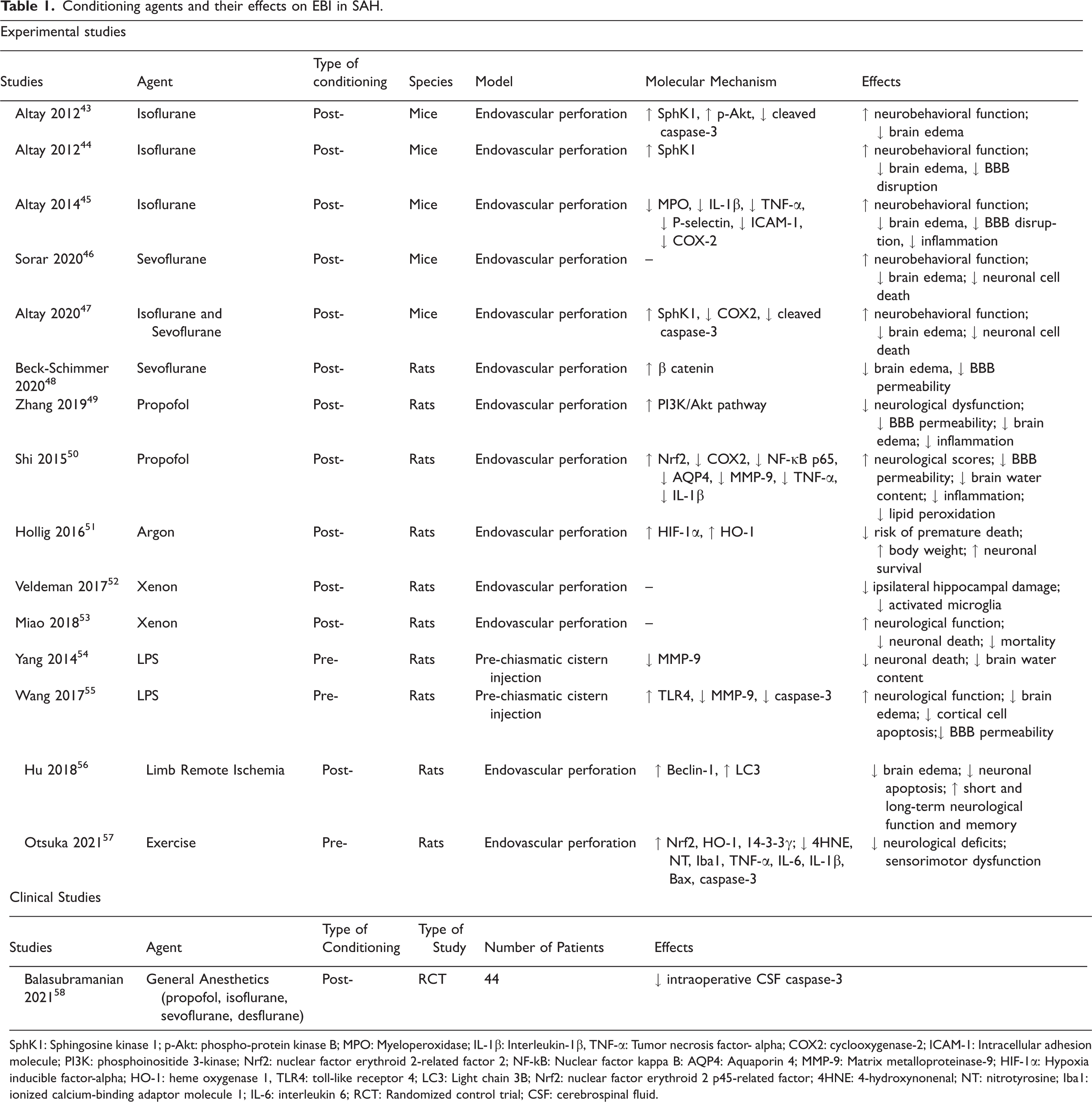
SphK1: Sphingosine kinase 1; p-Akt: phospho-protein kinase B; MPO: Myeloperoxidase; IL-1β: Interleukin-1β, TNF-α: Tumor necrosis factor- alpha; COX2: cyclooxygenase-2; ICAM-1: Intracellular adhesion molecule; PI3K: phosphoinositide 3-kinase; Nrf2: nuclear factor erythroid 2-related factor 2; NF-kB: Nuclear factor kappa B: AQP4: Aquaporin 4; MMP-9: Matrix metalloproteinase-9; HIF-1α: Hypoxia inducible factor-alpha; HO-1: heme oxygenase 1, TLR4: toll-like receptor 4; LC3: Light chain 3B; Nrf2: nuclear factor erythroid 2 p45-related factor; 4HNE: 4-hydroxynonenal; NT: nitrotyrosine; Iba1: ionized calcium-binding adaptor molecule 1; IL-6: interleukin 6; RCT: Randomized control trial; CSF: cerebrospinal fluid.
Conditioning agents and their effects on DCI in SAH.
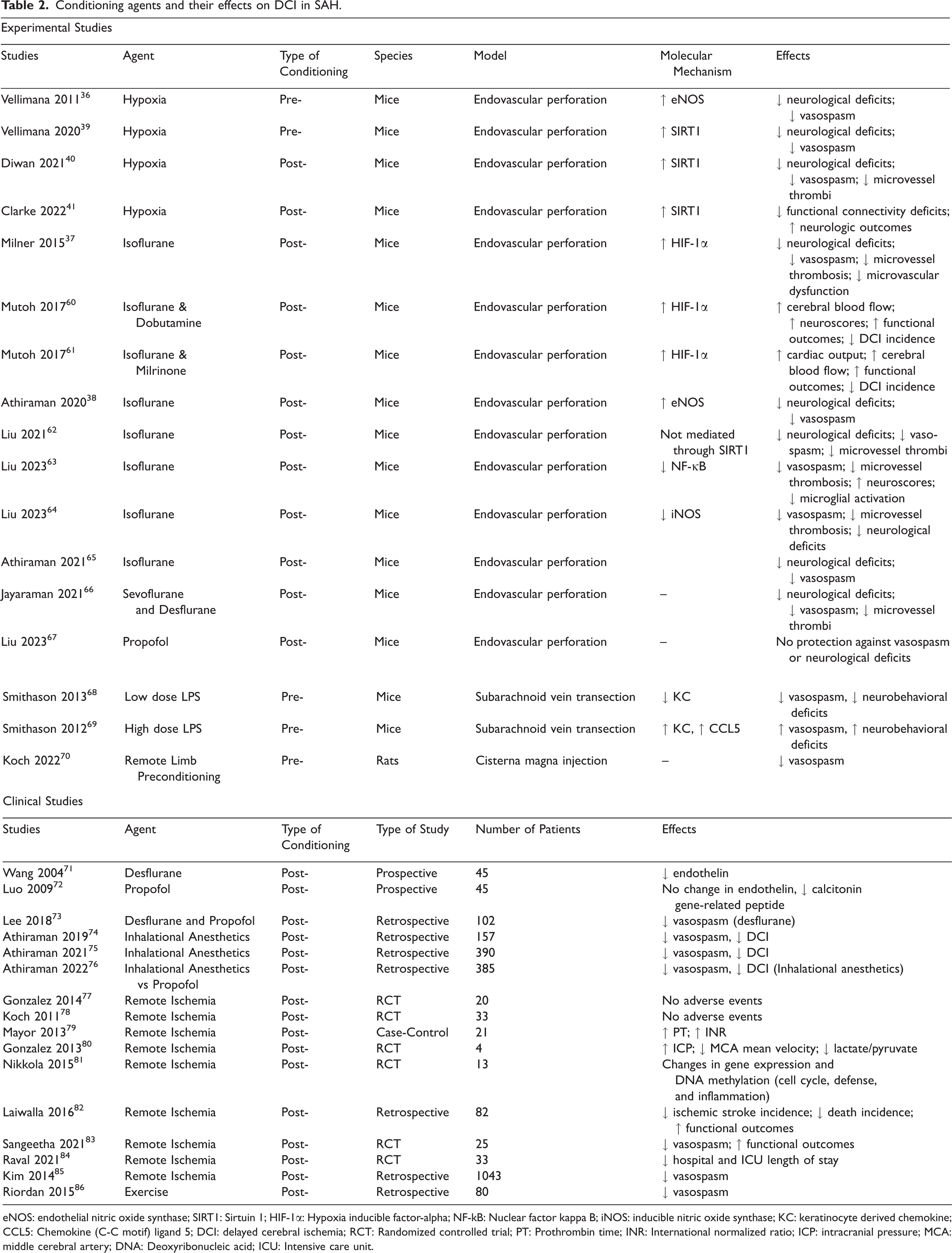
eNOS: endothelial nitric oxide synthase; SIRT1: Sirtuin 1; HIF-1α: Hypoxia inducible factor-alpha; NF-kB: Nuclear factor kappa B; iNOS: inducible nitric oxide synthase; KC: keratinocyte derived chemokine; CCL5: Chemokine (C-C motif) ligand 5; DCI: delayed cerebral ischemia; RCT: Randomized controlled trial; PT: Prothrombin time; INR: International normalized ratio; ICP: intracranial pressure; MCA: middle cerebral artery; DNA: Deoxyribonucleic acid; ICU: Intensive care unit.
Anesthetic conditioning
Numerous preclinical studies from independent laboratories have shown
Hypoxic conditioning
Because application of a hypoxic stimulus to SAH patients is not practical, identifying the molecular pathways underlying hypoxic conditioning-induced neurovascular protection in SAH is essential in order to translate this powerful conditioning strategy to the clinic. As such, our group has performed extensive animal studies elucidating SIRT1 as a critical mediator of hypoxic conditioning induced DCI protection in SAH.39 –41 Currently, studies examining the impact of hypoxic conditioning in EBI and long-term neurologic outcomes are lacking. Future studies casually implicating hypoxic conditioning and SIRT1 in EBI and long-term neurologic protection could provide a momentum to study SIRT1 as a potential therapy to attenuate secondary brain injury and improve long-term patient outcomes after SAH.
Remote limb ischemic conditioning
Recent phase 1 clinical trials demonstrate that remote limb ischemic conditioning (RIC) is a highly feasible, safe, well-tolerated and a potentially effective strategy to reducing DCI and improving neurologic outcome in SAH patients.77,78,83,84 Based on these promising preliminary results in relatively small patient cohorts, larger prospective, randomized, multi-institutional controlled trials are needed to validate the efficacy of RIC in SAH patients and identify optimal RIC protocols to maximize the protective effect in SAH patients. Importantly, there are very limited preclinical studies demonstrating the utility of RIC in animal models of SAH.56,70 Such an experimental model would be crucial for the continued translational success of this conditioning paradigm for several reasons: first, it would demonstrate the feasibility of using a noninvasive intervention like RIC as a treatment capable of affecting multiple endpoints. Second, it would allow fine-tuning of the specific parameters of RIC, both in terms of the window of therapeutic efficacy as well as the duration and number of cycles of limb ischemia. Third, animal studies would afford insight into underlying molecular mechanisms of RIC, results from which could be used to identify novel, more potent therapeutic targets for SAH.
Limitations and future directions
While experimental results to date strongly suggest conditioning strategies for SAH are promising, several limitations exist. 1) The majority of experimental studies have used younger male animals to evaluate SAH outcomes and hence future SAH studies should include female and older animals. 2) Most of the above-mentioned studies have examined the impact of conditioning strategies on either EBI
Conclusion
Conditioning-based therapeutics are a promising novel strategy to reduce SAH-induced brain injury and improve long-term patient outcome. In particular, the diverse neuro- and vasculo-protective epigenetic changes mediated by conditioning appear powerful in combatting SAH-induced neurovascular deficits, as demonstrated in preclinical and limited clinical studies. While work examining the utility of conditioning in SAH is still in its infancy, some clinical efforts are already underway to translate a conditioning-based therapy to SAH patients. Certainly, these early studies have proven promising, and continued translational success will likely benefit from a bench to bedside approach and vice versa.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by a K08 grant (K08NS125038), and Brain aneurysm foundation grant (GR0026849) awarded to Dr. Athiraman, and a R01 grant (NS128082-01A1) awarded to Dr. Zipfel. Figures 1 and 2 were created with BioRender.com.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.