
Abstract
Aneurysmal subarachnoid hemorrhage (aSAH) patients develop delayed cerebral ischemia and delayed deficits (DCI) within 2 weeks of aneurysm rupture at a rate of approximately 30%. DCI is a major contributor to morbidity and mortality after SAH. The cause of DCI is multi-factorial with contributions from microthrombi, blood vessel constriction, inflammation, and cortical spreading depolarizations. Platelets play central roles in hemostasis, inflammation, and vascular function. Within this review, we examine the potential roles of platelets in microthrombi formation, large artery vasospasm, microvessel constriction, inflammation, and cortical spreading depolarization. Evidence from experimental and clinical studies is provided to support the role(s) of platelets in each pathophysiology which contributes to DCI. The review concludes with a suggestion for future therapeutic targets to prevent DCI after aSAH.
Introduction
Aneurysmal subarachnoid hemorrhage (aSAH) has a high initial mortality rate, and approximately 30% of aSAH patients develop delayed cerebral ischemia and neurological deficits (DCI).1,2 Furthermore, roughly half of aSAH survivors will sustain persistent neurological deficits. 1 Not only is DCI a critical contributor to morbidity and mortality during the first few weeks following aSAH, it is also the main cause of long-term poor outcomes in aSAH patients. 1 The progression of DCI usually begins between the 4th and 7th day after aSAH and is characterized by either a new ischemic lesion or neurological decline. 3 It is currently accepted that DCI is multi-factorial and the mechanisms include microthrombi formation, blood vessel constriction, inflammation, and cortical spreading depolarization.1,2,4,5 However, it is not well-known if these processes are independent 6 or if there is a crucial mediator which induces these pathological events. This review will examine the potential role(s) of platelets in each of the pathological events of DCI after aSAH (Figure 1) and discuss future therapeutic targets.
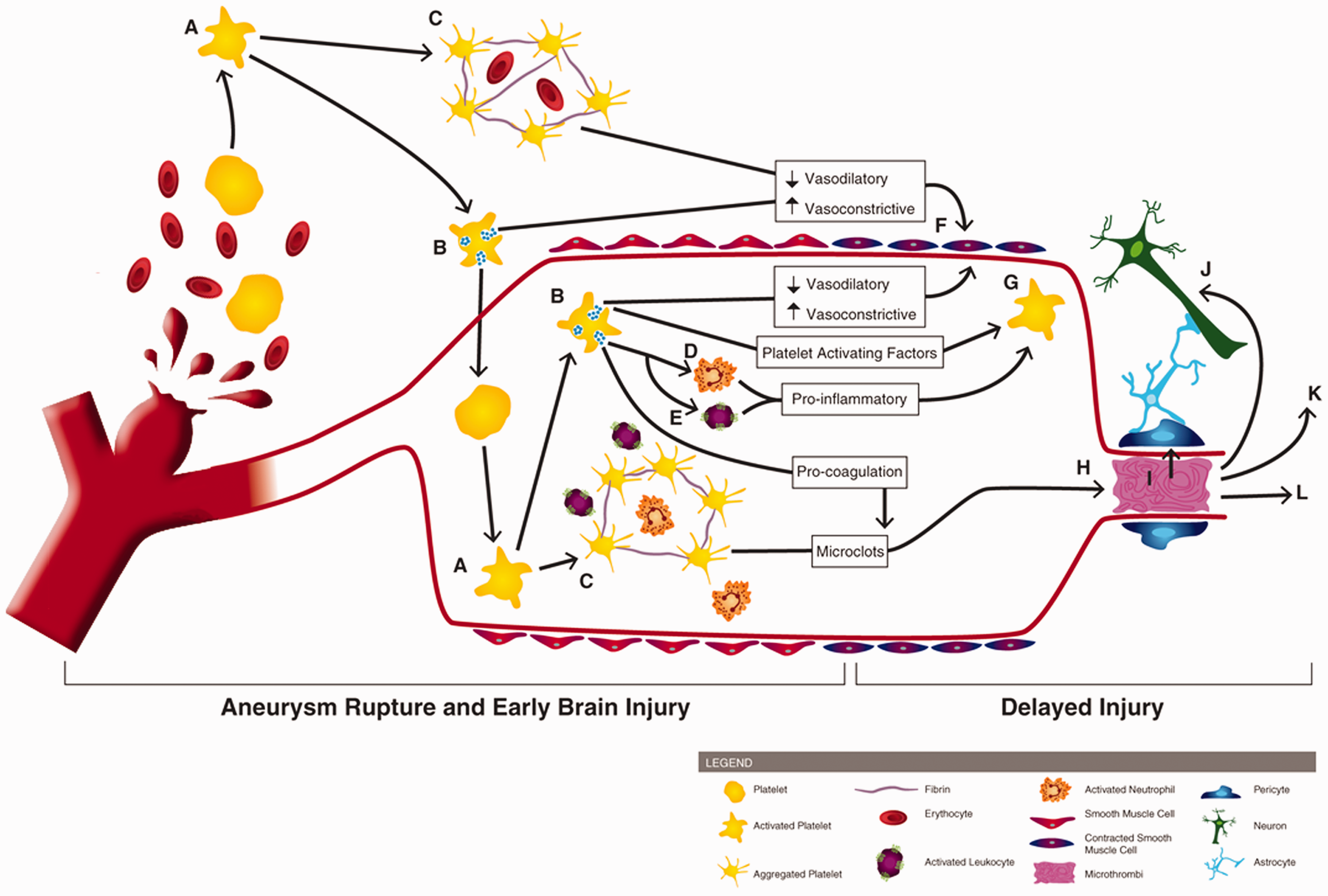
Involvement of platelets in early brain injury and the pathogenesis of delayed cerebral ischemia. Following aneurysm rupture, erythrocytes and platelets are spilled into the subarachnoid space. Upon exposure to the extracellular space (and its matrix proteins, collagen and laminin), platelets become activated (A) Activated platelets degranulate (B) and form aggregates which can contain erythrocytes and immune cells (C). Platelet degranulation can activate intraluminal platelets (thereby propagating platelet activation, degranulation, and aggregation), neutrophils (D) and leukocytes (E). Activated platelets reduce their vasodilation mechanisms and release vasoconstrictive molecules, promoting large artery vasospasm (F). Degranulating platelets, along with activated leukocytes/neutrophils, exacerbate platelet activation (G). Pro-coagulant factors, released by platelets, promotes microclot formation which can deposit within brain microvessels (H). Platelet aggregates and microthrombi in the microvessels can induce pericyte constriction (I), cause neurotoxicity (J), initiate cortical spreading depolarization (K), and occlude blood flow, thereby reducing cerebral blood flow and causing development of delayed infarctions (L).
Platelets are critical mediators of hemostasis, inflammation, and vascular tone. 7 Platelets promote clotting via self-aggregation and interactions with fibrin(ogen) and endothelial cells, participate in inflammation through the release of chemokines/cytokines and by interactions with immune cells, and initiate blood vessel constriction via the release of vasoconstricting factors.
Delayed platelet activation
In the days following aneurysm securement, aSAH patients remain hypercoagulable which contributes to DCI. 8 More specifically, platelet reactivity is increased in aSAH patients who go on to develop DCI compared to aSAH patients who did not develop DCI. 9
Platelets coursing through the vasculature are subject to numerous activation signals released during in response to SAH (Figure 2). In humans, aSAH leads to an increase in serum inflammatory cytokines/chemokines/lipid-mediators (CXCL12, 10 platelet activating factor (PAF) 11 ), von Willebrand factor (vWF),11,12 as well as platelet-derived factors (thrombin,11,13 thromboxane14,15), all of which have roles in platelet activation (Figure 2). Additionally, experimental SAH studies have observed platelet activation via direct interactions with activated leukocytes (via P-selectin and PSGL-1 interactions) 16 and disrupted endothelial cells (by a yet unknown mechanism). 17
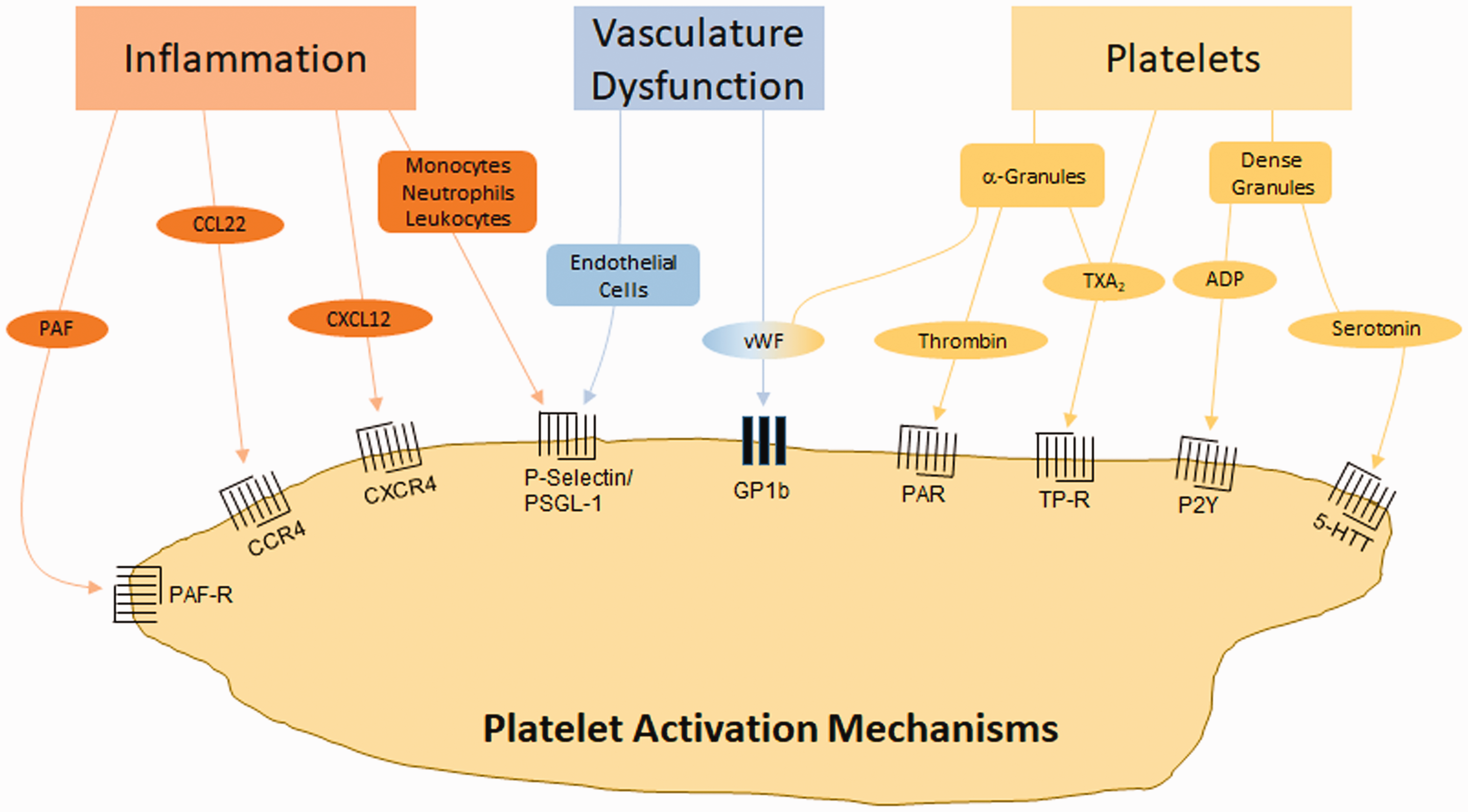
Mechanisms of platelet activation by SAH. Inflammation can induce platelet activation through receptors which respond to inflammatory cytokines (PAF, CCL22, CXCL12) or inflammatory cell binding (monocytes, neutrophils, leukocytes). Vascular dysfunction can induce platelet activation via endothelial cell adherence or through vWF. Platelets are also activated through molecules released from α-granules and dense granules. CCL22 (CC chemokine ligand 22): (ligand of CCR4 (CC chemokine receptor 4)), CXCL12 (CXC motif chemokine ligand 12): (ligand of CXCR4 (CXC motif chemokine receptor 4)). 5-HTT: serotonin receptor; ADP: adenosine diphosphate; GP: glycoprotein; P2Y: purinergic receptor; PAF: platelet activating factor; PAF-R: PAF receptor; PAR: protease activated receptor; PSGL-1: P-selectin glycoprotein ligand-1; TP-R: thromboxane receptor; TXA2: thromboxane A2; vWF: von Willebrand factor.
Upon activation, platelets degranulate, releasing platelet activating factors (thromboxane A2 (TXA2), adenosine diphosphate (ADP), thrombin, vWF, serotonin) and increasing platelet expression of activation and aggregation receptors (glycoprotein (GP) Ib, GPVI, integrins), thus propagating platelet activation. Additionally, activated platelets release extracellular vesicles stimulating coagulation,18,19 inflammation, and activating neighboring platelets (Figure 3), 7 although this has not been explicitly shown for SAH.
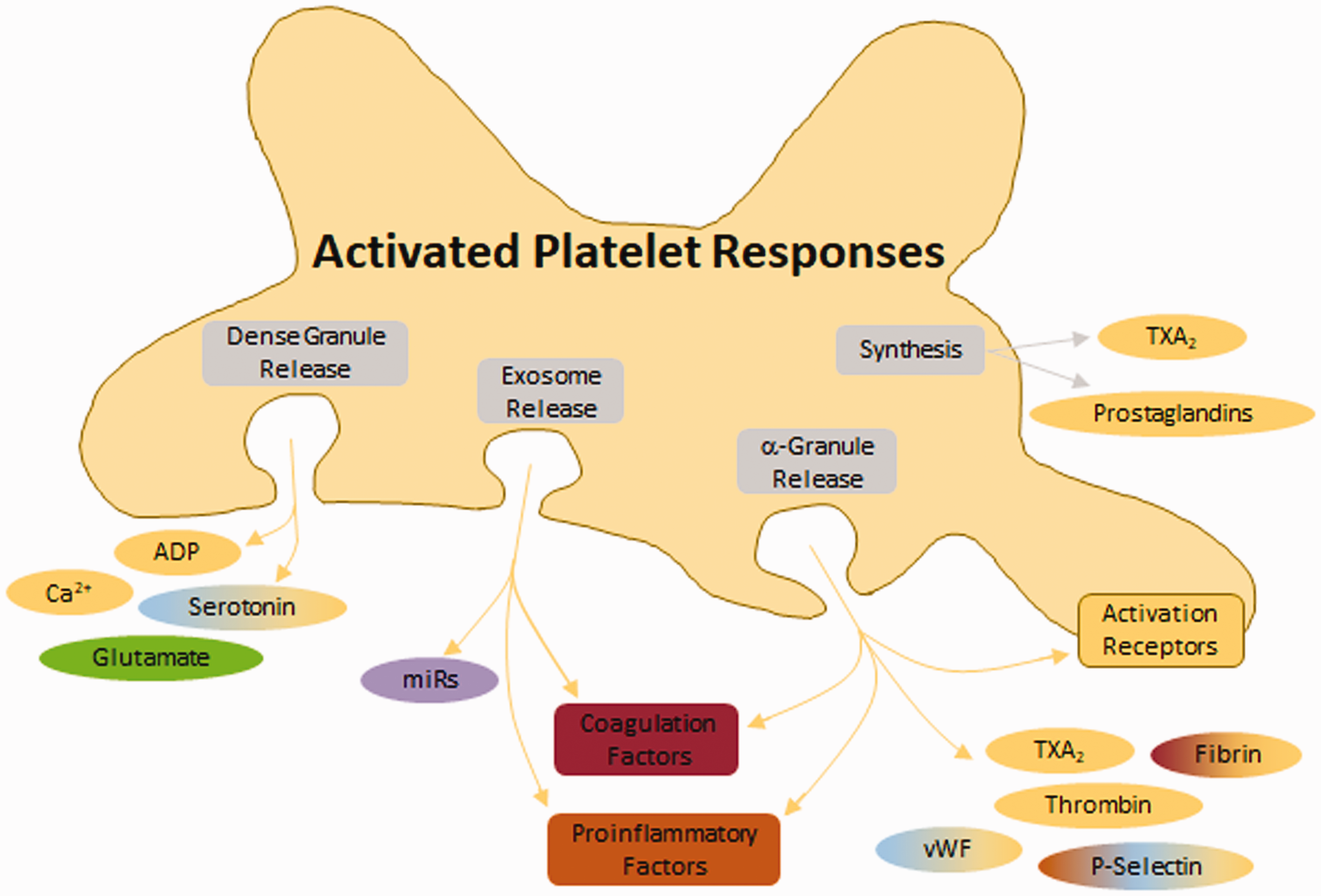
Response of platelets to activation. Following activation, platelets release several molecules from dense granules, exosomes, α-granules, and via synthesis. Dense granules release small molecules. Exosomes release miRs which have functions in coagulation, inflammation, and vasoconstriction. α-granules release proteins which promote coagulation, inflammation, and vasoconstriction. Additionally, α-granules deposit several platelet activation/aggregation receptors on the membrane surface (including glycoprotein (GP)VI, GP1b, P-selectin, platelet-endothelial cell adhesion molecule 1 (PECAM), and integrins).
Platelets cause microthrombi formation
Following SAH, platelets are subject to brief adhesion events to the aggravated vasculature tissue (including in distal microvessels)16,20 which leads to aggregation and microthrombi formation. 20 Deposition of platelet aggregates/microthrombi (which can contain leukocytes) within the microvasculature reduces cerebral blood flow. 20 Consequently, lower blood flow propagates intravascular microthrombosis and capillary obstruction either directly 21 or by reduced platelet inhibition, 22 and prolonged lowering of cerebral blood flow leads to the development of DCI.
In humans, microthrombi have been reported as an associative cause of DCI and worse neurological outcome.23,24 In aSAH autopsy studies, microthrombi (which are diffuse throughout the brain) accompany regions of infarct23,24 and develop with a timing similar to DCI. 25 Analysis of the cerebral microthrombi composition in aSAH patients indicates that the microthrombi consist of aggregated platelets and fibrin, sometimes mixed with leukocytes. 24
In animals subjected to SAH, microthrombi are observed in brain vasculature both locally and distally from the insult,26–32 as well as in both hemispheres,33–38 and microthrombi and occluded blood vessels occur throughout the brain from 2 days 32 to 7 days post-SAH.33,35,37,39,40 Furthermore, after experimental SAH, microthrombi count positively correlates with the number of apoptotic neurons, 32 infarction volume, 34 and delayed deficits. 34
A potential mechanism which platelets lead to microthrombosis is via P-selectin and nitric oxide (NO). Sabri et al. reported that mice with SAH had increased expression of membrane P-selectin on endothelial cells and that microthrombi co-localized with P-selectin staining. 32 Additionally, it was reported that P-selectin negatively correlates with brain NO. 32 As NO is a potent inhibitor of platelet activation, and P-selectin is involved in platelet-cell interactions, the authors concluded that the increased P-selectin and decreased NO may drive platelets towards microthrombosis. 32
Platelets induce vessel constriction
Platelets/microthrombi are found near arteries experiencing vasospasm.41–43 Upon adhering to the intraluminal surface of blood vessel endothelial cells, platelets rapidly change the local environment by releasing vasoconstricting agents (i.e. thromboxane A2 (TXA2),44–46 prostaglandin endoperoxides (e.g. PGG2, PGH2),47,48 platelet-derived growth factor (PDGF)-β,49,50 and serotonin51,52).
Large artery vasospasm
In aSAH patients with large artery vasospasm, platelets are hyper-active and have higher aggregability than in patients not experiencing vasospasm.53,54 Experimentally, platelets are reported to cause prolonged cerebral vasospasm,52,55–58 and several studies have even suggested that platelet activation precedes vasospasm after aSAH.9,49,50,53,54,59,60 The mechanism by which platelets induce large artery vasospasm may be via elevated levels of the vasoconstricting factors, TXA2 and PDGF-β.
A number of clinical studies have highlighted TXA2 as being involved in the development of vasospasm and pathogenesis of DCI.53,54,61–63 In a rabbit model of SAH, platelets were shown to cause contraction of the basilar artery which was effectively prevented when platelets were pre-incubated with a thromboxane synthetase inhibitor. 60 The study by Tanaka et al. provides evidence that platelets are a cause of large artery vasospasm. 60
PDGF-β has also been reported to positively correlate with incidence and severity of vasospasm after aSAH,49,50 and in animals models.55,58 Experimentally, vasospasm increases as plasma levels of PDGF-β rise 55 and use of a PDGF antagonist can prevent vasospasm. 58 However, the evidence for PDGF-β being a possible mechanism is correlational and does not provide insight into causality. Furthermore, the source of PDGF-β was not reported.
Three additional preclinical studies which further support platelets as a major contributor to vasospasm following SAH were those of Pisapia et al. and Sonobe and Suzuki. Pisapia et al. utilized a vasodilating drug and a microthrombi antagonist after SAH in mice and found that the vasodilating drug had no effect on microthrombi count. 35 Unfortunately the study did not report on vasospasm, so it is unclear if the microthrombi antagonist had any effect on vasospasm. However, this study highlights two important ideas: (1) large artery vasospasm does not cause microthrombi, and (2) the events (microthrombi and vasospasm) may either be distinct pathophysiological mechanisms or platelets have a causal relationship with vasospasm. Direct evidence of platelets causing vasospasm after SAH was provided by Sonobe and Suzuki; the authors applied blood directly to feline basilar arteries and concluded that the observed vasospasm was due to platelet-derived serotonin 57 which has been supported by another study. 52
Taken together, these clinical and experimental models argue that platelets play a significant role in the cause of large artery vasospasm. While the mechanism remains unknown, platelets may be a therapeutic target which can attenuate the burden of large artery vasospasm after aSAH.
Microvessel constriction
Direct observation of pial arterioles after SAH in mice led to a very prominent correlation between platelet aggregates and microvessel constriction,27,32,64 which lead to cerebral blood flow deficits. 64 Studies have shown that microvessel constriction and formation of platelet aggregates/clots began within seconds after SAH 27 and that platelet aggregates were observed within the most constricted microvessels, 27 leading to a striking correlation between platelet aggregates/clots and microvessel constriction. 27 In the study by Friedrich et al., it was concluded that the microvessels containing platelet aggregates have perfusion deficits, and that these platelet aggregates may initiate or propagate local microvessel constriction. 64 In a follow-up study, the supplemental video shows that after the microthrombus forms the microvessel constricts even further, as well as a new constriction beginning near the microthrombi. 27 Additional imaging studies are needed since antagonists for either platelets or vasospasm were not administer, so it is not possible to determine a causal relationship.
Two studies allude to mechanisms by which platelets are linked to microvessel constriction, however causality cannot be determined. First, Sabri et al. reported that decreased NO and increased P-selectin on arteriole endothelium may be a mechanism for microthrombosis and microvessel constriction. 32 Of note, Sabri et al. observed microthrombi within microvessels experiencing constriction. Increased P-selectin on the endothelium promotes platelet binding and subsequent thrombi formation, whereas decreased NO will not be able to inhibit platelets. In the second study, Li et al. observed an elevated expression of the PDGF-β receptor on capillary pericytes. 65 Although PDGF-β levels were not measured by Li et al., since others report that PDGF-β is elevated following SAH and that PDGF-β positively correlates with DCI,49,50 this may be a mechanism of platelet-induced microvessel constriction. The role of PDGF-β in platelet-mediated microvessel constriction is a yet unexplored area.
Although the mechanisms by which platelets cause microvessel constrictions remains unknown, there is evidence that platelets play a role,6,13 possibly through release of vasoactive factors, 23 or P-selectin binding. 32 Additional studies are needed before a causal relationship can be confirmed.
Platelets promote inflammation
The inflammatory role of platelets is well-documented; platelets can activate or propagate inflammation via release of pro-inflammatory factors and through direct interactions with leukocytes and neutrophils. 7
After SAH, although a causal relationship cannot be determined, both platelets and inflammation are associated with worse early brain injury, DCI, and poor functional outcomes.66–69 The work of Frontera et al. in aSAH patients found that brain injury severity correlated with more platelet activation and inflammation (e.g. C-reactive peptide), and that both platelet activation and C-reactive peptide were associated with worse 3-month functional outcomes.66,67 Since platelet reactivity and C-reactive peptide increased concomitantly, a causal relationship could not be identified.66,67 However, in aSAH studies by Ray et al., platelet activation (e.g. coated platelets) correlated with DCI,68,69 and an increase in platelet activation preceded an increase in the number of circulating neutrophils. 68 Ray et al. only reported one inflammatory cell measurement (i.e. inflammatory cytokines were not measured), so it is not possible to say definitively that platelet activation preceded inflammation after SAH. However, the evidence is clear that platelet activation correlated with a delayed increase in the number of neutrophils. More studies are needed to attempt to unveil a causal relationship between inflammation and platelets following SAH.
Using intravital microscopy, Ishikawa et al. observed significant platelet-leukocyte interactions after SAH in mice within 24 h after injury. 16 Use of a platelet antagonist (P-selectin antibody) prevented platelet-leukocyte interactions, as well as reduced leukocyte adherence to endothelium, suggesting that platelets can induce leukocyte activation after SAH. Although general inflammation (such as cytokine levels) was not assessed, the finding that a platelet antagonist can reduce leukocyte activation provides some evidence of a causal relationship.
Can platelets cause cortical spreading depolarization/ischemia?
Cortical spreading depolarizations/ischemia have been correlated with DCI. 70 Evidence is limited on platelets directly causing cortical spreading depolarization/ischemia. However, microthrombi, vasospasm, and microvessel constriction each promote cortical spreading depolarization/ischemia. 71
A mechanism by which platelets induce cortical spreading depolarization/ischemia may be via glutamate. Glutamate levels are high after aSAH in patients, 72 and excessive amounts of glutamate cause excitotoxicity, spreading depolarizations, and neuronal death. 73 Interestingly, platelets induce glutamate-mediated neuronal toxicity and death. Bell et al. showed that activated platelets are very potent sources of glutamate (which can exceed 300 µm in vitro), and the concentration of glutamate released by activated platelets is neurotoxic in cell culture and after SAH in rats. 29 Further, seven days post-SAH, there was a strong correlation between the proximity of microthrombi and the reduction of glutamate receptors on neurons, suggesting platelets can induce neuronal dysfunction and death. Although cortical spreading depolarizations/ischemia were not examined, this study warrants investigation into platelets as an initiator of cortical spreading depolarizations/ischemia after SAH.
Platelets as a therapeutic target to prevent DCI
Within the above sections, platelets were shown to have potential roles in the pathological events which contribute to DCI after SAH. Thus, platelets may be a therapeutic target to prevent DCI. One reservation of using anti-platelet therapies in aSAH patients is the potential of new bleeds (i.e. rebleeding or microbleeds). However, recent aSAH clinical trials have shown that the risk of new bleeds for anti-platelet drugs is minimal. 74 Of course, caution in patients with intraventricular drains or those in need of surgical intervention is the rule,75,76 it should be noted that there exists a patient population which receives anti-platelet drugs following coil embolization with minimal complications.77–80 So why are anti-platelets not more widely used for aSAH patients? Despite their safety in use, trials of anti-platelet agents have not shown a clear clinical benefit. Two reasons may account for this: (1) only a portion of patients with SAH will develop DCI and so far we are unable to predict this cohort and (2) we have limited understanding of the mechanism for DCI including the most potent targets, as well as the timing and duration of pharmacologic treatment. Another possible reason anti-platelets are not widely used or studied for aSAH is that there are many different targets on platelets (i.e. inhibiting activation, inhibiting aggregation, inhibiting interactions with other cells) and no one knows which target to choose.
To date, the use of anti-platelet therapies have had marginal success in clinical trials. In the 1970s, a clinical study investigated the potential of serotonin antagonists for treating aSAH patients. The study had small sample sizes (32 treated patients, 99 untreated patients), but the results suggested that serotonin antagonists may be useful in preventing delayed vasospasm, temporary deficits, and permanent deficits. 81 Despite promising findings, serotonin antagonists were not investigated in large, randomized clinical trials.
Trials using classical anti-platelet therapies (inhibitors of cyclooxygenase (acetylsalicylic acid),82–84 thromboxane synthetase,85,86 phosphodiesterase 3, 87 and P2Y12 88 ) had a tendency towards less secondary cerebral ischemia and better outcome, however the studies were generally under-powered. 89 Only one study examining ticlopidine observed significantly improved outcome for aSAH patients treated with an anti-platelet. 88 A meta-analysis of these 7 anti-platelet trials indicated that there was no conclusive evidence that single anti-platelet therapy was beneficial for preventing DCI after aSAH. 89 More recent clinical trials have focused on investigating the potential for dual anti-platelet therapy in SAH. The data from studies using dual anti-platelet therapy have been promising for reducing DCI. 90 Below we discuss other potential targets for therapeutic design.
Alternative anti-platelet targets
Platelet activation receptors
Several platelet activation receptors (protease activated receptors (PAR), thromboxane receptors, purinergic receptors (P2Y)) have been tested as therapeutic targets for aSAH.74,78,91–95 Two activation receptors which may be promising are the GPIb-IX-V complex and GPVI. The GPIb-IX-V complex interacts with vWF and thrombin, and is crucial in platelet adhesion to endothelial cells and stabilizing microclots. GPVI interacts with fibrinogen and is important in platelet-platelet interaction/aggregation. To date, neither of these platelet activation receptors have been tested for experimental nor clinical SAH.
Endogenous platelet inhibition
Platelets have endogenous inhibitory mechanisms, so augmenting these mechanisms may have therapeutic potential. Prostacyclin (acts via prostaglandin I2 receptor) and NO, which are platelet inhibitors and vasodilators, 7 were promising in recent clinical studies.96–98 In a small study with 90 aSAH patients, prostacyclin lowered the incidence of DCI (21%–23% for prostacyclin treatment vs 38% for placebo) although this was not statistically significant since the study was not powered to investigate a difference in DCI incidence. 96 Two studies using a NO donor, molsidomine, observed a significantly lower incidence of DCI (13.8% molsidomine plus nimodipine vs 48% nimodipine only, p < 0.01), improved 3 month outcome, and lower mortality in aSAH patients receiving the NO donor plus nimodipine.97,98 Large, randomized trials should be considered to determine if either of these agents can prevent DCI and improve outcome after aSAH.
Another endogenous inhibitory mechanism, glucagon-like peptide 1, was tested in a preclinical SAH study. The experimental study investigated the therapeutic benefit of glucagon-like peptide 1 on early brain injury after SAH. 99 The results suggested that glucagon-like peptide 1 is promising for early brain injury, but its benefit in the delayed phase is not known.
Platelet aggregation
Although there has not yet been a large, randomized clinical trial to prevent platelet aggregation, a number of small clinical studies and case reports have shown that antagonizing platelet aggregation via GP IIb/IIIa may be therapeutically beneficial.100–109 Overall, the conclusions from these studies indicate that GP IIb/IIIa antagonists are safe and effective, with minimal risk of hemorrhagic complications. Large, randomized trials need to be performed to determine if GP IIb/IIIa antagonists can reduce DCI and improve outcome after SAH.
Other targets
Targeting either P-selectin or its receptor P-selectin glycoprotein ligand 1 would limit platelet adherence to leukocytes and endothelial cells. As demonstrated in experimental SAH models, P-selectin has some therapeutic potential in preventing microthrombi, 32 platelet-endothelial binding, 32 and platelet-leukocyte interactions. 16 Additional experimental studies would need to investigate the efficacy of P-selectin or its receptor in the context of delayed injury after experimental SAH prior to thinking about clinical translation. vWF is free in the blood, released by activated platelets and endothelial cells, and expressed on endothelial cells.7,110 Upon activation and release of the Weibel-Palade bodies, endothelial cells rapidly increase free vWF (promoting platelet-platelet binding) and their surface vWF (promoting platelet adhesion). Thus, targeting either vWF or its receptors are potential therapeutic targets. Support for targeting vWF comes from experimental SAH studies which found that ADAMTS13 (which cleaves multimers of vWF) effectively reduces the thrombotic potential of vWF. Additionally, ADAMTS13 is a thrombolytic. 111 Several experimental SAH studies have reported that recombinant ADAMTS13 reduces microthrombi,33,112–114 improves short-term functional outcome, 33 attenuates delayed microthrombi, 112 and reduces delayed neuronal death/ischemia. 112 Additionally, in mice, ADAMTS13 treatment did not increase the amount of subarachnoid hematoma. 112 Thus, the dual function of ADAMTS13 makes it an attractive option as a therapy for SAH and it may be time to investigate ADAMTS13 in small clinical studies.
Summary
Platelets are a key player in the pathogenesis of DCI following SAH since platelets are major contributors to microthrombosis, vasospasm, microvessel constriction, and inflammation after SAH, and may also be important initiators of cortical spreading depolarization/ischemia and neurotoxcitiy. Although early clinical trials on anti-platelet therapies were inconclusive, newer anti-platelets and alternative targets on platelets should be pursued and tested for preventing DCI after SAH. At this time, GP IIb/IIIa antagonists, prostacyclin, and NO are ready for large, randomized clinical trials, while ADAMTS13 should be considered for small clinical studies.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Funding support was provided by the Brain Aneurysm Foundation (DWM) and NIH (SLB).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.