
Abstract
Cerebral arteriovenous malformations (AVMs) entail a significant risk of intracerebral hemorrhage owing to the direct shunting of arterial blood into the venous vasculature without the dissipation of the arterial blood pressure. The mechanisms involved in the growth, progression and rupture of AVMs are not clearly understood, but a number of studies point to inflammation as a major contributor to their pathogenesis. The upregulation of proinflammatory cytokines induces the overexpression of cell adhesion molecules in AVM endothelial cells, resulting in enhanced recruitment of leukocytes. The increased leukocyte-derived release of metalloproteinase-9 is known to damage AVM walls and lead to rupture. Inflammation is also involved in altering the AVM angioarchitecture via the upregulation of angiogenic factors that affect endothelial cell proliferation, migration and apoptosis. The effects of inflammation on AVM pathogenesis are potentiated by certain single-nucleotide polymorphisms in the genes of proinflammatory cytokines, increasing their protein levels in the AVM tissue. Furthermore, studies on metalloproteinase-9 inhibitors and on the involvement of Notch signaling in AVMs provide promising data for a potential basis for pharmacological treatment of AVMs. Potential therapeutic targets and areas requiring further investigation are highlighted.
Keywords
Introduction
Cerebral arteriovenous malformations (AVMs) are comprised of vessels that directly shunt blood from the arterial to the venous system because of the absence of a capillary bed. The incidence of AVMs is 0.94 to 1.34 per 100,000 person-years with a mean age of presentation at 30 to 35 years.1, 2, 3
The prevailing theory of AVM rupture risk is the lack of intervening capillaries leading to an abnormally high-pressure blood flow through the AVM nidus. This high pressure is then transmitted to the venous vasculature, resulting in venous hypertension by impaired outflow, eventually resulting in rupture.4, 5
Arteriovenous malformation rupture is very common, making intracerebral hemorrhage (ICH) the major cause of AVM-related morbidity and mortality. The ICH risk for untreated AVMs is 2% to 6% per year.2, 6, 7, 8 However, the annual hemorrhage risk may be as high as 34% if any additional risk factors are present. 9 History of hemorrhage, high blood pressure, intranidal aneurysm, venous stenosis, and deep venous drainage are the main factors that increase the likelihood of AVM rupture.4, 10, 11
Although there are several therapeutic options to eradicate AVMs, the treatment-related morbidity is significant, especially for large, deep-seated, or eloquently located nidi.
6
The recently published ARUBA trial (A Randomized trial of Unruptured Brain Arteriovenous malformations) showed that medical management alone is superior to medical management with interventional therapy for the prevention of death or stroke in patients with unruptured brain AVMs.
12
Thus, there is a pressing need to better understand the pathogenesis of AVMs and the molecular mechanisms that destabilize these lesions to develop novel medical therapies, which may reduce the need for invasive intervention. Studies have shown that inflammation plays a major role in vascular dysmorphogenesis by weakening the AVM vessel walls. Certain genetic polymorphisms have been shown to significantly upregulate the expression of angiogenic and proinflammatory proteins, which may, in turn, contribute to AVM destabilization and rupture. In this review, we critically analyze the existing data from
The common pathway
The common pathway for AVM formation and rupture appears to involve an inflammatory reaction (cytokines, neutrophils, macrophages, etc.) in the AVM walls, triggered by genetic/hemodynamic factors (Figure 1). Inflammation is then followed by increased angiogenesis and concomitant breakdown of extracellular matrix by various proteinases and cell death, all of which contribute to AVM wall weakening and rupture. These pathways will be discussed in this review.
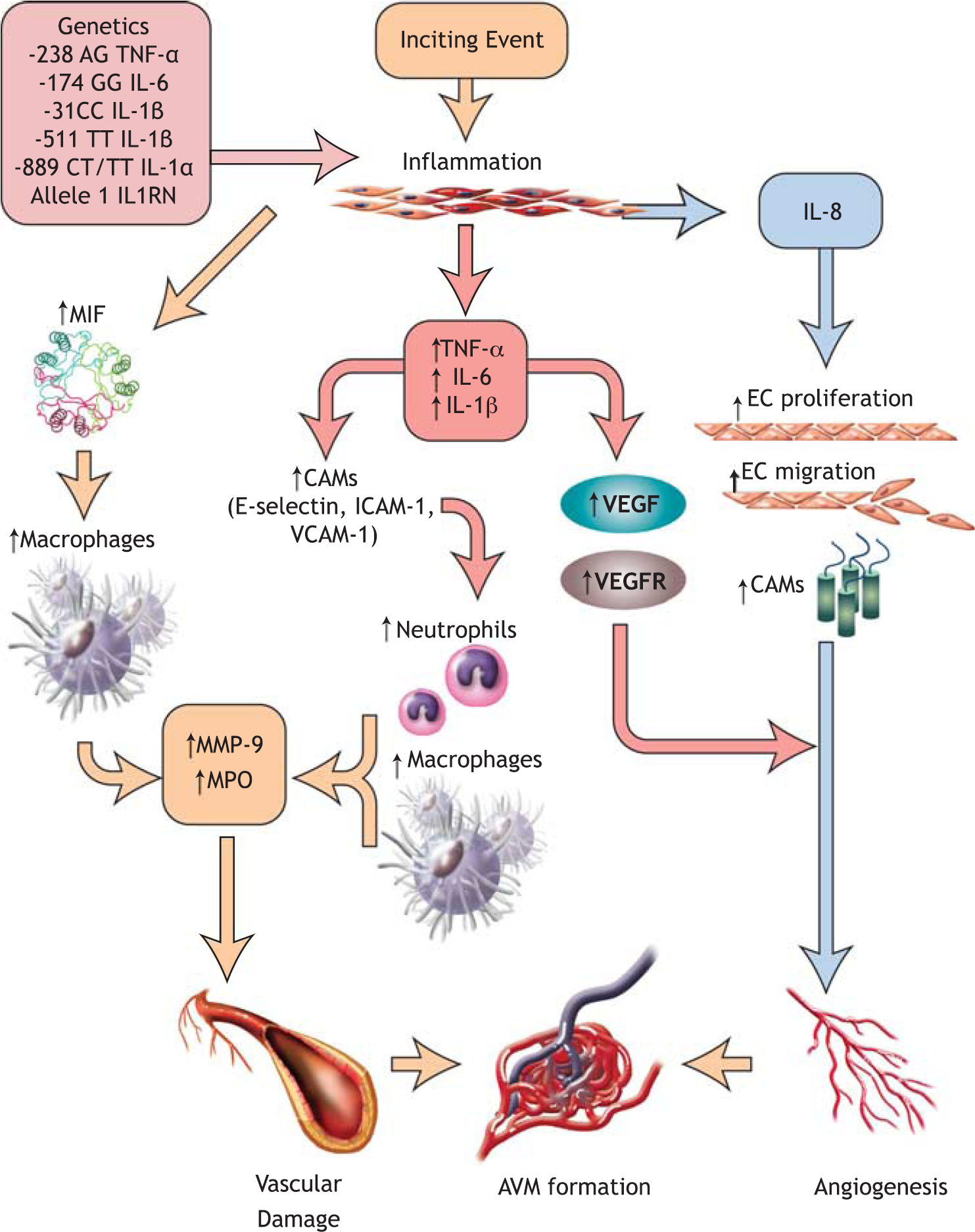
Contributors to AVM pathogenesis. The SNPs shown above upregulate the inflammatory response to inciting events and lead to the overexpression of proinflammatory cytokines, which in turn exacerbate the inflammation, recruit leukocytes and activate the endothelial cells of the AVM. As a result, increased vascular damage and angiogenesis are observed, resulting in AVM formation and expansion. AVM, arteriovenous malformation; CAMs, cell adhesion molecules; EC, endothelial cell; ICAM-1, intercellular cell adhesion molecule-1; IL1RN, interleukin-1 receptor antagonist; IL-6, interleukin-6; IL-1α, interleukin-1-alpha; IL-1β, interleukin-1-beta; IL-8, interleukin-8; MIF, macrophage migration inhibitory factor; MMP, matrix metalloproteinase; MPO, myeloperoxidase; SNPs, single-nucleotide polymorphisms; TNF-α, tumor necrosis factor-alpha; VCAM-1, vascular cell adhesion molecule-1; VEGF, vascular endothelial growth factor; VEGFR, vascular endothelial growth factor receptor.
Genetic predisposition
Genetics play a major role in the magnitude of the inflammatory response upon an inciting event. Several single-nucleotide polymorphisms (SNPs) have been identified in the genes of major proinflammatory cytokines that result in varying levels of inflammatory responses, thereby altering the extent of vascular dysmorphogenesis.13, 14 The cytokines, tumor necrosis factor-α (TNF-α), interleukin-6 (IL-6), interleukin-1α and 1β (ΙL-1α and IL-1β), and IL-1 receptor antagonist (IL1RN) each have several SNPs, some of which are associated with greater likelihood of pathogenesis than others (Table 1).
Effect of gene polymorphisms on inflammation in AVMs

AVMs, arteriovenous malformations; ICH, intracranial hemorrhage; IL-6, interleukin-6; IL-1, interleukin-1; IL-8, interleukin-8; IL-1β, interleukin-1-beta; IL-1α, interleukin-1-alpha; IL-1RN, interleukin-1 receptor antagonist; MMP, matrix metalloproteinase; TNF-α, tumor necrosis factor-alpha.
Two SNPs have been identified in the IL-6 gene, −174 G>C and −572 G>C, and their effects on the rate of ICH were showed by Pawlikowska
Two SNPs were also identified in the gene of the proinflammatory cytokine TNF-α in AVM patients, TNF-α −238 G>A and −308 G>A. The AG genotype of the −238 G>A SNP was associated with a 6.4% rate of ICH in patients carrying this SNP presenting with a new ICH likely through overexpression of TNF-α and IL-6. 17 In contrast, the TNF-α 308 G>A SNP was not found to increase the risk of new ICH.
The effect of the proinflammatory cytokine IL-1 on brain AVMs has been studied extensively as well. Three SNPs on the IL-1β gene, two SNPs on the IL-1α gene, and five alleles of the IL1RN gene have been shown to be associated with alterations in AVM hemorrhage risk.18, 19 AVM patients with the IL-1β −31 CC and the −511 TT genotypes had an increased hemorrhage risk and patients with the IL-1α −889 C>T genotype (either CT or TT) or IL1RN allele 1 had a greater AVM susceptibility (Table 1).18, 19, 20 Thus, IL-1 may be a promising therapeutic target for patients with AVMs.
Effect of increased proinflammatory cytokines on AVMs: a central role for IL-6
Various cytokines have been implicated in the pathogenesis of AVMs; most notable is the contribution of IL-6. When IL-6 is expressed in large amounts, there is a significant increase in the mRNA levels of IL-1β, TNF-α, and IL-8 in human AVM tissues. 21 Through the upregulation of these cytokines, IL-6 indirectly stimulates leukocyte recruitment, endothelial activation, and vascular smooth muscle cell (SMC) proliferation (Table 2). Leukocyte recruitment occurs when the cellular adhesion molecules (CAMs) of ECs bind circulating leukocytes and affix them to the site of inflammation. Indeed, resected AVMs were found to have increased expression of CAMs: namely E-selectin, intercellular CAM-1 (ICAM-1), and vascular CAM-1 (VCAM-1). E-selectin concentration was increased to the greatest extent relative to ICAM-1 and VCAM-1. 22
Factors involved in AVM pathogenesis
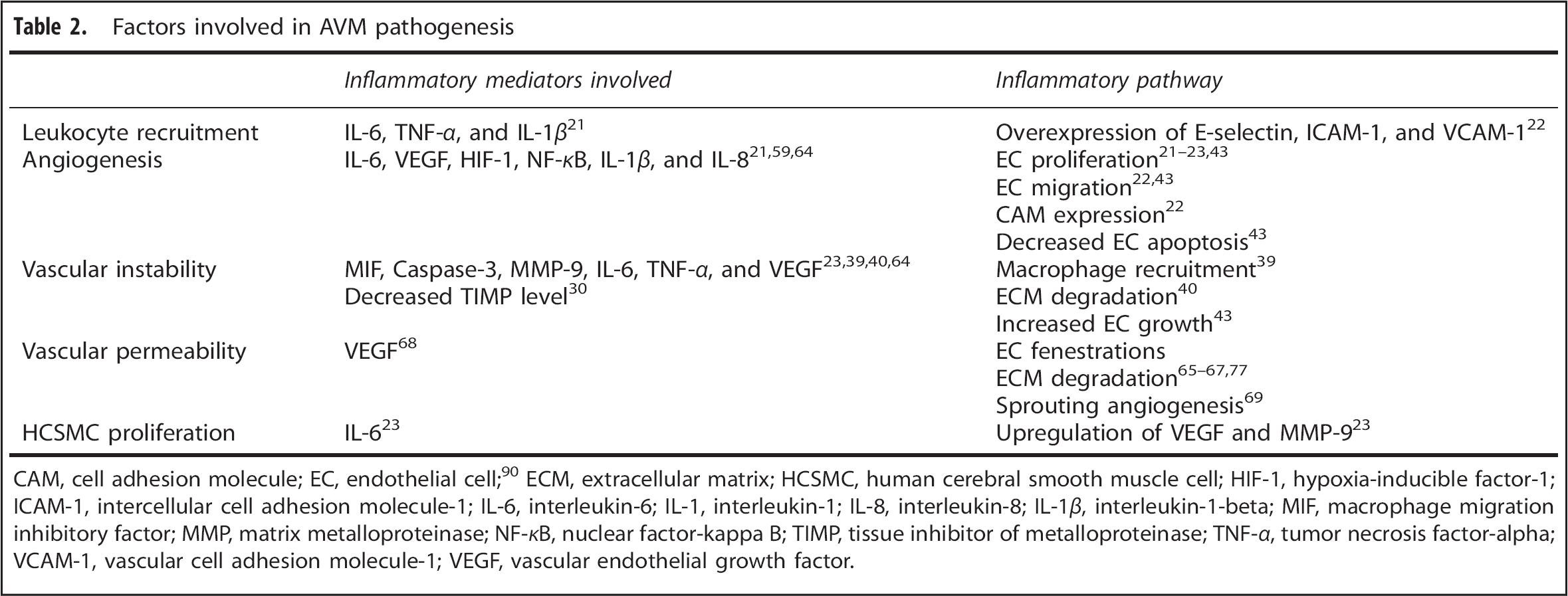
CAM, cell adhesion molecule; EC, endothelial cell; 90 ECM, extracellular matrix; HCSMC, human cerebral smooth muscle cell; HIF-1, hypoxia-inducible factor-1; ICAM-1, intercellular cell adhesion molecule-1; IL-6, interleukin-6; IL-1, interleukin-1; IL-8, interleukin-8; IL-1β, interleukin-1-beta; MIF, macrophage migration inhibitory factor; MMP, matrix metalloproteinase; NF-κB, nuclear factor-kappa B; TIMP, tissue inhibitor of metalloproteinase; TNF-α, tumor necrosis factor-alpha; VCAM-1, vascular cell adhesion molecule-1; VEGF, vascular endothelial growth factor.
In addition to leukocyte recruitment, the increased expression of IL-1β and IL-8 secondary to elevated IL-6 has a significant upregulatory effect on angiogenesis. The magnitude of angiogenesis was measured in cultured human cerebral ECs exposed to IL-6, where 100 ng/ml of IL-6 was shown to have a similar effect on human cerebral ECs as 20 ng/ml of vascular endothelial growth factor (VEGF). 21 Additionally, an increase in IL-6 concentration stimulates VEGF release, VEGF receptor II activation along with human cerebral SMC proliferation and greater MMP-9 expression. 23 Hence, the combination of increased angiogenesis and breakdown of extracellular matrix initiated in part by IL-6 may lead to vascular instability and AVM rupture.
Neutrophil and macrophage recruitment to AVMs leads to release of matrix metalloproteinases and vascular remodeling
The upregulation of inflammatory cytokines and the overexpression of CAMs on ECs result in the enhanced recruitment of leukocytes to the AVM tissue. Recruited leukocytes secrete myeloperoxidase, MMPs, cytokines, and other proteolytic enzymes, all of which cause damage to the AVM vessel walls, leading to rupture of the nidus.
24
Chen
Given the role of MMP-9 in the pathogenesis of AVMs, investigators have attempted to use MMP-9 level as a screening tool to determine the likelihood of rupture. Starke
Macrophage migration inhibitory factor (MIF) appears to be a key factor in macrophage-induced vascular degradation in AVMs. The MIF protein has been found to be elevated in AVMs—a twofold increase compared with control human brain tissue has been observed.
39
Macrophage migration inhibitory factor is associated with the upregulation of MMP-2, MMP-9, MMP-12, and downregulation of TIMPs, causing dilation of the arterial wall and eventual rupture.15, 40, 41 Based on studies in abdominal aortic aneurysms, MIF has been shown to induce MMP expression and leads to increased breakdown of many types of collagen, elastin, fibrillin, fibronectin, tenascin, and osteonectin.15, 42, 43 It is noteworthy that the extracellular form of MIF was mostly found in the tunica intima and adventitia of the vasculature, while the intracellular form was found in the tunica media and activated caspase-3 was also detected in the tunica media of AVMs. This co-localization led Chen
Endoglin and its role in inflammation and vascular remodeling
Endoglin is a membrane glycoprotein located on cell surfaces and is part of the transforming growth factor-β (TGF-β) receptor complex. It is involved in hereditary hemorrhagic telangiectasia, which can also result from a loss-of-function mutation in activin-like kinase receptor-1 (ALK1) or SMAD4 genes. Based on several studies, 9% to 16% of hereditary hemorrhagic telangiectasia patients had an AVM, 27% of whom had a history of ICH.44, 45, 46, 47 Homozygous loss of these genes leads to death
As part of the TGF-β receptor complex, ENG is also involved in the neovascularization of damaged tissue during inflammation. It binds activated circulating leukocytes, enabling them to adhere to damaged endothelium or ischemic tissues. Based on myocardial infarction studies, mice with underexpressed ENG—resembling the vasculature in patients with hereditary hemorrhagic telangiectasia—had a decreased ability to stimulate vessel repair, but this ability was restored with the injection of wild-type mononuclear cells.
49
Rossi
Angiogenesis in AVMs and the link to inflammation: the role of nuclear factor-kappa B and hypoxia-inducible factor-1
Excessive angiogenesis and vascular remodeling likely contribute to the formation and progression of cerebral AVMs.
11
Most of the cerebral vasculature is formed during embryogenesis, with minimal postnatal vasculogenesis. AVMs are generally considered congenital vascular lesions that form in the late stages of fetal development.
Inflammation stimulates lymphocytes to release several inflammatory cytokines, including IL-6, IL-1β, and ΤNF-α, all of which upregulate the expression of VEGF in a multitude of pathways. These three cytokines induce the expression of NF-κB, which binds the promoter regions of the VEGF and IL-8 genes and upregulates their expression, thereby enhancing AVM angiogenesis.64, 63 VEGF in turn increases capillary permeability and upregulates ICAM-1 and VCAM-1 on ECs, thus enhancing leukocyte accumulation, inflammation, and the degradation of the vascular walls via leukocyte-derived MMPs.65, 66, 67, 68, 69 Moreover, VEGF can create fenestrations in the endothelium previously linked by tight junctions.
70
Using
The NF-κB-VEGF pathway in AVMs is also stimulated under hypoxic conditions by HIF-1. Owing to the shunting of blood directly from the arterial to the venous circulation, perinidal brain tissue may receive relatively lower amounts of oxygen, causing HIF-1 to accumulate and upregulate the expression of VEGF by binding to hypoxia responsive element.74, 75 The induction of VEGF by HIF-1 promotes EC proliferation and migration, while also inhibiting EC apoptosis (this is partly the result of upregulation of factors, such as Flt-1 and Flk-1)
74
In fact, Wautier
The role of inflammatory molecules such as IL-6, MMP-9, and NF-κB, and factors such as HIF-1 and VEGF in the pathogenesis of AVMs has direct implications on AVM treatment, embolization in particular. Even though embolization is a commonly used preoperative or preradiosurgical adjunct, it induces HIF-1a, VEGF, and MMP-9 upregulation among other inflammatory mediators.38, 78, 79 In fact, many studies have shown that embolized AVMs have significantly higher amounts of HIF-1 and VEGF than nonembolized ones.78, 80 Partial AVM occlusion leads to hypoxia, which in turn upregulates VEGF, resulting in elevation of MMP-9 expression via NO- and NF-κB-dependent pathways and angiogenesis.28, 29, 38, 72, 79 These findings suggest that incomplete embolization could have a destabilizing effect on AVMs and may increase the subsequent risk of hemorrhage, warranting further investigation into what the impact of embolization is on the complication risk after surgical resection and on recovery.
The role of VEGF in AVM pathogenesis has prompted investigators to study the relationship between local and systemic VEGF levels. Although elevated local VEGF expression has been linked to an increased likelihood of ICH,
33
quantifying the local amount of VEGF and thus the risk of ICH is only feasible once the AVM has been resected. To be able to predict presence of weakened vasculature from early on, Kim
The notch signaling pathway in cerebral AVMs
Murphy
After experiments on mice showed that Notch4 signaling is involved in AVM pathogenesis and Notch4 repression can lead to hemodynamic normalization, Murphy
It is not yet clear whether Notch initiates the AVM pathogenesis, or if it is an epiphenomenon of which expression is requisite for the maintenance of the AVM angioarchitecture. In contrast to data from animal studies, experiments on human AVMs do not suggest that increased Notch signaling can form AVM lesions or contribute to their development. 89 It is unknown whether pharmacological inhibition of Notch activity would lead to AVM regression in humans.
Potential therapeutic targets
The degradative activity of MMP-9 is believed to be one of the primary mechanisms through which inflammation damages the AVM vasculature and often leads to hemorrhage.15, 24, 27, 36, 91 MMP-9 inhibitors have showed utility in protection from ICH.92, 93, 94 Inhibiting MMP-9 may also negate the harmful effects of AVM-related inflammatory responses. Lee
Most studies of MMP-9 inhibitors for AVM stabilization have been in animal models. Hashimoto
A promising approach to treat cerebral AVMs medically is the use of VEGF inhibitors such as bevacizumab. In a cerebral AVM model in mice by focal Alk1 gene deletion combined with VEGF stimulation, Walker
The Notch pathway in AVMs may be another promising target for medical treatment. Inhibition of Notch using doxycycline treatment reversed the AVM-like lesion, preventing vessel growth and hemorrhage.28, 29, 86, 97 Murphy
Limitations
Many of the cell culture, animal, and human studies described in this review provide important insights into the development, progression, and rupture of AVMs; however, there are several limitations to be taken into consideration. We acknowledge that co-occurrence has been used as rationale for the cause-effect relationship both in this review as well as in the original reports. Therefore, certain assumptions may have been made regarding the role of interleukins, VEGF, and Notch in AVM pathogenesis despite insufficient scientific data.
Perhaps the most important gap in AVM research that has hindered advances in understanding of the disease is the absence of a true experimental model. Currently, there is no available true animal model for cerebral AVMs. A reliable animal model is key for studying disease mechanisms and testing new therapies. Some models have been suggested and used, yet these only resemble some but not all aspects of human cerebral AVMs.
96
The first cerebral AVM model was recently developed by the distinguished University of California—San Francisco Group by combining a focal
The second major limitation of AVM research is the relative rarity of the disease in the general population. For example, the recent ARUBA trial initially called for an enrollment of 800 patients but was reduced to 400 after slow trial recruitment, prompting a reassessment of the design. At the conclusion of the trial, outcome data were available for only 223 subjects. 12 Further adding to the complexity of the problem is the heterogeneous nature of the disease. Specifically, there are five different groups of AVMs based on the Spetzler—Martin classification, each with a distinct natural history. Also, hemorrhagic versus nonhemorrhagic presentation, embolized versus nonembolized lesions, and time from hemorrhage to treatment are all important variables that can affect the profile of inflammatory gene expression.
The most pressing priority in AVM research is the conception of an animal model that completely resembles human brain AVMs. This will undoubtedly pave the way for development of safe and efficient medical therapies. Studying the role of Notch is crucial in the search for a treatment to achieve regression of AVM lesions solely by pharmacological means. Another avenue warranting further exploration is the use of MMP inhibitors for rupture risk reduction. A very promising strategy appears to involve VEGF blockade as well. VEGF inhibition has preliminarily proven to inhibit angiogenesis and reduce vessel density in cerebral AVMs as discussed above. 96 Also, VEGF blockade could prove useful to reduce the size of the AVM making resection easier and radiosurgery safer. Furthermore, bevacizumab has been shown to have radiation-sensitizing effects. 100
A promising future area in AVM research is the use of targeted imaging to assess the inflammatory status of lesions and their stability. Ferumoxytol-enhanced MRI is one such technique that is based on the critical role of inflammation and macrophages in the formation and rupture of AVMs. 101 As with intracranial aneurysms, the technique may provide important information regarding the risk of AVM rupture, or may be used to monitor the effects of antiinflammatory drugs on these lesions.
Conclusions
Inflammation plays a fundamental role in the progression and rupture of cerebral AVMs via the effects of cytokines on leukocytes, ECs, and SMCs. Several genetic polymorphisms have been identified that exacerbate lesion progression and increase the risk of AVM rupture owing to increased expression of IL-6, IL-1β, IL-1α, TNF-α, and IL-8. These cytokines promote the recruitment of neutrophils and macrophages to the site of injury, and the resulting leukocyte-derived metalloproteinase release damages the vessel walls of AVMs. In addition, increased production of MIF and VEGF in AVMs may contribute to their destabilization by enhancing inflammation and angiogenesis, respectively. A multitude of therapeutic strategies have been investigated with promising results, most notably VEGF blockade and Notch inhibition. As data accrue concerning which constituents of the inflammatory response appear critical in AVM formation, progression, and/or rupture, more specific and efficacious therapeutic strategies can be devised and tested.
Footnotes
The authors declare no conflict of interest.