
Abstract
Intracerebral hemorrhage (ICH) mobilizes circulating leukocytes that contribute to neuroinflammation and neural injury. However, little is known about the endogenous regulatory immune mechanisms to restrict neuroinflammation following ICH. We examined the role of group 2 innate lymphoid cells (ILC2) that are a specialized subset of innate immune modulators in a mouse model of ICH. We found accumulation of ILC2 in the brain following acute ICH and a concomitant increase of ILC2 within the peripheral lymph nodes. Depletion of ILC2 exacerbated neurodeficits and brain edema after ICH in male and female mice. This aggravated ICH injury was accompanied by augmented microglia activity and leukocyte infiltration. In contrast, expansion of ILC2 using IL-33 led to reduced ICH injury, microglia activity and leukocyte infiltration. Notably, elimination of microglia using a colony stimulating factor 1 receptor inhibitor diminished the exacerbation of ICH injury induced by depletion of ILC2. Brain-infiltrating ILC2 had upregulation of IL-13 after ICH. Results from in vitro assays revealed that ILC2 suppressed thrombin-induced inflammatory activity in microglia-like BV2 cells. Thus, our findings demonstrate that ILC2 suppress neuroinflammation and acute ICH injury.
Introduction
Intracerebral hemorrhage (ICH) is a devastating type of stroke with high morbidity and mortality, and afflicts approximately 2 million people worldwide annually. 1 The damage from ICH includes not only the primary tissue injury that is due to the mass effects of the hematoma but also the development of perihematomal edema (PHE), which induces a severe secondary injury and the destruction of adjacent tissues, in addition to the impairment of blood-brain-barrier (BBB). While clinically critical, PHE in ICH patients remains mechanistically ill-defined and therapeutically non-targetable with available medications.
Inflammation has emerged as a critical contributor to PHE formation and subsequent neurological deterioration. Recent evidence has demonstrated that leukocyte infiltration into the brain are critical contributors to BBB disruption and PHE formation after ICH.2 –4 Among brain-infiltrating leukocytes, cells from the arm of innate immunity such as neutrophils, monocytes and natural killer cells arrive the ICH brain as early as 6 h after onset.5 –9 Subsequently, brain infiltration of multiple lymphocyte subsets such as CD4+ T, CD8+ T and B cells can be observed within 24 h after ICH, and peaked around day 3 post-ICH.10,11 While previous studies were largely focused on the proinflammatory leukocytes in acute ICH,8,12,13 a few studies reported a beneficial role of regulatory T cells to confer protection during later stage of ICH. 14 Given that the majority of PHE formation occurs within 24 h after ICH, well before the activation of antigen-specific T cells, it is therefore challenging to understand how adaptive lymphocytes participate in acute ICH injury, before becoming primed with brain antigens, a process taking weeks to months in the setting of neuroimmune disorders. It is also not clear how regulatory T cells provide protection in later stages of ICH.
Group 2 innate lymphoid cells (ILC2) are a population of innate lymphoid effector cells of type 2 immunity. Different from T or B lymphocytes, ILC2 can sense alarmins such as IL-33 released from damaged tissues and rapidly become activated to fine tune the inflammatory response because they do not need classic antigen presentation for activation. 15 As a critical innate source of type 2 effector cytokines, ILC2 produce cytokines such as IL-13 to control the initiation and resolution of tissue inflammation.16 –18 Given the prompt nature of the ILC2 in response to tissue injury without need for antigen presentation and the rapid evolution of PHE, we postulated that ILC2 would play a major role in control of neuroinflammation following ICH.
Materials and methods
Animals
All animal procedures conformed to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were approved by Animal Care and Use Committees at the Tianjin Neurological Institute (protocol approval number: 2022-DWFL-148). Male and female C57BL/6 mice (8 to 10 weeks old, 20–25 g) were purchased from the Beijing Vital River Laboratory Animal Technology Co., Ltd. A total of 180 mice of both sexes were used in the study. Mice were randomly assigned to each experiment. All mice were housed in pathogen-free conditions at the animal facilities at Tianjin Neurological Institute under a standardized light–dark cycle and had free access to food and water. Animal data reporting followed the ARRIVE 2.0 guidelines. 19
ICH model
ICH was induced by intracerebral injection of collagenase as we previously described. 20 After anesthesia, mice were placed in a stereotactic frame and a 1 mm hole in diameter was drilled on the right side of skull (2.3 mm lateral to midline, 0.5 mm anterior to bregma). We injected mice using an infusion pump (Kd Scientific Inc., Holliston, MA) in the right striatum with 0.03 U bacterial collagenase (Type IV-S, Sigma, St. Louis, MO) in 0.5 ml saline at a rate of 0.5 µL/min at a depth of 3.5 mm beneath the skull. After injection completion, the incision was closed with sutures following surgery. Throughout the procedure, animal body temperature was maintained at 37 °C with a homeothermic blanket. After surgery, animals remained under observation with free access to food and water.
Neurological assessment
Neurological assessment was performed at day 1 and 3 after ICH by two investigators masked to the treatment assignment. The modified Neurological Severity Score (mNSS) and corner turn test were conducted to evaluate neurological deficits mice. In the neurological deficit scoring system, mice were evaluated for motor function (muscle and abnormal movement), sensory function (visual, tactile, and proprioceptive), and reflexes (pinna, corneal, and startle). The range of scores is from 0 to 18, defined as follows: a score of 13–18 indicates severe injury, 7–12 indicates moderate injury, and 1–6 indicates mild injury. The corner-turning test assesses sensorimotor and postural asymmetries. In this process, each mouse is allowed to proceed into a corner with an angle of 30° and then must turn right or left. Each mouse repeated this procedure 10 times with an interval of ≥30 s between trials. The percentage of ipsilateral turns was then calculated.
Brain water content
Brain water contents were measured at day 3 after ICH. Briefly, without perfusion, brains were removed from mice the wet weight of contralateral hemisphere, ipsilateral hemisphere, and cerebellum was weighted separately. The separated brain tissue parts were dried for 24 hours at 100 °C, after which the dry weight of each part was weighed again. The following formula was used to calculate brain water content: (wet weight − dry weight)/wet weight × 100%.
Drug administration
Anti-CD90.2mAb (105302, BioLegend) or anti-CD4 mAb (100402, BioLegend) was injected intraperitoneally at a dose of 300 μg every two days as previously described.21,22 IL-33 (HY-P70460, MedChemExpress) was injected intraperitoneally 600 ng/2 days as previously described [19, 20]. Rat IgG2b k (14-4031-82, eBioscience) was used as the control. PLX5622 was formulated by SYSE Bio (D20010801). C57BL/6J mice were fed with PLX5622-formulated AIN-76A diet for 2 weeks to fully ablate CNS-resident microglia. One adult mouse (25 g) consumes about 3.5 g chow diet per day.
Assessment of hematoma volume
At day 3 after ICH, brains were removed and perfusion-fixed in 4% paraformaldehyde overnight at 4 °C. The brains were then sliced transversely around the hematoma at a thickness of 1 mm and placed onto slides. After photographing these slices, hematoma volumes were calculated by using Image Pro Plus software (Media Cybernetics, USA).
Flow cytometry
Mouse brains were removed after perfusion with cold PBS and mechanically cut into small pieces using sharp scissors. 1 ml of papain solution (LK003176, Worthington Biochemical Corporation) at a concentration of 0.125 U/ml was used to digest the brain tissue at 37 °C for 30 min. After washing with PBS, cell pellets were collected by centrifugation at 2000 rpm for 5 min, then resuspended in 30% percoll (17089101, Cytiva) which myelin debris were removed by centrifuging at 700 g for 10 min. Cell pellets were then washed by PBS and resuspended in 1% bovine serum albumin (BSA), counted and for antibody staining.
To obtain lymph nodes cells, lymph nodes were grinded through a 70 μm cell strainer. After washing with PBS, cells were resuspended in 1% BSA, counted and for staining analysis. For spleen, tissues were grinded through a 70 μm cell strainer. After washing with PBS, Red Blood Cell Lysis Buffer (R1010, Solarbio) was then added to remove red blood cells. Followed by washing with PBS, cells were then resuspended in 1% BSA. Fluorescence-conjugated antibodies were then added to cell suspensions and stained for 30 min on ice. The following antibodies were used: CD3 (17A2, 100235, Biolegend), CD45 (30-F11, 103131, Biolegend), CD90.2 (30-H12, 105312, Biolegend), IL7R (135040, Biolegend), GATA3 (L50-823, 563349, BD), CD11b (M1/70, 101216, Biolegend), Ly-6G (1A8, 127607, Biolegend), Ly6C (HK1.4, 128015, Biolegend), F4/80 (BM8, 123115, Biolegend), CD4 (GK1.5, 100408, Biolegend), CD19 (6D5, 115511, Biolegend), NK1.1 (PK136, 108706, Biolegend), CD8a (53-6.7, 100738, Biolegend), ST2 (DIH9, 145303, Biolegend), Lineage markers include CD3e (1995367, eBioscience), CD45R (B220, 2247476, eBioscience), CD11c (1937214, Biolegend), Ter119 (2342104, eBioscience), Ly6G/Ly6C (2116628, eBioscience), NK1.1 (108704, Biolegend), CD4 (100404, Biolegend), CD5 (100604, Biolegend), CD8a (100704, Biolegend), TCR-β (109204, Biolegend), TCR-γ/δ (118103, Biolegend), IL-13 (eBio13A, 47-7133-82, eBioscience). For intracellular staining, cells were fixed in and permeabilized with Fixation and Permeablization Solution (00-5523, Invitrogen) for 20 min after surface marker staining, and then washed in permeabilization buffer (00-8333, Invitrogen). Antibodies targeting intracellular molecules were then added and stained for 45 min. Cells were then washed and suspended in flow Flow cytometric data were acquired on a FACSAria flow cytometer (BD Biosciences, San Jose, CA) and analyzed with Flow Jo software version 10.8.1 (Informer Technologies).
Cell culture
BV-2 cells, an immortalized mouse microglia line, from the Department of Neurology, Tianjin Neurological Institute, were cultured in Dulbecco's Modified Eagle Medium (DMEM, 10566016, Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (FBS, 2053264, Biological Industries), 1% Penicillin-Streptomycin-Gentamicin Solution (P1410, Solarbio). Cells were seeded on 6-well plates (1 × 105 cells/mL/well) and maintained in 95% humidified air and 5% CO2 at 37 °C for 2 days. Then, cells were administrated with thrombin (T8021-1000U, Solarbio) or IL-13 (HY-P70460 MedChemExpress) for 24 h.
Quantitative real-time PCR (qRT-PCR)
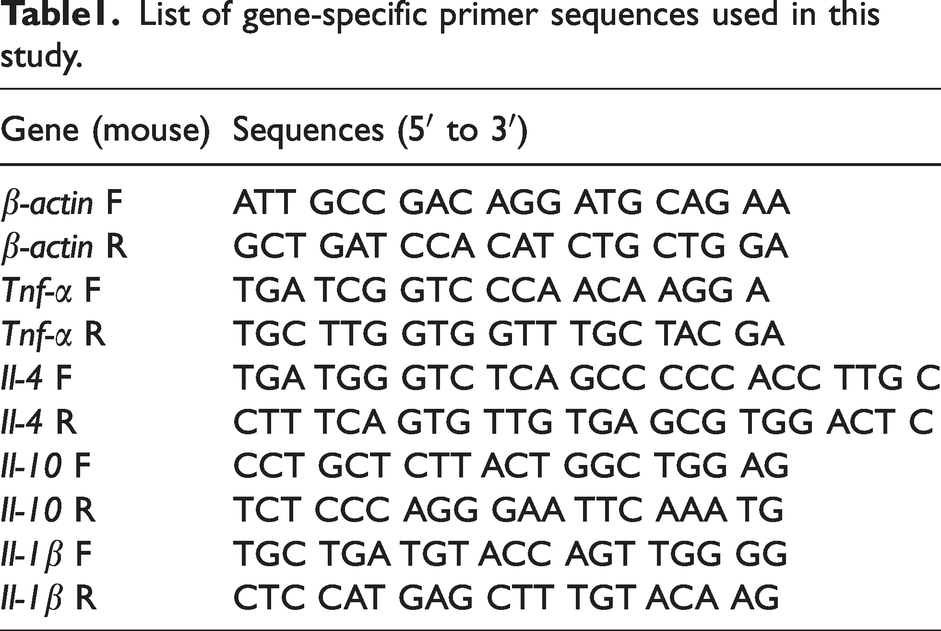
The isolation of total RNA from samples was performed by TransZol Up (ET111, TransGen Biotech) strictly in line with manufacture’s specification. Subsequently, 2 μg of RNA was synthesized to cDNA using the One-Step gDNA Removal and cDNA Synthesis SuperMix (AT311, TransGen Biotech). The operation of Green qPCR SuperMix (AQ601, TransGen Biotech) quantitative PCR of Il-1β, Tnf-α, Il-4 and Il-10 was performed with the adoption of Applied Bio systems 7500 Fast Real-Time PCR System. The mRNA level of Il-1β, Tnf-α, Il-4 and Il-10 was calculated by 2−ΔΔCt and normalized to β-actin. The gene list and primers we present in flowing Table 1.
List of gene-specific primer sequences used in this study.
Statistical analysis
Sample size was determined using Gpower3.1 software at a power of 0.8 and to a significance level of 0.05. The experimental design was based on our previous publications with similar mechanistic studies completed in our laboratory.23 –25 Randomization was based on the random number generator function in Microsoft Excel. Normal distribution was assessed using D'Agostino-Pearson normality test. Statistical analyses were performed by using GraphPad Prism9 software. A two tailed unpaired Student t test was used to compare the differences between two groups. One-way ANOVA followed by Tukey post hoc test or 2-way ANOVA followed by Bonferroni post hoc test were used for three or more groups. Data are expressed as mean ± SD. P < 0.05 was considered significant.
Results
Accumulation of ILC2 in the brain and peripheral lymph nodes in ICH mice
Using a mouse model of ICH induced by intracerebral injection of collagenase, we determined the dynamics of ILC2 counts in the brain lymph nodes and blood. Mouse ILC2 were identified as Lin−CD45highCD90.2+ ST2+ cells, in which Lin = CD3e, CD45R, CD11b, Ter119, Ly-6G, CD11c, NK1.1, CD4, CD5, CD8a, TCR-β, TCR-γδ. Flow cytometry analysis revealed that the counts of ILC2 were dramatically increased in the brain at day 1 and day 3 after ICH (Figure 1(a) and (b)). Similarly, we also found an increase of ILC2 in the mesenteric lymph nodes in the periphery (Figure 1(c)). As compared to sham mice, the counts of ILC2 were decreased at day 1 and increased at day 3 in blood after ICH (Figure 1(d)). In addition, CD4+ T, CD8+ T and B cells were reduced in blood at day 1 and day 3 after ICH (Supplementary Figure 1A). Immunostaining revealed that the number of ILC2 within perihematomal regions was increased at day 1 after ICH (Supplementary Figure 1B). These results suggest that ILC2 can be mobilized and transmigrate into the injured brain during early stage of ICH.
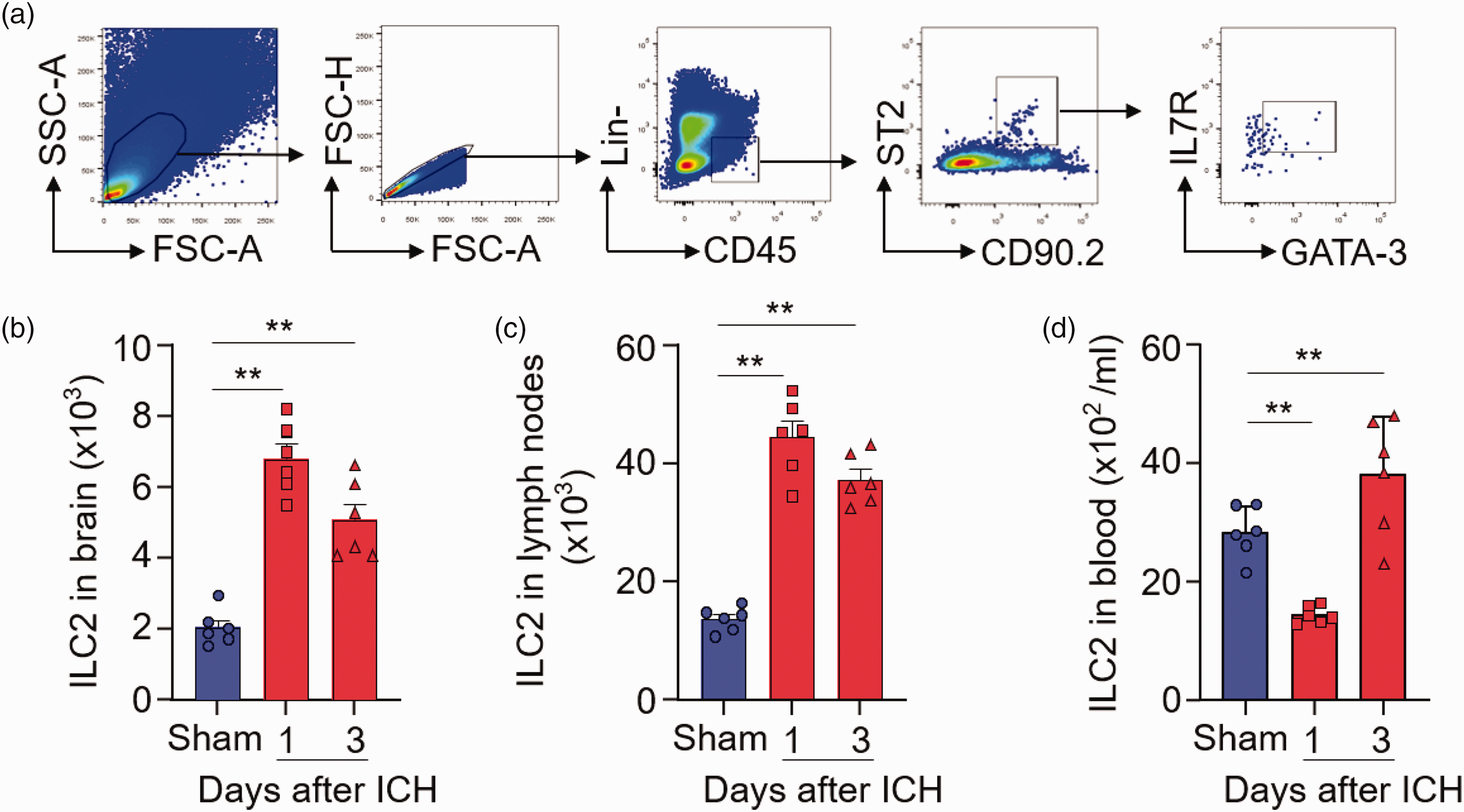
Accumulation of ILC2 in the brain of ICH mice. ICH was induced in C57BL/6 mice by intracerebral injection of collagenase. At day 1 and 3 after ICH, brain tissues and mesenteric lymph nodes were collected for flow cytometry analysis. (a) Gating strategy of ILC2 (CD45+Lin−CD90.2+ST2+ GATA-3+ IL7R+). (b) Bar graph showing counts of ILC2 in the brains of ICH mice. n = 6 mice per group. (c) Bar graph showing counts of ILC2 in the mesenteric lymph nodes of ICH mice. n = 6 mice per group and (d) Bar graph showing counts of ILC2 in blood of ICH mice. n = 6 mice per group. Data are presented as mean ± SD. **P < 0.01.
Depletion of ILC2 exacerbates ICH injury, microglia activity and leukocyte invasion
To determine whether ILC2 affect ICH injury, we used antibody to deplete ILC2 in vivo. Flow cytometry analysis revealed no significant alterations of NK cells of naïve mice receiving anti-CD90.2 mAb (Supplementary Figure 2A). Anti-CD90.2 mAb depletes both ILC2 and CD4+ T cells. In contrast, anti-CD4 mAb only depletes CD4+ T cells. Groups of mice received IgG control, anti-CD90.2 mAb or anti-CD4 mAb prior to ICH induction (Figure 2(a)). We measured the changes of CD4+ T cells, CD8+ T cells, B cells and ILC2 in the brain and periphery of ICH mice receiving anti-CD90.2 mAb or anti-CD4 mAb injection (Supplementary Figure 2B-D). We found augmented neurological deficits, brain edema and lesion volumes in ICH mice receiving anti-CD90.2 mAb as compared to ICH mice receiving IgG or anti-CD4 mAb (Figure 2(b) to (e)). Similar neurodeficits, brain water content and lesion volume were seen in male versus female MCAO mice receiving IgG control or anti-CD90.2 mAb (Supplementary Figure 3). Next, we measured the level of IL-13 and IL-10 in the brain from ICH mice receiving IgG or anti-CD90.2 mAb. We found the level of IL-13 and IL-10 were reduced in the brains from ICH mice receiving anti-CD90.2 mAb (Supplementary Figure 4A-B). In addition, we found an increase of microglia (CD11b+CD45int) and infiltrating neutrophils (CD11b+CD45high Ly6G+) in the brain from ICH mice receiving anti-CD90.2 mAb as compared to ICH mice receiving IgG or anti-CD4 mAb (Figure 3(a) and (b)). These results demonstrate that depletion of ILC2 leads to augmented ICH injury and neuroinflammation.
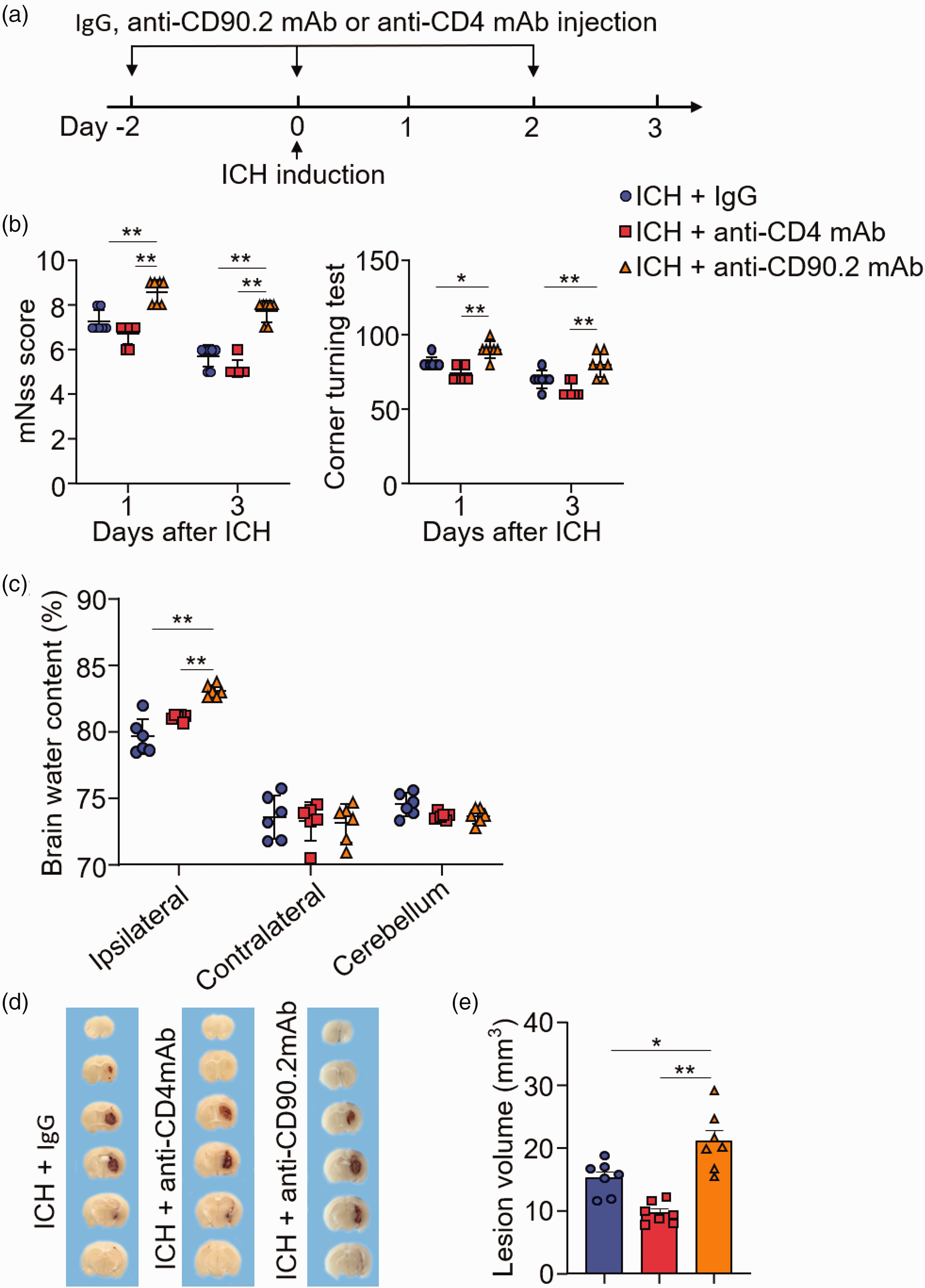
Depletion of ILC2 exacerbates hemorrhagic brain injury and neuroinflammation. ICH was induced in C57BL/6 mice by intracerebral injection of collagenase. (a) Flow chart illustrates drug administration and experimental design. Mice received intraperitoneal (i.p.) injections of 300 μg anti-CD90.2mAb, 300 μg anti-CD4mAb or IgG at 2 days prior to ICH surgery and every 2 days thereafter until the end of experiment. At day 3 after ICH, neurodeficits, brain water content or lesion volume were assessed. (b) Summarized results of modified Neurological Severity Score (mNSS) and corner-turning test scores in ICH mice receiving indicated treatments at day 1 and 3. n = 7 mice per group. (c) Brain water content in groups of mice receiving indicated treatments at day 3 after ICH. n = 6 mice per group. (d) Coronal sections from groups of mice receiving IgG, anti-CD4mAb or anti-CD90.2mAb and analysis of hematoma volume at day 3 after ICH and (e) Quantification of hematoma volume in groups of mice receiving IgG (n = 6), anti-CD4mAb (n = 6) or anti-CD90.2mAb (n = 7). Data are presented as mean ± SD. *P < 0.05, **P < 0.01.
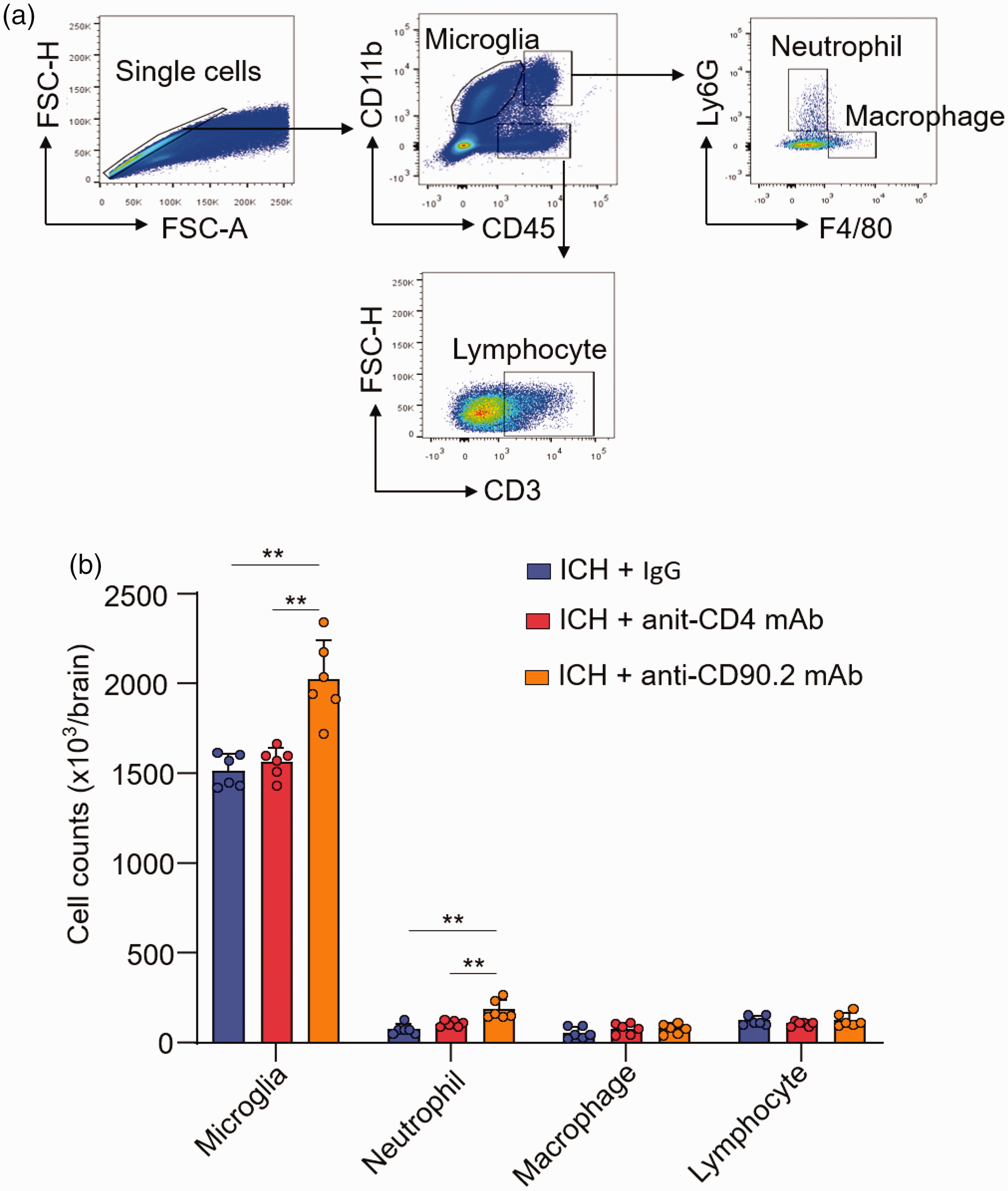
Depletion of ILC2 exacerbates microglia activity and leukocyte invasion. ICH was induced in C57BL/6 mice by intracerebral injection of collagenase. At day 3 after ICH, immune cells were isolated from brain tissues for flow cytometry analysis. (a) Gating strategy of microglia (CD11b+CD45int), brain-infiltrating neutrophils (CD11b+CD45highLy6G+ F4/80−), macrophages (CD11b+CD45high Ly6G− F4/80+), and T cells (CD11b−CD45highCD3+) and (b) Counts of microglia and brain-infiltrating leukocyte subsets in the brains from indicated groups of ICH mice. n = 6 mice per group. Data are presented as mean ± SD. *P < 0.05, **P < 0.01.
Expansion of ILC2 attenuates ICH injury, microglia activity and leukocyte invasion
IL-33 is an alarmins that can swiftly expand ILC2 in vivo. 26 We next tested whether expansion of ILC2 can influence ICH injury. Groups of mice received IgG control or IL-33 prior to ICH induction (Figure 4(a)). In ICH mice receiving IL-33, we found reduced neurological deficits, brain edema and lesion volumes versus ICH mice receiving IgG control (Figure 4(b) to (d)). Additionally, we found an increase of IL-13 and IL-10 in the brains from ICH mice receiving IL-33 versus IgG control (Supplementary Figure 4B–C). Flow cytometry analysis revealed an increase of brain-infiltrating ILC2 in ICH mice receiving IL-33 versus IgG control (Figure 4(e)), together with a decrease of microglia (CD11b+CD45int) and infiltrating neutrophils (CD11b+CD45highLy6G+) (Figure 4(f)). These results suggest that expansion of ILC2 ablates ICH injury and neuroinflammation.
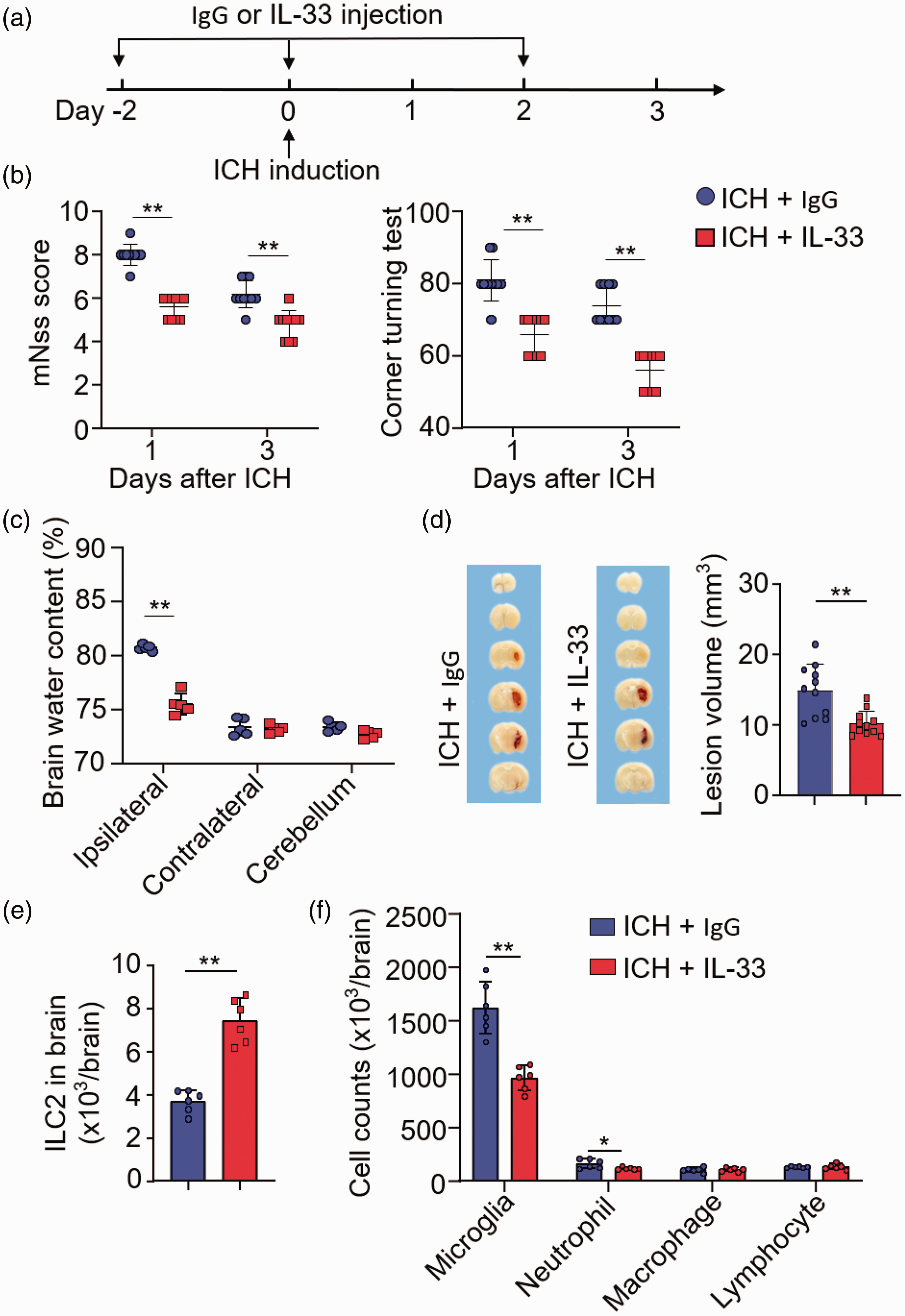
Expansion of ILC2 attenuates hemorrhagic brain injury and neuroinflammation after ICH. ICH was induced in C57BL/6 mice by intracerebral injection of collagenase. (a) Flow chart illustrates drug administration and experimental design. Mice received i.p. injections of 600 ng IL-33 or an equal volume of IgG 2 days prior to ICH surgery and every 2 days thereafter until the end of experiment. At day 3 after ICH, neurodeficits, brain water content or lesion volume were assessed. (b) Summarized results of modified Neurological Severity Score (mNSS) and corner-turning test scores in ICH mice receiving indicated treatments at days 1 and 3. n = 10 mice per group. (c) Brain water content in groups of mice receiving indicated treatments at day 3 after ICH. n = 6 mice per group. (d) Coronal sections from ICH mice receiving IgG or IL-33 to measure hematoma volume at day 3 after ICH. n = 10 mice per group. (e) Bar graph showing counts of ILC2 in the brains of ICH mice. n = 6 mice per group and (f) At day 3 after ICH, immune cells were isolated from brain tissues for flow cytometry analysis. Bar graphs showing counts of microglia and brain-infiltrating leukocyte subsets from indicated groups of ICH mice. n = 6 mice per group. Data are presented as mean ± SD. **P < 0.01.
Microglia depletion abolishes the protective effect of ILC2 following ICH
As microglia are the first responder to ICH, we next examined the role of microglia in ILC2-mediated protection against ICH injury. For this purpose, groups of mice received a colony stimulating factor 1 receptor (CSF1R) inhibitor PLX5622 prior to ICH induction (Supplementary Figure 5A). Treatment was continued until the end of experiments (Figure 5(a)). The count of ILC2 in the brain after microglia depletion was not significantly altered (Supplementary Figure 5B-C). Of interest, we found that the exacerbated ICH injury induced by antibody depletion of ILC2 was abolished in groups of mice PLX5622 (Figure 5(b) and (c)), suggesting that microglia are involved in the protective effect of ILC2 against ICH injury.
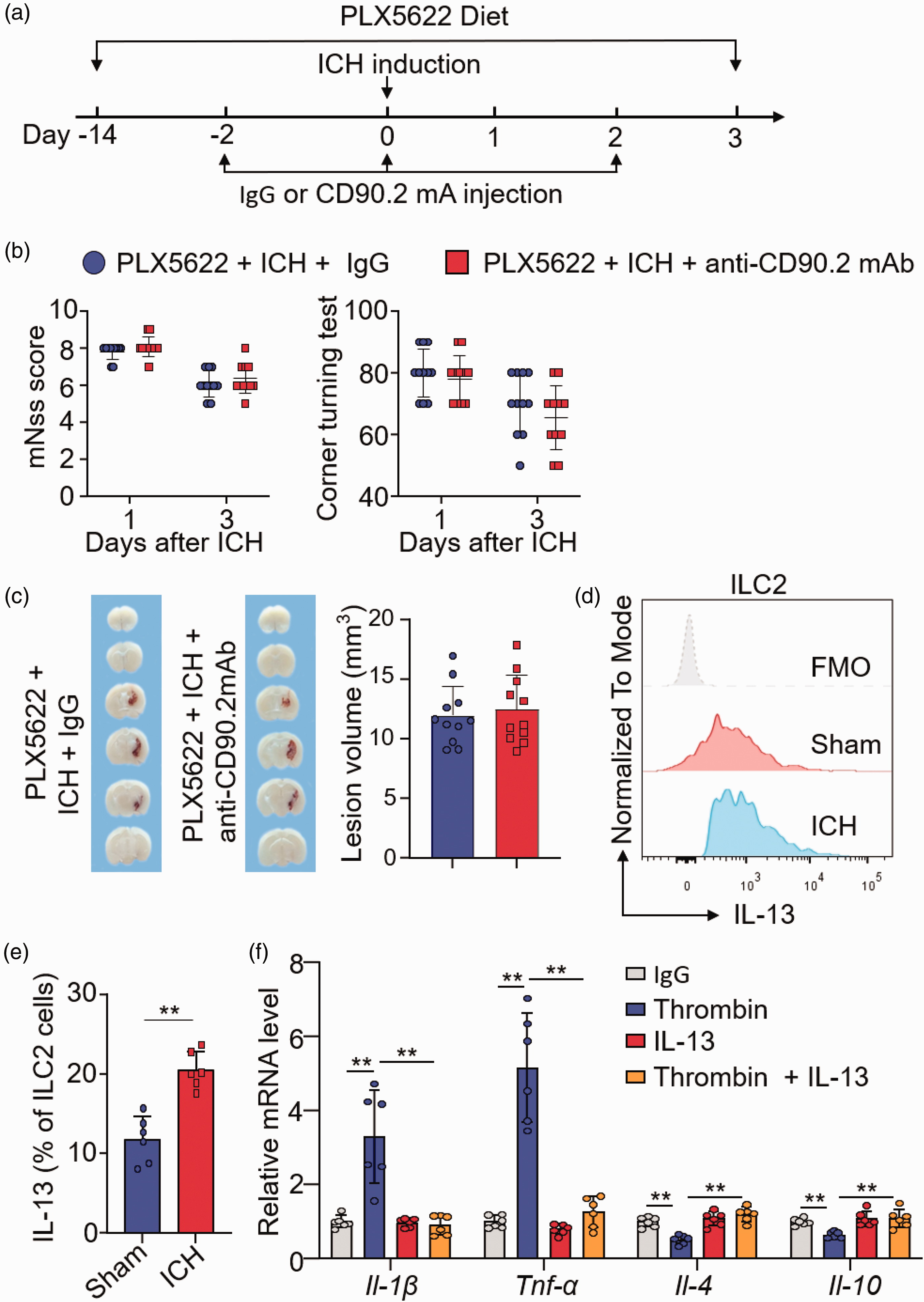
Microglia depletion abolishes the protective effect of ILC2 following ICH. ICH was induced in C57BL/6 mice by intracerebral injection of collagenase. (a) Flow chart illustrates drug administration and experimental design. C57BL/6 mice received PLX5622 in chow for 14 days and continuously until the end of experiment. ICH was induced in PLX5622-treated mice by injection of collagenase. Groups of ICH mice received i.p. injections of 300 μg anti-CD90.2mAb or an equal volume of IgG 2 days before ICH induction and every 2 days thereafter until the end of experiment. At day 3 after ICH, neurodeficits and lesion volume were assessed. (b) Summarized results of modified Neurological Severity Score (mNSS) and corner-turning test scores in ICH mice receiving indicated treatments at day 1 and 3. n = 11 mice per group. (c) Coronal sections from ICH mice receiving IgG or anti-CD90.2 mAb to measure hematoma volume at day 3 after ICH. n = 11 mice per group. At day 3 after ICH, brain tissues were collected for flow cytometry analysis. (d) Expression of IL-13 in brain-infiltrating quantified by flow cytometry analysis. FMO, fluorescence minus one. (e) Quantification of IL-13 expression in ILC2. n = 6 mice per group and (f) Microglia-like BV-2 cells were seeded in six-well tissue culture plates (2 × 105 cells per well). After 24 h, cells were treated with IgG PBS, 40 U/mL thrombin, 20 ng/mL IL-13 or 40 U/mL thrombin + 20 ng/mL IL-33. After 24 h, cells were collected and mRNA expression of Il-1β, Tnf-α, Il-4 and Il-10 was measured by quantitative real-time RT-PCR. n = 6 per group. Data are presented as mean ± SD. **P < 0.01.
ILC2 suppress thrombin-induced inflammatory activity in microglia-like BV2 cells
To test the functional profile of ILC2 in ICH, we measured type 2 cytokine expression in ILC2 harvested from the ICH brain. Intracellular staining revealed an upregulation of IL-13 in ILC2 from the ICH brain (Figure 5(d) and (e)). To understand whether ILC2-derived type 2 cytokine can modulate microglia activity, we added IL-13 into the medium of cultured microglia-like BV2 cells that were subjected to thrombin stimulation. As a result, real-time RT-PCR assay revealed that IL-13 treatment ablated thrombin-induced production of inflammatory factors such as Il-1β and Tnf-α in cultured microglia-like BV2 cells (Figure 5(f)). In contrast, the production of immunoregulatory factors such as Il-4 and Il-10 was augmented in cultured microglia-like BV2 cells receiving IL-13 (Figure 5(f)). Taken together, these results demonstrate that ILC2 can suppress the inflammatory activity of microglia in ICH.
Discussion
In this study, we demonstrate that ICH-mobilized peripheral ILC2 infiltrate into the injured brain. As documented here, brain-infiltrating ILC2 exhibit upregulation of IL-13, suppress neuroinflammation and reduce ICH injury, a process involving microglia. Additionally, expansion of ILC2 using IL-33 suppressed ICH injury. Notably, IL-13 is a prominent type 2 effector cytokine that can directly suppress the inflammatory activity of microglia.27,28 Together, our findings further extend the understanding of ILC2 to restrict CNS inflammatory and brain edema after ICH, suggesting that immune modulation targeting ILC2 may provide new avenues to restrain inflammatory CNS injury.
ILC2 express an array of chemokine receptors, including CXCR6, CCR1, CCR4, CCR6, CCR8 and CCR9.29,30 These machineries may allow ILC2 to respond to various signals and cues that are released to the periphery following brain injury and migrate into the brain. The accumulation of ILC2 in the brain and increase of ILC2 in the lymph nodes following ICH suggest that these ILC2 can swiftly sense acute brain injury and mobilized to home into the brain. The location of ILC2 within the perihematomal regions together with their immunomodulatory features suggest that these cells may serve as a key player to orchestrate local inflammation and ICH injury. As potent responders to alarmins such as IL-33,31 –33 ILC2 can be expanded in vivo by administration of IL-33 in ICH mice, in which ICH injury and neuroinflammation was reduced. IL-33 may act on a variety of cells in the periphery other than ILC2, including T cells and macrophages.26,30 Considering the beneficial role of ILC2 to reduce ICH injury, it’s reasonable to postulate that the accumulation of ILC2 in the brain and an increase of ILC2 in the lymph nodes may represent an endogenous immunoregulatory machinery to counteract inflammatory brain insult in ICH, although further investigations are required in a reasonable cohort of ICH patients to test this postulation.
CD4+ T cells are detrimental to ICH and can exacerbate acute neural injury as we and others previously reported.34,35 The finding that depletion of ILC2 led to augmented neuroinflammation and ICH injury suggests a beneficial role of ILC2 to reduce hemorrhagic brain injury. In support of this view, we showed that expansion of ILC2 suppressed inflammatory activity of microglia, leukocyte infiltration, neurological deficits and brain edema. In line with these findings, a previous study reported that ILC2 can accumulate in the mouse meningeal space in response to spinal cord injury in an IL-33-dependent manner and produce type 2 cytokines, possibly act a modulator to restrain neuroinflammatory conditions and promote neuronal survival. 36 In another study using a mouse model of traumatic brain injury (TBI), an expansion and brain infiltration of IL-10-expressing ILC2 were noted after TBI onset, 37 suggesting an anti-inflammatory role of ILC2 in acute brain injury, although functional studies remain to be explored using TBI models subjected to ILC2 depletion or expansion. Together, these findings support a beneficial role of ILC2 in ICH injury and its potential to serve as a biomarker or treatment target for hemorrhagic brain injury. In addition, we found similar extent of exacerbated acute ICH injury in male versus female mice subjected to antibody depletion of ILC2, although sexual dimorphism has been reported in stroke.38 –42 Nevertheless, these results cannot exclude the potential involvement of sex differences in ILC2 function and stroke long-term outcome, which warrant future investigations. Reportedly, aging is another factor that influences stroke outcomes.43,44 As only young mice were used in our study, whether aging could affect the effects of ILC2 on ICH injury awaits future investigations.
We found increased number of microglia in ICH mice receiving ILC2 depletion. The increased microglia number may result from following reasons. First, depletion of ILC2 leads to reduced suppression of microglia because ILC2 can produce regulatory cytokines such as IL-13 that can directly limit microglia activity. Second, depletion of ILC2 may lead to increased inflammatory factors in the environment that promote microglia activity after ICH. Third, depletion of ILC2 results in increased lesion volume and increased tissue damage leading to stronger stimulation and microglia activation.28,45 The involvement of microglia in the benefit of ILC2 to restrict neuroinflammation following ICH suggests a previously unrecognized interaction between ILC2 and brain-resident microglia. Although we found that depletion of microglia diminished the benefit of ILC2, these results suggest the involvement of microglia in the beneficial effects of ILC2. However, these results cannot exclude the contributions from other cell types such as neutrophils to the protective effects of ILC2. In support of this view, we found increased infiltration of neutrophils in ICH mice after depletion of ILC2, accompanied by reduced IL-10 and IL-13 levels in ICH brains. Reportedly, IL-10 and IL-13 can suppress the activity of neutrophils. 46 These results suggest that neutrophils could also be a contributing factor to the benefit of ILC2 in ICH, which awaits future investigations. BV-2 cells are a murine microglia cell line and have been used as an alternative for primary microglia, which is supported by genetic profiling showing that BV-2 cells shared 90% of genes with primary microglia. 47 Indeed, we found that brain-infiltrating ILC2 had upregulation of IL-13 that can directly suppress the inflammatory activity of microglia and switch on their expression of anti-inflammatory factors. Although this modulatory effect of ILC2 on microglia has not been covered by previous reports, previous studies demonstrate that ILC2 can modulate macrophages in the periphery. One recent study has revealed that ILC2 are essential for the maintenance of alternatively activated macrophages in the fat tissues. 26 The other two studies also reported that ILC2 can interact with macrophages in the lung to modulate their alternative activation.48,49 Although the specific effector molecules derived from ILC2 vary among these studies, i.e., IL-5, IL-10 or IL-13, the impact of ILC2 on tissue-resident microglia or macrophages suggest ILC2 as an indispensable modulator the initiation, progression and resolution of tissue inflammation.
In summary, this study provides novel evidence that ILC2 can suppress neuroinflammation and hemorrhagic brain injury. Future design of immune therapies targeting ILC2 may serve as a potential therapy or at least a complementary approach to restrict inflammatory CNS injury.
Supplemental Material
sj-pdf-1-jcb-10.1177_0271678X231208168 - Supplemental material for Group 2 innate lymphoid cells suppress neuroinflammation and brain injury following intracerebral hemorrhage
Supplemental material, sj-pdf-1-jcb-10.1177_0271678X231208168 for Group 2 innate lymphoid cells suppress neuroinflammation and brain injury following intracerebral hemorrhage by Mingming Liu, Danni Wang, Lin Xu, Yan Pan, Huachen Huang, Minshu Li and Qiang Liu in Journal of Cerebral Blood Flow & Metabolism
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported in part by National Science Foundation of China (82225015, 82171284, 82200279), National Key Research and Development Project of China (2021ZD0202400), National Postdoctoral Program for Innovative Talents (BX20220227), Postdoctoral Science Foundation (2022M712390), Science and Technology Development Fund of Tianjin Education Commission for higher Education grant (2021ZD035) and Tianjin Key Medical Discipline (Specialty) Construction Project.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
Q.L. formulated the concept and designed the studies. M.L. and D.W. performed the experiments. M.L., D.W., L. X. and Y.P. analyzed the results. M.L., L. X., H.H., M.L. and Q.L. interpreted the results. M.L., D.W. and Q.L. wrote and edited the manuscript.
Supplementary material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.