
Abstract
Vascular cognitive impairment and dementia (VCID) is a series of cognitive dysfunction associated with cerebrovascular diseases and currently lacks effective treatments. The white matter, which is essential for neuronal information processing and integration, is nourished by a network of capillaries and is vulnerable to chronic hypoperfusion. Here, we show that metformin, a widely used drug for the treatment of type 2 diabetes, alleviates the white matter damage and improves cognitive impairment in a mouse model of VCID established by bilateral carotid artery stenosis (BCAS)-induced chronic hypoperfusion. Mechanistically, metformin restores the dysfunctions of oligodendrocyte precursor cells (OPCs) under hypoxia. Metformin up-regulates prolyl hydroxylases 2 via activating the AMP-activated protein kinase pathway, leading to hypoxia-inducible factor-1α (HIF-1α) degradation in OPCs. These findings suggest that metformin may have a promising therapeutic role in alleviating cognitive abnormalities by ameliorating white matter damage of VCID.
Introduction
Vascular cognitive impairment and dementia (VCID) refers to the spectrum of cerebral vascular pathologies, ranging from subjective cognitive decline to dementia. 1 Vascular dementia is the second leading cause of dementia after Alzheimer's disease, accounting for 15–30% of all dementia cases.1,2 VCID can be caused by a variety of pathological processes, the most common of which is chronic cerebral hypoperfusion resulting from either large or small cerebral vessel diseases.3,4
The white matter of the central nervous system is vulnerable to ischemia. 3 Pathological studies reveal that patients with vascular dementia had significantly lower myelin density in the white matter compared to age-matched controls, which is associated with cerebral hypoperfusion. 5 White matter hyperintensities (WMH), the typical neuroimaging features of cerebral small cerebral vessel diseases (CSVD), are considered the direct manifestation of chronic cerebral hypoperfusion.6,7 Patients with lower cerebral blood flow (CBF) display a higher WMH burden, while CBF is lower in WMH than in normal-appearing white matter. 8 The WMH progression is linked to a decline in cognitive function in the elderly and has been suggested to serve as a predictor of mild cognitive impairment.9,10 White matter lesions affecting cognitive function may be due to demyelination-induced dysfunction of neural circuitry and synaptic activity. 11 Therefore, the strategies to promote myelin repair may benefit white matter integrity and cognitive improvement in VCID.
Metformin is the most widely used oral hypoglycemic drug for the treatment of type 2 diabetes and metabolic syndrome. 12 Type 2 diabetes is characterized by accelerated cognitive decline and higher dementia risk. Among individuals with diabetes, long-term treatment with metformin was shown to reduce the risk of cognitive decline and dementia.13,14 Recently, the beneficial effects of long-term metformin use on cognitive function were partially attributed to the alleviation of CSVD burden. 15 Emerging evidence hints that metformin has a protective effect on white matter integrity. Metformin has been shown to reduce demyelination and promote remyelination in lysophosphatidylcholine-induced demyelination in mice.16,17 Metformin can also accelerate myelin recovery and ameliorate behavioral deficits in the animal model of multiple sclerosis. 18 Moreover, metformin treatment is demonstrated to effectively prevent brains from ischemia-induced injury by alleviating inflammation 19 and to improve motor and cognitive performance following neonatal hypoxia-ischemia. 20 These findings arouse the interest to determine whether metformin can restore VCID-related white matter lesions and cognitive impairment.
In the present study, using the murine bilateral carotid artery stenosis (BCAS) model, we found that metformin could attenuate white matter injury and cognitive impairment induced by chronic cerebral hypoperfusion. Moreover, metformin was demonstrated to restore dysfunctions of oligodendrocyte precursor cells (OPCs) under hypoxic conditions via the AMPK-PHD-HIF-1α pathway. The findings shed light on re-purposing metformin to treat VCID.
Material and methods
Animals
All animal experiments were performed in adherence with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Naval Medical University Committee, the Shanghai Zhongshan Hospital Committee on Animal Care, and the ARRIVE guidelines (Animal Research: Reporting In Vivo Experiments, www.nc3rs.org.uk/arrive-guidelines). C57BL/6 mice were purchased from Shanghai Jihui Laboratory Animal Co., Ltd. (Shanghai, China), maintained under specific pathogen-free conditions, and used at 10–12 weeks of age. We anesthetized mice with pentobarbital, and then the whole brains were collected following transcardial perfusion and overnight post-fixation with 4% paraformaldehyde. The corpus callosum between 0.86 mm and 0.62 mm from the bregma was used for analysis.
Bilateral carotid artery stenosis (BCAS) surgery
The BCAS model was established as previously described. 21 Briefly, male mice were deeply anesthetized with 1.5% isoflurane in 30% oxygen. After exposing both common carotids from their sheaths, a microcoil with a 0.18 mm inner diameter (Bejing Cinontech Co. Ltd., China) was wound around the common carotid. Another microcoil of the same size was wound around the other common carotid 30 minutes later.
Drug administration
Metformin administration refers to a previous study. 22 In the BCAS + Met group, mice received 5 mg/ml metformin (Sigma, Catalog#D150959) in the drinking water, and metformin administration began one day following surgery. The sham group and BCAS+Veh group were treated with drinking water without metformin. Drinking bottles were replaced with fresh metformin solution every week. For use of inhibitors in vivo, 0.9% saline was used as the vehicle. DMOG (Sigma, Catalog#400091) and dorsomorphin (Sigma, Catalog#P5499) were dissolved in 5% DMSO in saline, respectively, stored in −20°C, and diluted in saline immediately before intraperitoneal injection (i.p.). Mice were administrated with DMOG 8 mg/kg and dorsomorphin 2.5 mg/kg via the i.p. route at 2-day intervals for 60 days.
Cerebral blood flow (CBF) measurement and behavioral test
CBF was measured using a laser-speckle blood flow imaging system (RFLSI Pro, RWD Life Science, China) as described. 23 Open field test, 24 Y‐Maze test 25 and novel object recognition test 26 was performed as previously reported. More detailed description was provided in supplementary information for methods and material.
Cell culture, Western blotting and immunostaining
The primary culture and purification of OPCs were performed as previously described with some modifications. 27 To mimic a chronic mild-hypoxic condition, OPCs were incubated with cobalt chloride. 28 Luxol Fast Blue (LFB) staining was performed as previously described. 29 The Western blotting analysis, immunofluorescence staining and BrdU incorporation were followed standard protocols. More detailed information for antibodies and material was included in supplementary information for methods and material.
Statistical analysis
The data were analyzed using One-way ANOVA with Tukey post hoc test or Dunnett’s multiple comparisons test for pairwise comparisons in multiple groups (for variables with homogenous variance) and Games-Howell (for variables with non-homogenous variance). Student's t-test was used for two groups. The data are presented as mean ± SD. The 95% confidence intervals were set and value of p < 0.05 was considered statistically significant. All statistical analyses were made using Prism 6 (GraphPad) and PASW Statistics 18 (SPSS, IBM).
Results
Metformin alleviates cognitive impairment after chronic cerebral hypoperfusion
Bilateral common carotid artery stenosis (BCAS) model are commonly used to simulate chronic cerebral hypoperfusion, as previously reported.21,29 To test the effectiveness of BCAS modeling, we monitored the cerebral blood flow (CBF) of mice before and after surgery (Figure 1(a) to (c)). At 10 minutes after modeling, the CBF of the mice decreased significantly (Baseline: 364.27 ± 18.50 PU, Immediately: 120.60 ± 13.67 PU, Figure 1(d), confidence interval: [−268.93, −218.41], p < 0.001). Subsequently, the CBF gradually recovered on 2, 7 and 14 days after surgery. By 30 days after surgery, CBF had returned to about 80% of the level before surgery (Day 30: 293.25 ± 13.36 PU, confidence interval: [−96.28, −45.76], p < 0.001). To examine the effect of metformin on chronic hypoperfusion in mice, we performed BCAS surgery on adult mice, which were then treated which metformin (BCAS + Met) or vehicle control (BCAS + Veh) for 60 days (Figure 1(a)). At 30 and 60 days after surgery, the CBF of mice in both groups was lower than that in the sham group (Figure 1(e)), but there was no significant difference between the BCAS + Veh group and the BCAS + Met group (Day 30, BCAS + Veh vs BCAS + Met: 291.58 ± 12.52 PU vs 302.12 ± 12.81 PU, confidence interval: [−10.78, 31.85], p > 0.05; Day 60, BCAS + Veh vs BCAS + Met: 331.02 ± 9.45 PU vs 337.88 ± 7.93 PU, Figure 1(f) confidence interval: [−9.95, 23.67], p > 0.05). These results suggest that metformin treatment has no significant effect on long-term blood flow restoration in BCAS.
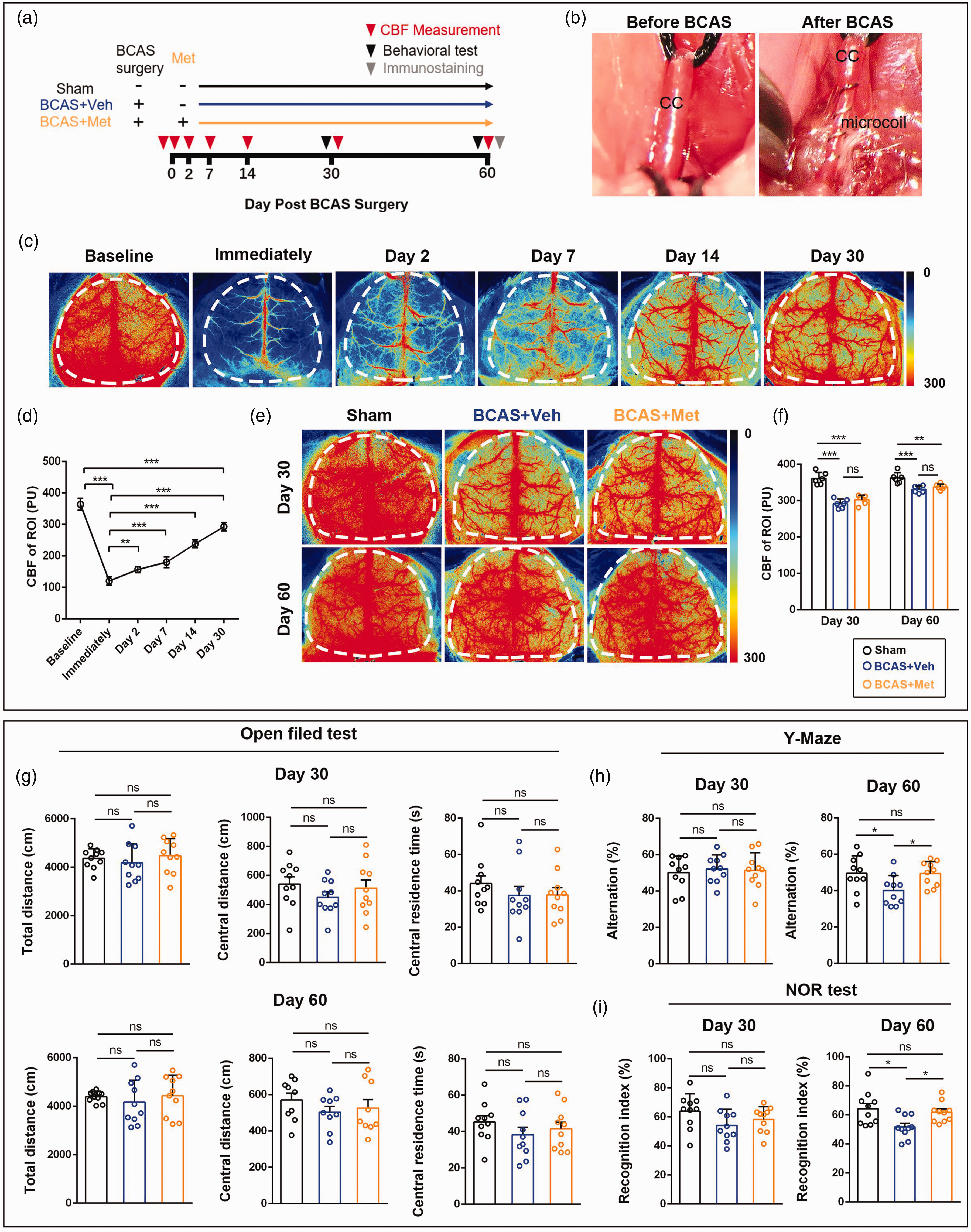
Metformin administration attenuates cognitive impairment in BCAS mice. (a) Schematic diagram of BCAS modeling and metformin treatment. (b) BCAS was established by encircling the bilateral common carotid with microcoils. CC, Common Carotid. (c, d) Representative images and quantitative analysis of CBF at different time points after BCAS. Regions of interest were identified with dotted lines. (e, f) Representative images and quantitative analysis of CBF on Day 30 and Day 60 in different groups. Data are presented as mean ± SD (n = 6 mice). (g) Open field test. (h) Y-maze test and (i) Novel object recognition test. Data are presented as mean ± SD (n = 10 mice). *p < 0.05, ***p < 0.001, ***p < 0.001.
To assess the effect of metformin on the cognitive function of BCAS mice, we performed a set of behavioral tests on mice 30 days and 60 days after surgery (Figure 1(a)). We first detected the spontaneous activity of mice in the open field. The results showed that there was no significant difference in the total movement distance among different groups, suggesting that BCAS or metformin treatment did not affect the motor activity of mice (Figure 1(g)). In addition, no significant changes in the movement distance and time in the central area were observed among the three groups, indicating that BCAS or metformin treatment did not induce anxiety-like behaviors (Figure 1(g)). Next, we performed the Y-maze test to evaluate the short-term working memory of mice. The results showed that there was no significant difference in the alternations among the three groups 30 days after surgery (Figure 1(h)). However, the alternation rate in the BCAS + Veh group (40.12 ± 8.23%) was significantly reduced compared with that in the sham group (49.51 ± 9.67%) 60 days after surgery (Figure 1(h), confidence interval: [−18.59, −0.17], p < 0.05). At the same time, metformin treatment significantly increased the alternation rate in BCAS + Met mice (49.34 ± 6.80%) compared with BCAS + Veh mice (Figure 1(h), confidence interval: [0.01, 18.44], p < 0.05). We also examined the recognition ability of mice by the novel object recognition test. The BCAS + Veh group had a lower recognition index (51.78 ± 7.90%) compared with the sham group (64.28 ± 11.42%) 60 days after surgery (Figure 1(i), confidence interval: [−22.45, −2.54], p < 0.05). Notably, the recognition index significantly increased in BCAS + Met mice (61.82 ± 7.01%) compared with BCAS + Veh mice (Figure 1(i), confidence interval: [0.08, 19.99], p < 0.05). These results suggest that chronic cerebral hypoperfusion cause cognitive decline and that metformin treatment could alleviate impairment.
Metformin ameliorates white matter lesions induced by chronic cerebral hypoperfusion
Myelin is vulnerable to ischemia leading to white matter damage and cognitive dysfunction.3,30 To examine the influence of BCAS on the white matter, we performed Luxol Fast Blue (LFB) staining in the corpus callosum of the mice 60 days after surgery. The results showed that the integrity of white matter in the corpus callosum was impaired after BCAS, mainly manifested as myelin loss, nerve fiber disturbance, and increased vacuolization (Figure 2(a)). The white matter damage score in the BCAS + Veh group (2.17 ± 0.75) was significantly increased compared with the sham group (Figure 2(b), confidence interval: [1.32, 3.02], p < 0.001). However, the white matter damage score in the BCAS + Met group (1.00 ± 0.63) was lower than that in the BCAS + Veh group (Figure 2(b), confidence interval: [−2.02, −0.32], p < 0.01). FluoroMyelin staining confirmed that the myelin density in the corpus callosum of the BCAS + Veh group was only 52.03 ± 3.03% of the sham group (Figure 2(c) and (d), confidence interval: [−52.75, −43.20], p < 0.001). Notably, the decrease of myelin density after BCAS was improved via metformin treatment (71.14 ± 4.60% of the sham group, Figure 2(d), confidence interval: [14.34, 23.89], p < 0.001). In addition, we performed immunofluorescence staining for myelin basic protein (MBP), the marker for mature myelin (Figure 2(e)). We found that the mean fluorescence intensity (MFI) of MBP decreased in the BCAS + Veh group (56.39 ± 3.08% of the sham group, Figure 2(f), confidence interval: [−47.70, −39.53], p < 0.001). After metformin treatment, the MFI of MBP recovered to 75.25 ± 4.78% of the sham group, which was higher than that of the BCAS + Veh group (Figure 2(f), confidence interval: [12.31, 25.41], p < 0.001). The MFI of neurofilament-200 (NF200) decreased after BCAS, suggesting mild axon loss, but there was no difference between the BCAS + Veh group (85.65 ± 3.25% of the sham group) and the BCAS + Met group (89.58 ± 2.90% of the sham group, Figure 2(e) and (f), confidence interval: [−0.96, 8.82], p > 0.05). Meanwhile, the MFI of MBP normalized to NF200 further confirmed that the density of myelinated axons decreased after BCAS (65.98 ± 5.48% of the sham group, Figure 2(f), confidence interval: [−41.30, −26.73], p < 0.001). Although the loss of myelinated axons did not recover to normal in the BCAS + Met group (81.91 ± 5.48% of the sham group, confidence interval: [−22.84, −13.33], p < 0.001), it was still higher than that in the BCAS + Veh group (confidence interval: [8.40, 23.46], p < 0.001). Taken together, these results indicated that metformin could alleviate the white matter damage induced by chronic cerebral hypoperfusion.
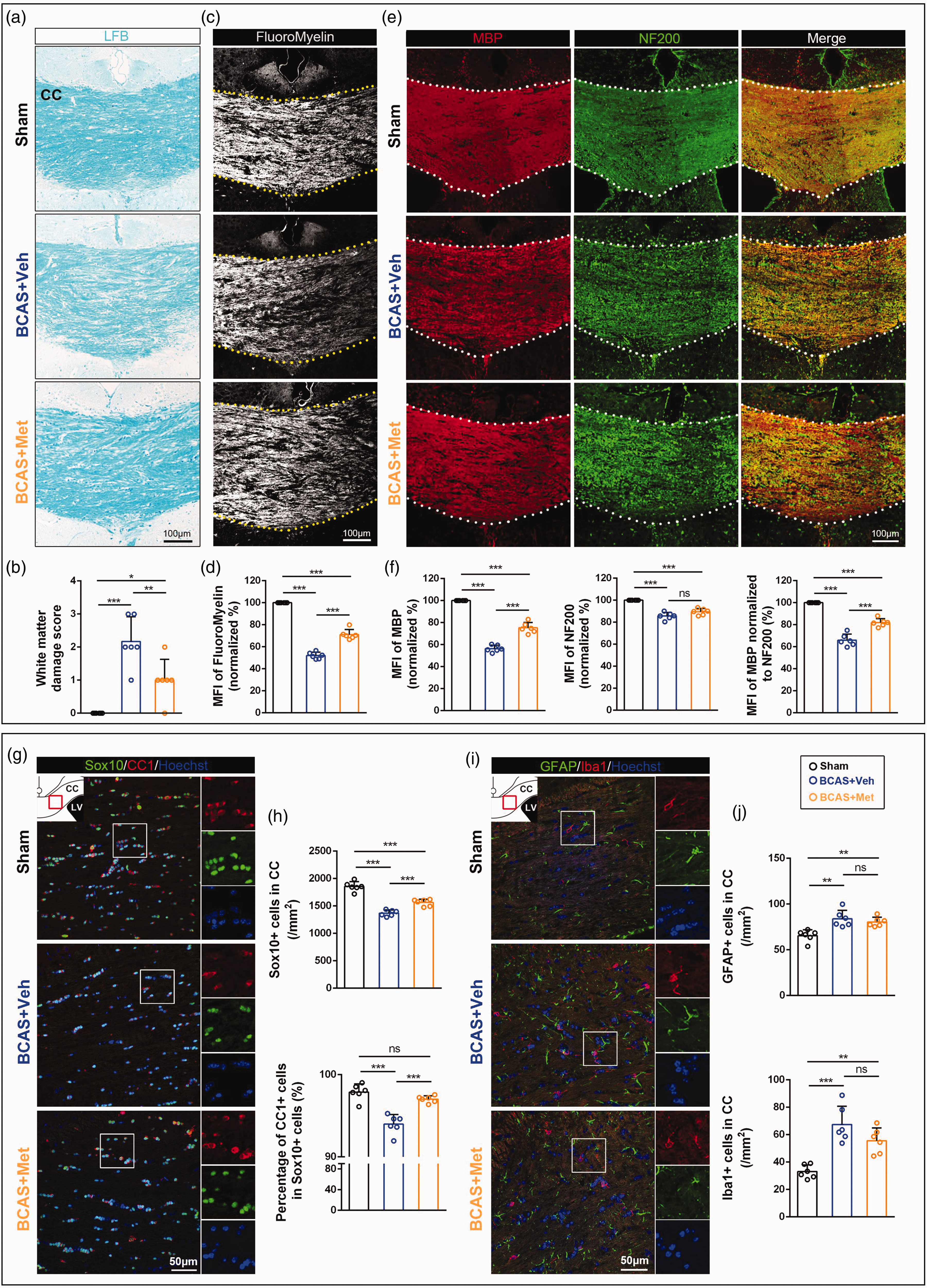
Metformin ameliorates white matter lesions in the corpus callosum at 60 days post-BCAS. (a) Representative images of LFB staining at the corpus callosum. CC, corpus callosum. (b) Quantitative analysis of the severity of white matter damage. (c) Representative graphs of FluoroMyelin staining at the corpus callosum. (d) Quantitative analysis of the MFI of FluoroMyelin. MFI, mean fluorescence intensity. (e) Representative images of MBP (red) and NF200 (green) fluorescence at the corpus callosum. (f) Quantitative analysis of the MFI of MBP, NF200, and MBP normalized to NF200. (g) Representative images of Sox10+ (green) and CC1+ (red) cells in the corpus callosum. (h) Quantitative analysis of the number of Sox10+ cells per unit area in the corpus callosum and the proportion of CC1+ cells in Sox10+ cells. (i) Representative images of GFAP+ (green) and Iba1+ (red) cells in the corpus callosum and (j) Quantitative analysis of the number of GFAP+ cells and Iba1+ cells per unit area in the corpus callosum. All data are presented as mean ± SD (n = 6 mice). *p < 0.05, **p < 0.01, ***p < 0.001.
In the central nervous system, myelin is formed by oligodendrocytes which are derived from the differentiation and maturation of oligodendrocyte precursor cells (OPCs). To understand the underlying mechanism of metformin effect on white matter lesions, we further explored the oligodendroglial basis in the corpus callosum of mice 60 days post-BCAS (Figure 2(g)). We used adenomatous polyposis coli (APC aka CC1) and SRY-Box Transcription Factor 10 (Sox10) to mark mature oligodendrocytes and found that the total number of oligodendrocyte linage cells (Sox10+ cells) was significantly decreased in the BCAS + Veh group (1370.54 ± 55.38 cells/mm2) compared with the sham group (1855.94 ± 87.07 cells/mm2, Figure 2(h), confidence interval: [−589.49, −381.32], p < 0.001), and the percentage of mature oligodendrocytes (CC1+/Sox10+ cells) also reduced significantly after BCAS (93.98 ± 1.17%) compared with the sham group (97.89 ± 0.99%, Figure 2(h), confidence interval: [−5.28, −2.54], p < 0.001). Intriguingly, metformin treatment could increase the density of Sox10+ cells (1562.51 ± 61.67 cells/mm2) versus the BCAS + Veh group (Figure 2(h), confidence interval: [87.89, 296.06], p < 0.001), and the proportion of CC1+ cells in Sox10+ cells also increased (97.02 ± 0.41%, Figure 2(h), confidence interval: [1.67, 4.42], p < 0.001), indicating the involvement in the regulation of oligodendroglial differentiation and maturation. Astrogliosis and microglial activation are characteristic pathologies in BCAS-induced white matter lesions. 23 In our results, the BCAS + Veh group showed more Glial fibrillary acidic protein (GFAP) positive cells (83.92 ± 9.17 cells/mm2) and Ionized calcium-binding adaptor protein-1 (Iba1) positive cells (67.46 ± 13.26 cells/mm2) than the sham group (GFAP+ cells: 65.48 ± 6.37 cells/mm2, Figure 2(i) and (j), confidence interval: [7.81, 29.07], p < 0.01; Iba1+ cells: 33.05 ± 4.87 cells/mm2, confidence interval: [19.77, 49.06], p < 0.001). Although the density of these cells numerically lowered in the BCAS + Met group (GFAP+ cells: 80.47 ± 5.10 cells/mm2; Iba1+ cells: 55.65 ± 9.30 cells/mm2) compared with the BCAS + Veh group, a significant statistical difference was not observed (Figure 2(j), GFAP+ cells: confidence interval: [−14.08, 7.18], p > 0.05; Iba1+ cells: confidence interval: [−26.46, 2.83], p > 0.05), suggesting sustained glial activation in the lesions.
Metformin promotes OPCs differentiation, survival, and proliferation in vitro
To further investigate the effect of metformin on OPCs in vitro, we treated the primary cultured OPCs with different concentrations of metformin in the differentiation medium for 72 hours. 10 ng/ml thyroid-hormone 3 (T3) was used as a positive control. The results showed that 10 µM metformin could effectively promote the expression of MBP and myelin associated glycoprotein (MAG) in the primary cultures compared with the metformin-free group (2.59 ± 0.67-fold for MBP, Figure 3(a) and (b), confidence interval: [0.61, 2.58], p < 0.001; 1.46 ± 0.09-fold for MAG, confidence interval: [0.02, 0.89], p < 0.05), so we chose 10 µM metformin concentration in the following study. Consistently, MBP+ staining was greatly increased in the cultures treated with 10 µM metformin (1.30 ± 0.04 mm2) compared with the control group (0.69 ± 0.06 mm2, Figure 3(c) and (d), confidence interval: [0.51, 0.72], p < 0.001). We also found that metformin inhibited OPCs apoptosis during differentiation, as the proportion of Caspase3+ cells in the metformin group (1.43 ± 0.19%) was lower than that in the control group (3.04 ± 0.28%, Figure 3(c) and (d), confidence interval: [−1.92, −1.31], p < 0.001). In addition, we examined the effect of metformin on OPCs proliferation and found that the number of BrdU+ cells in the metformin group (51.85 ± 2.35%) was increased compared with the control group (46.53 ± 1.71%, Figure 3(e) and (f), confidence interval: [2.67, 7.97], p < 0.01).
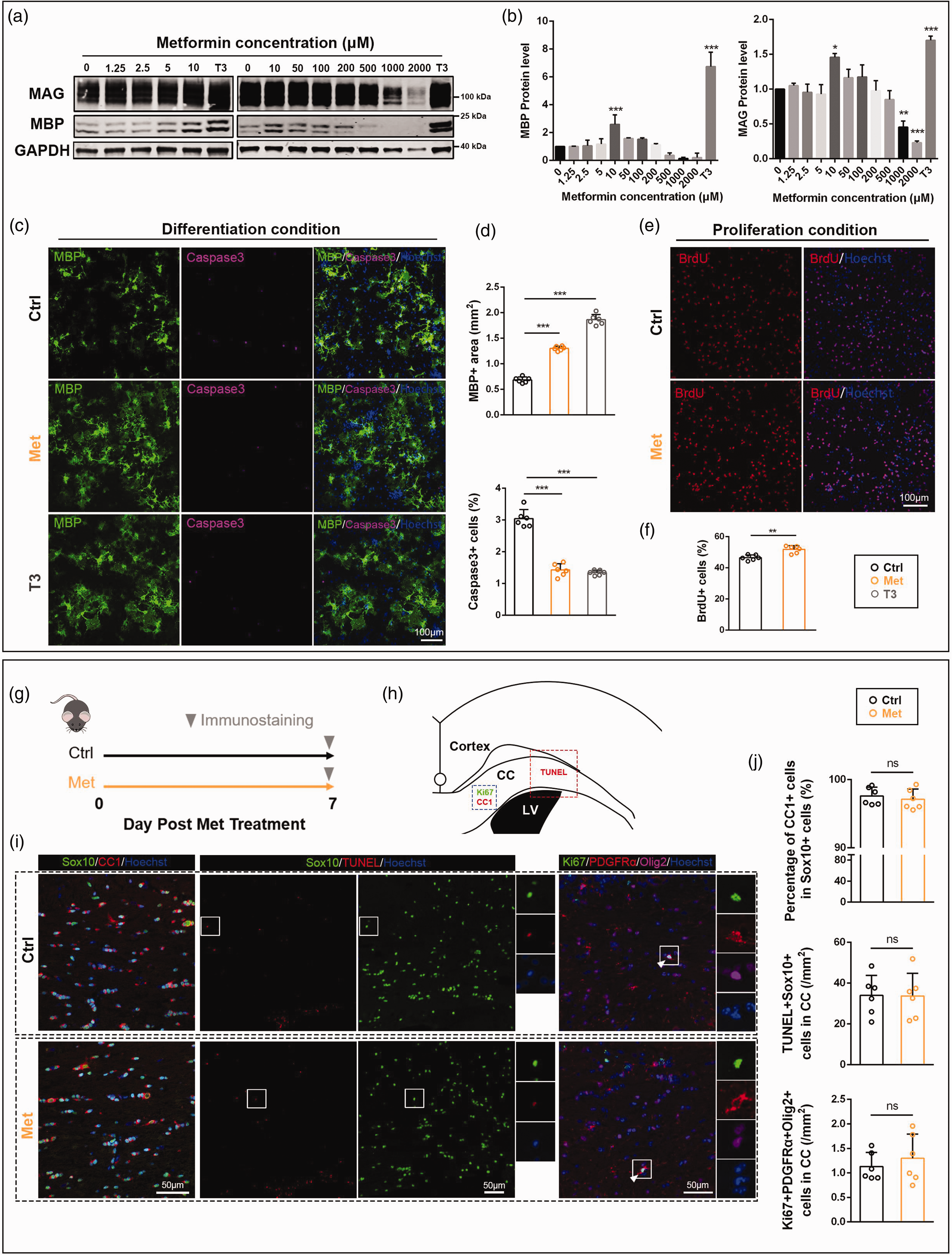
The effect of metformin on OPCs in vitro and in vivo. (a) Western blotting analysis of MBP and MAG expression in OPCs under differentiation conditions. GAPDH severs as an internal positive control. (b) Quantitative analysis of MBP and MAG expression. (c) Representative images of MBP+ (green) and Caspase3+ (purple) cells under differentiation conditions. (d) Quantitative analysis of the MBP+ staining signals and the proportion of Caspase3+ cells. (e) Representative images of BrdU+ (red) cells under proliferative conditions. (f) Quantitative analysis of the proportion of BrdU+ cells. (g) Schematic diagram of metformin treatment. (h) Immunofluorescence region of the corpus callosum. (i) Representative images of CC1+ (red) and Sox10+ (green) cells; TUNEL+ (red) and Sox10+ (green) cells; Ki67+ (green), PDGFRα+ (red) cells, and Olig2+ (purple) cells and (j) Quantitative analysis of the proportion of CC1+ cells in Sox10+ cells, the number of TUNEL+Sox10+ cells per unit area, and the number of Ki67+PDGFRα+Olig2+ cells per unit area. All data are presented as mean ± SD (n = 3 biological replicates for WB, n = 6 biological replicates or 6 mice for IF). *p < 0.05, **p < 0.01, ***p < 0.001.
Furthermore, normal control mice were treated with metformin for 7 consecutive days to observe the effect of metformin on OPCs in vivo (Figure 3(g)). Then, we used CC1 and Sox10 to mark mature oligodendrocytes, Ki67, PDGF Receptor α (PDGFRα) and oligodendrocyte lineage transcription factor 2 (Olig2) to mark proliferative OPCs, and TUNEL and Sox10 to mark apoptotic oligodendrocyte lineage cells (OLs) in the immunostaining study. Our results showed that metformin treatment had little effect on differentiation, proliferation and apoptosis of OPCs in white matter of adult mice, which was different from the effect of metformin on OPCs in vitro (Figure 3(h) to (j)).
Metformin ameliorates the dysfunction of OPCs under hypoxic conditions
To further examine the effect of metformin on OPCs under hypoxic conditions, we added 100 µM CoCl2 to primary cultured OPCs to mimic the hypoxic state of prolonged cerebral hypoperfusion. The western blotting analysis showed that hypoxia inhibited OPCs differentiation (Figure 4(a)), as manifested in a significant decrease in the expression of MBP and MAG (0.10 ± 0.06-fold for MBP, Figure 4(b), confidence interval: [−1.04, −0.76], p < 0.001; 0.07 ± 0.01-fold for MAG, confidence interval: [−1.27, −0.58], p < 0.001), while metformin treatment could improve OPCs differentiation under hypoxia (0.56 ± 0.07-fold for MBP, Figure 4(b), confidence interval: [0.32, 0.60], p < 0.001; 0.59 ± 0.24-fold for MAG, confidence interval: [0.17, 0.86], p < 0.01). Consistently, metformin treatment also increased MBP+ signal area under hypoxia (CoCl2 +Met vs CoCl2: 0.31 ± 0.02 mm2 vs 0.18 ± 0.04 mm2, Figure 4(c) and (d), confidence interval: [0.07, 0.19], p < 0.001).
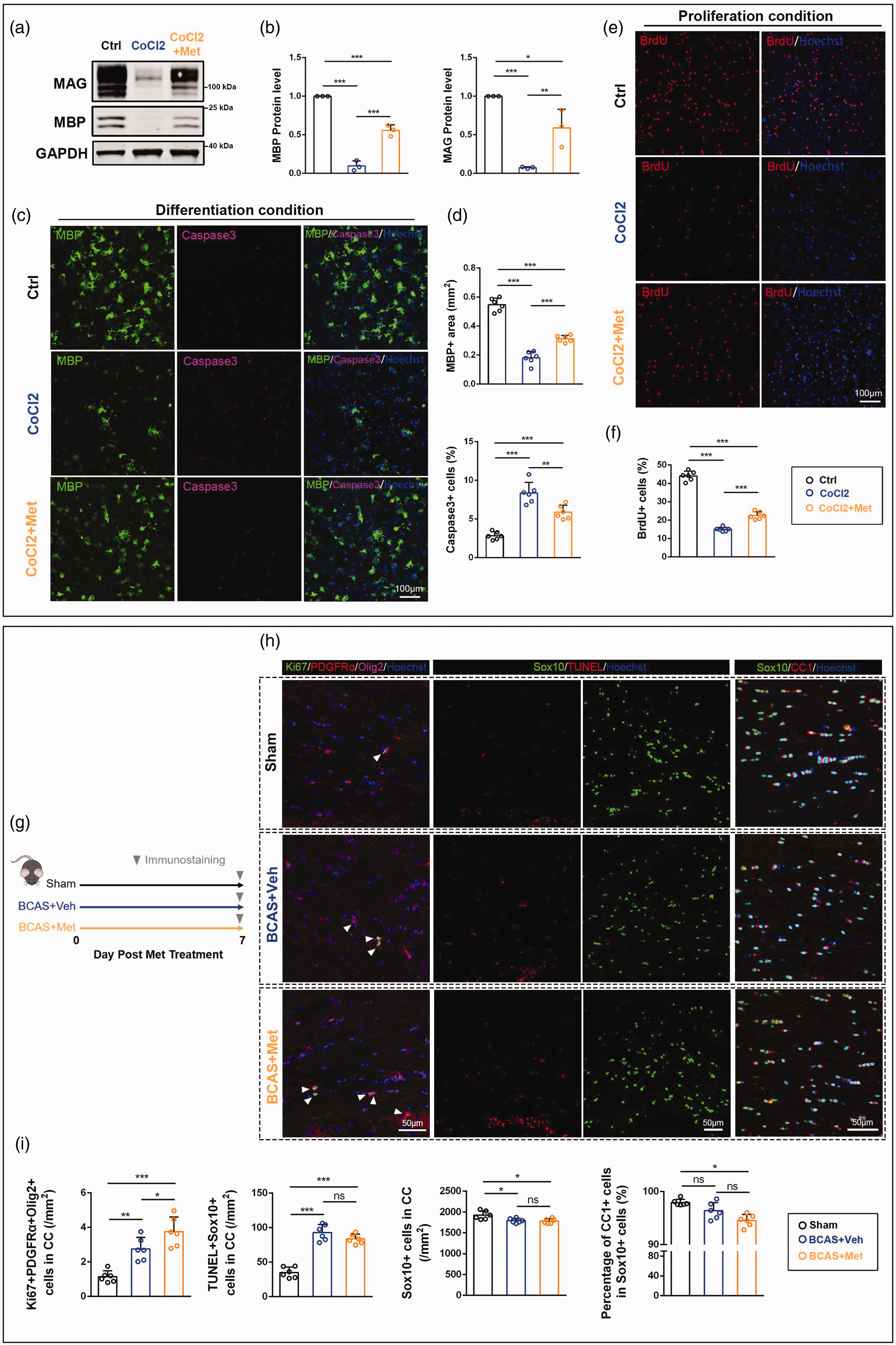
Metformin attenuates hypoxia-induced OPCs dysfunction. (a) Western blotting of MBP and MAG in OPCs under differentiation conditions. CoCl2, cultures with 100 μM CoCl2; CoCl2+Met, cultures with 100 μM CoCl2 and 10 μM metformin. GAPDH Continued.severs as an internal positive control. (b) Quantitative analysis of MBP and MAG expression. (c) Representative images of MBP+ (green) and Caspase3+ (purple) cells under differentiation conditions. (d) Quantitative analysis of MBP+ staining signals and the proportion of Caspase3+ cells. (e) Representative images of BrdU+ (red) cells under proliferative conditions. (f) Quantitative analysis of the proportion of BrdU+ cells. (g) Schematic diagram of BCAS modeling and metformin treatment. (h) Representative images of Ki67+ (green), PDGFRα+ (red) cells, and Olig2+ (purple) cells; TUNEL+ (red) and Sox10+ (green) cells; CC1+ (red) and Sox10+ (green) cells and (i) Quantitative analysis of the number of Ki67+PDGFRα+Olig2+ cells per unit area, the number of TUNEL+Sox10+ cells per unit area, the number of Sox10+ cells per unit area, and the proportion of CC1+ cells in Sox10+ cells. All data are presented as mean ± SD (n = 3 biological replicates for WB, n = 6 biological replicates or 6 mice for IF). *p < 0.05, **p < 0.01, ***p < 0.001.
Meanwhile, hypoxia also enhanced OPCs apoptosis, as revealed by the increased proportion of Caspase3+ cells (8.37 ± 1.38%) compared with the control group (2.85 ± 0.50%, Figure 4(c) and (d), confidence interval: [4.03, 7.01], p < 0.001). Metformin could reduce the proportion of Caspase3+ cells under hypoxia (5.88 ± 0.90%, confidence interval: [−3.97, −1.00], p < 0.01). Under proliferative conditions, hypoxia resulted in a significant decrease in the proportion of BrdU+ cells (CoCl2 vs Ctrl: 14.99 ± 1.13% vs 44.04 ± 2.59%, Figure 4(e) and (f), confidence interval: [−32.09, −26.02], p < 0.001), while it could be improved by metformin (22.52 ± 2.06%, confidence interval: [4.50, 10.57], p < 0.001).
Subsequently, we observed the effect of metformin on OPCs function in vivo at 7 days post-BCAS (Figure 4(g)). At the beginning of BCAS, white matter OLs reacted with increased number of proliferating OPCs (BCAS + Veh vs Sham: 2.75 ± 0.67 cells/mm2 vs 1.15 ± 0.34 cells/mm2, Figure 4(h) and (i), confidence interval: [0.63, 2.58], p < 0.01) and apoptosis of OLs (BCAS + Veh vs Sham: 92.90 ± 11.62 cells/mm2 vs 35.13 ± 7.76 cells/mm2, Figure 4(h) and (i), confidence interval: [41.68, 73.87], p < 0.001). At the same time, more OPCs proliferated in the BCAS + Met group (3.76 ± 0.85 cells/mm2) than in the BCAS + Veh group (confidence interval: [0.03, 1.98], p < 0.05). Metformin treatment did not seem to reduce the BCAS-induced apoptotic OLs which was most likely the main reason for the decrease in Sox10+ cell numbers (Figure 4(h) and (i)). However, the proportion of mature oligodendrocytes decreased in both BCAS + Veh group and BCAS + Met group at 7 days post-BCAS (Figure 4(h) and (i)), which may be related to the increase of reactive OPCs.
Taken together, these results indicated that the hypoxia-induced dysfunction of OPCs could ameliorated by metformin treatment in vitro and in vivo.
Metformin restores the functions of OPCs under hypoxic conditions via the AMPK-PHD-HIF-1α pathway in vitro
Previous studies have shown that hypoxia-inducible factor-1α (HIF-1α) is the master regulator of the cellular response to hypoxia.31,32 Therefore, we subsequently assessed the expression of HIF-1α in OPCs under hypoxic conditions. Compared with the control group, the western blotting results showed that CoCl2 treatment enhanced HIF-1α expression (1.91 ± 0.35-fold, Figure 5(a) and (b), confidence interval: [0.36, 1.46], p < 0.01) and this increase was attenuated by metformin (1.19 ± 0.13-fold, confidence interval: [−1.27, −0.18], p < 0.05). Prolyl hydroxylases 2 (PHD2) is responsible for the hydroxylation of HIF-1α. 33 In agreement, we found that CoCl2 downregulated PDH2 (0.23 ± 0.02-fold, Figure 5(a) and (b), confidence interval: [−0.83, −0.71], p < 0.001). However, under metformin treatment, the level of PHD2 was increased (0.35 ± 0.04-fold, confidence interval: [0.06, 0.18], p < 0.01).
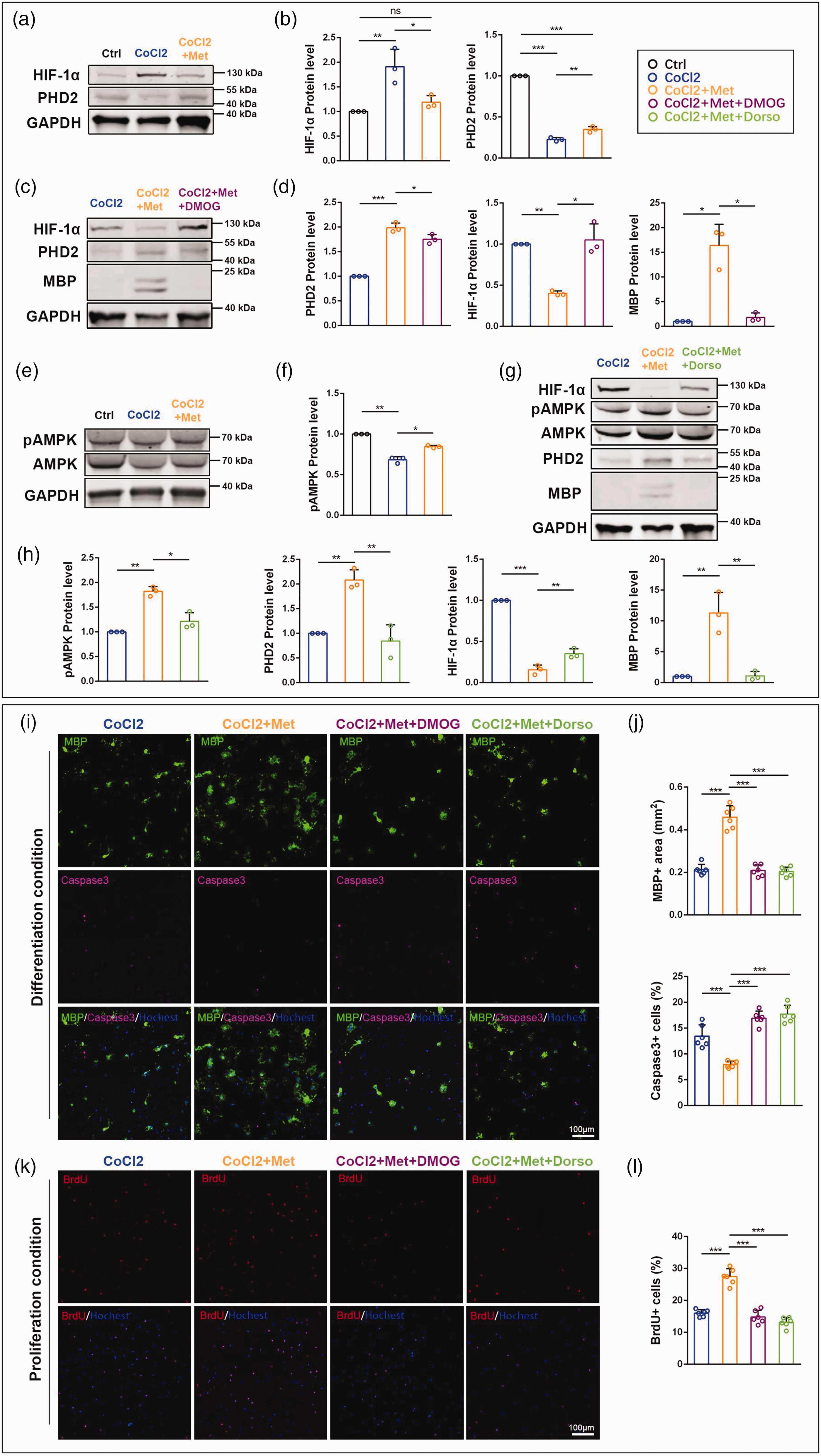
Metformin restores the functions of OPCs under hypoxic conditions by AMPK-PHD-HIF-1α pathway. (a) Western blotting of HIF-1α and PHD2. GAPDH severs as an internal positive control. (b) Quantitative analysis of HIF-1α and PHD2 expression. (c) Western blotting of PHD2, HIF-1α, and MBP. (d) Quantitative analysis of PHD2, HIF-1α, and MBP expression. (e) Western blotting Continued.of AMPK and pAMPK. (f) Quantitative analysis of pAMPK expression. (g) Western blotting of AMPK, pAMPK, PHD2, HIF-1α, and MBP. Dorso, dorsomorphin. (h) Quantitative analysis of pAMPK, PHD2, HIF-1α, and MBP expression. (i) Representative images of MBP+ (green) and Caspase3+ (purple) cells under differentiation conditions. (j) Quantitative analysis of MBP+ staining signals and the proportion of Caspase3+ cells. (k) Representative images of BrdU+ (red) cells under proliferative conditions and (l) Quantitative analysis of the proportion of BrdU+ cells. All data are presented as mean ± SD (n = 3 biological replicates for WB, n = 6 mice for IF). *p < 0.05, **p < 0.01, ***p < 0.001.
To investigate whether PHD2 is required in HIF-1α inhibition under metformin treatment in response to hypoxia, OPCs were treated with dimethyloxalylglycine (DMOG, an inhibitor of PHDs activity). Compared with the CoCl2 + Met group (1.99 ± 0.10-fold), the western blotting results showed a decrease of PHD2 in the CoCl2 + Met + DMOG group (1.75 ± 0.10-fold, Figure 5(c) and (d), confidence interval: [−0.43, −0.04], p < 0.05), suggesting the validity of DMOG. At the same time, DMOG treatment (1.05 ± 0.19-fold) also abrogated metformin-mediated HIF-1α reduction (0.40 ± 0.03-fold, Figure 5(c) and (d), confidence interval: [0.01, 1.29], p < 0.05), and the expression of MBP in the CoCl2 + Met + DMOG group (1.82 ± 0.89-fold) was significantly lower than that in the CoCl2 + Met group (16.41 ± 4.27-fold, Figure 5(c) and (d), confidence interval: [−28.20, −0.99], p < 0.05). These results suggested that metformin treatment elevated the PHD2 level, thereby leading to the HIF-1α decrease and relieving the blockage of OPCs differentiation.
Metformin functions through both AMP-activated protein kinase (AMPK) -dependent and AMPK-independent pathways. 12 As metformin has been reported to rejuvenate aging OPCs mainly via an AMPK-dependent mechanism, 34 we next examined whether metformin regulated AMPK activation in OPCs under hypoxic conditions (Figure 5(e)). Our results showed that metformin restored AMPK pathway inhibition in OPCs under hypoxia, as the levels of phosphorylated AMPK (pAMPK) was upregulated in the CoCl2 + Met group (0.85 ± 0.02-fold) compared with the CoCl2 group (0.68 ± 0.04-fold, Figure 5(f), confidence interval: [0.05, 0.27], p < 0.05).
To test whether AMPK signaling is involved in the regulation of the expression of HIF-1α and PHD2 in OPCs under hypoxic conditions, we treated the culture with dorsomorphin, a small-molecule inhibitor of AMPK activation. Western blotting results showed that inhibiting AMPK activation by dorsomorphin (CoCl2 + Met + Dorso vs CoCl2 + Met: 1.22 ± 0.17-fold vs 1.82 ± 0.10-fold, Figure 5(g) and (h), confidence interval: [−1.07, −0.14], p < 0.05) abrogated metformin-mediated PHD2 increase (CoCl2 + Met + Dorso vs CoCl2 + Met: 0.85 ± 0.33-fold vs 2.08 ± 0.21-fold, confidence interval: [−1.80, −0.67], p < 0.01), enhanced HIF-1α expression (CoCl2 + Met + Dorso vs CoCl2 +Met: 0.35 ± 0.06-fold vs 0.16 ± 0.06-fold, confidence interval: [0.08, 0.31], p < 0.01), and inhibited MBP expression (CoCl2 + Met + Dorso vs CoCl2 + Met: 1.09 ± 0.74-fold vs 11.28 ± 3.33-fold, confidence interval: [−15.12, −5.25], p < 0.01).
Subsequently, when cells were treated with DMOG or dorsomorphin, the immunofluorescence staining revealed that OPCs differentiation decreased (CoCl2 + Met + DMOG group vs CoCl2 + Met: 0.21 ± 0.03 mm2 vs 0.46 ± 0.05 mm2, Figure 5(i) and (j), confidence interval: [−0.33, −0.17], p < 0.001; CoCl2 + Met + Dorso group vs CoCl2 + Met: 0.20 ± 0.02 mm2 vs 0.46 ± 0.05 mm2, confidence interval: [−0.33, −0.18], p < 0.001), apoptosis increased (CoCl2 + Met + DMOG group vs CoCl2 + Met: 16.95 ± 1.35% vs 7.98 ± 0.62%, Figure 5(i) and (j), confidence interval: [6.42, 11.51], p < 0.001; CoCl2 + Met + Dorso group vs CoCl2 + Met: 17.74 ± 1.70% vs 7.98 ± 0.62%, confidence interval: [7.21, 12.30], p < 0.001), and proliferation was reduced (CoCl2 + Met + DMOG group vs CoCl2 + Met: 14.86 ± 2.05% vs 27.49 ± 2.50%, Figure 5(k) and (l), confidence interval: [−15.65, −9.60], p < 0.001; CoCl2 + Met + Dorso group vs CoCl2 + Met: 13.14 ± 1.55% vs 27.49 ± 2.50%, confidence interval: [−17.37, −11.33], p < 0.001). Taken together, these results suggested that metformin restored the OPCs functions under hypoxia through the AMPK-PHD-HIF-1α pathway.
AMPK-PHD-HIF-1α pathway is involved in the ameliorative effects of metformin on OPCs dysfunction induced by BCAS
To test whether the AMPK-PHD-HIF-1α pathway was involved in metformin-improved OPCs function in vivo, we injected DMOG and dorsomorphin intraperitoneally into mice treated with metformin after BCAS.
We performed immunofluorescence staining to detect the effect of inhibitors on OPCs function (Figure 6(a)). The results showed that the metformin-induced OPCs proliferation in corpus callosum was inhibited after the injection of DMOG and dorsomorphin, respectively (BCAS + Met + DMOG group vs BCAS + Met group: 2.53 ± 0.50 cells/mm2 vs 3.19 ± 0.35 cells/mm2, Figure 6(b) and (c), confidence interval: [−1.22, −0.10], p < 0.05; BCAS + Met + Dorso group vs BCAS + Met group: 2.07 ± 0.49 cells/mm2 vs 3.19 ± 0.35 cells/mm2, confidence interval: [−1.66, −0.56], p < 0.01). However, compared with metformin, DMOG or dorsomorphin administration did not seem to affect OLs apoptosis (Figure 6(b) and (d)). Regarding the OPCs differentiation, we found that the proportion of CC1+ cells in Sox10+ cells was reduced after DMOG injection (BCAS +Met + DMOG group vs BCAS + Met group: 95.42 ± 1.11% vs 96.96 ± 0.76%, Figure 6(b) and (e), confidence interval: [−2.76, −0.31], p < 0.05), while dorsomorphin injection was able to reduce mature oligodendrocytes more significantly (BCAS +Met + Dorso group vs BCAS + Met group: 94.29 ± 1.04% vs 96.96 ± 0.76%, Figure 6(b) and (e), confidence interval: [−3.84, −1.49], p < 0.001). Besides, the MFI of MBP was lower in both BCAS + Met +DMOG group (95.84 ± 3.53%) and BCAS + Met + Dorso group (90.81 ± 2.47%) than in BCAS + Met group (Figure 6(b) and (e), BCAS + Met + DMOG group vs BCAS + Met group: confidence interval: [−7.87, −0.45], p < 0.05; BCAS + Met + Dorso group vs BCAS + Met group: confidence interval: [−11.78, −6.60], p < 0.001). Together, inhibition of AMPK and PHD in vivo affected the ameliorative effects of metformin on OPCs proliferation and differentiation, with AMPK appearing to be more important for the therapeutic effects of metformin.
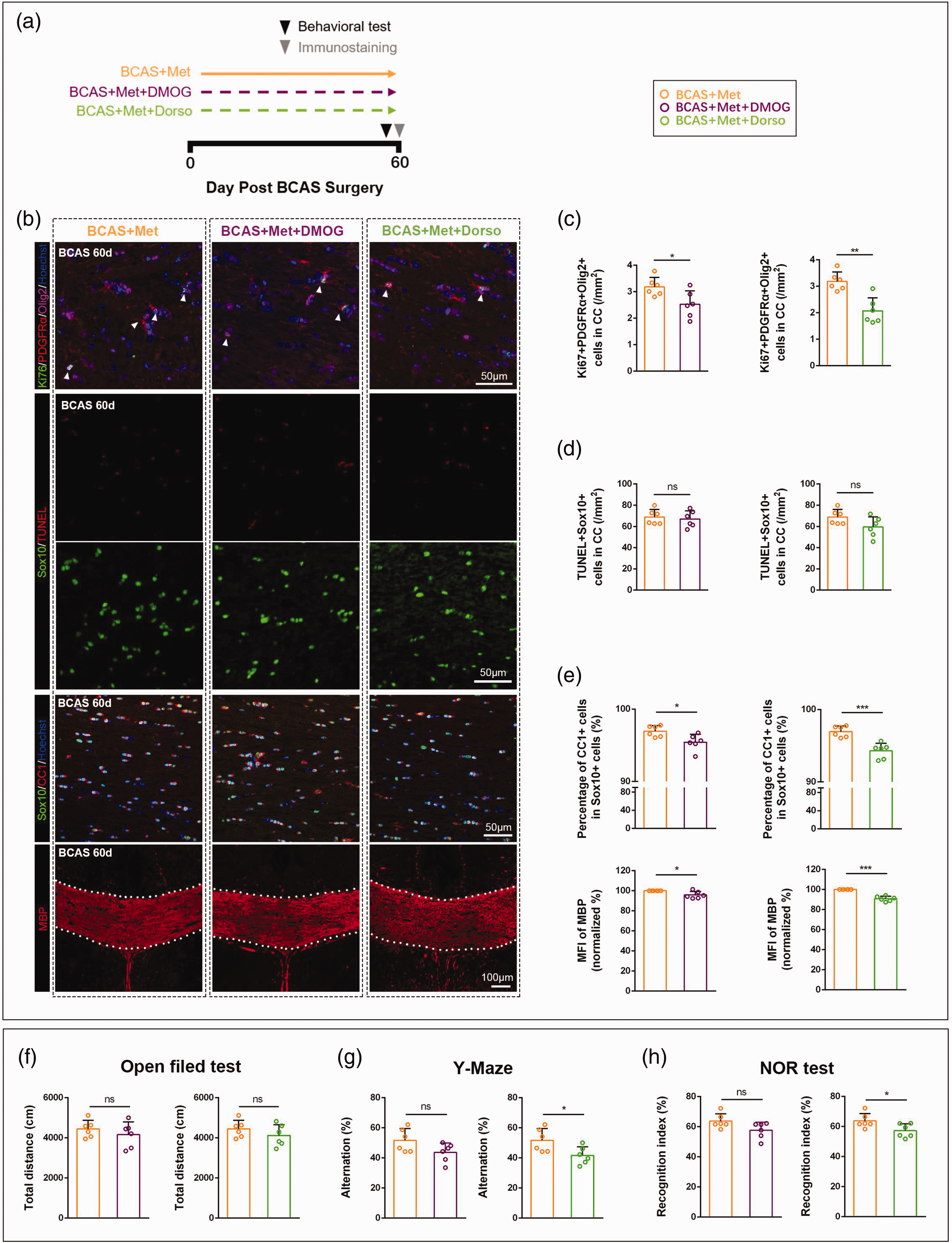
AMPK-PHD-HIF-1α pathway is involved in the ameliorative effects of metformin on OPCs dysfunction induced by BCAS. (a) Schematic diagram of metformin, DMOG, and dorsomorphin administration. (b) Representative immunofluorescence images. (c–e) Quantitative analysis of the number of Ki67+PDGFRα+Olig2+ cells per unit area, the number of TUNEL+Sox10+ cells per unit area, the proportion of CC1+ cells in Sox10+ cells and the MFI of MBP. (f) Open field test. (g) Y-maze test and (h) Novel object recognition test. Data are presented as mean ± SD (n = 6 mice). *p < 0.05, **p < 0.01, ***p < 0.001.
Next, we performed behavioral tests on mice administered with inhibitors. The open field test showed that DMOG or dorsomorphin administration did not affect the motor function of mice (Figure 6(f)). After DMOG administration, mice appeared to reduce short-term working memory capacity, but was not statistically significant. In contrast, after administration of dorsomorphin, the short-term working memory capacity of mice was significantly lower than that of BCAS + Met group (BCAS + Met + Dorso group vs. BCAS + Met group: 41.63 ± 5.78% vs. 51.71 ± 7.77%, Figure 6(g), confidence interval: [−18.89, −1.28], p < 0.05). Moreover, we found that the recognition index of BCAS + Met + Dorso group (57.25 ± 4.55%) was significantly lower than that of BCAS + Met group (63.74 ± 4.90%, Figure 6(h), confidence interval: [−12.58, −0.40], p < 0.05), while there was no difference between BCAS + Met + DMOG group and BCAS + Met group.
Taken together, metformin might regulate BCAS-induced OPCs dysfunction in part through the AMPK-PHD-HIF-1α pathway (Figure 7).
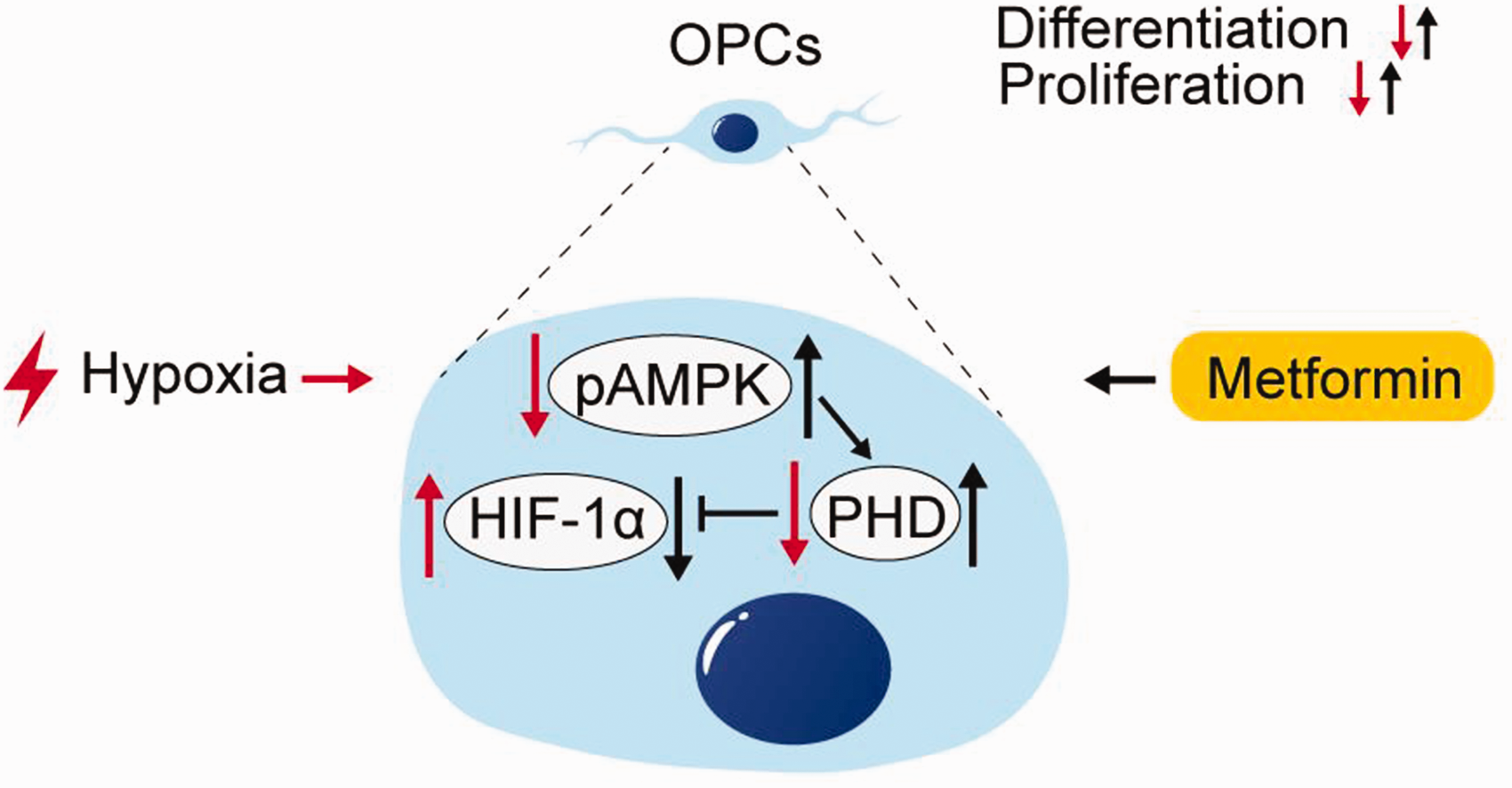
Schematic diagram of metformin restoring OPCs function under hypoxic conditions.
Discussion
The white matter, which connects different cerebral regions, is essential for information processing and integration. White matter comprises myelinated axons and various glial cells, which are nourished by a network of capillaries extending from the perforating or medullary end arteries. 3 The myelin sheaths are derived from the specialized oligodendroglia processes, wrap axons, facilitating saltatory conduction of action potentials and providing metabolically support for axons. 35 Due to the high demand for energy for maintaining the structure of myelin and providing axonal consumption, oligodendrocytes are vulnerable to ischemic insults. 3 White matter lesions caused by chronic cerebral ischemia are associated with a variety of cognitive dysfunction. 36 Indeed, in the BCAS model which is considered to highly mimic the clinical carotid stenosis-induced cerebral hypoperfusion, white matter lesions are demonstrated to be closely related to cognitive impairments.37–39 In this study, we found that working memory significantly declined in mice 60 days after BCAS as examined by the Y-maze test. Meanwhile, the recognition ability of the mice was greatly decreased as detected by the novel object recognition test. Pathological studies revealed that the integrity of white matter in the corpus callosum was impaired after BCAS, and significant myelin loss was shown by FluoroMyelin staining and MBP immunofluorescence staining. Demyelination affects the functioning of neural circuitry and causes neuronal degeneration due to the loss of metabolic supports. 40 As white matter injury is the common pathologic feature of VCID, these results support the notion as promoting myelin repair to benefit cognitive improvement in VCID patients with chronic cerebral ischemia.
In the central nervous system (CNS), in stark contrast to the poor regeneration that occurs following a neuronal injury, myelin repair can occur, although the regeneration process is difficult in many pathological conditions. 41 OPCs, as the major source for generating mature oligodendrocytes during development, are also the main cellular source for remyelination in demyelinating lesions. Upon activation by the demyelination, OPCs undergo proliferation, migration, and finally differentiation into mature oligodendrocytes. 41 However, OPCs in white matter lesions face hostile microenvironments created by hypoxia, activated glial cells, and disintegrated myelin sheath which impedes oligodendroglial differentiation and maturation, leading to failure of remyelination. 42 In the present study, we observed that the number of both OPCs and mature oligodendrocytes were significantly decreased in the corpus callosum of mice 60 days post-BCAS. Further in vitro study revealed that hypoxia enhanced OPCs apoptosis and hindered its proliferation and differentiation, suggesting the inhibition of remyelination in the whiter matter lesions after chronic hypoperfusion. Notably, metformin treatment was identified to ameliorate hypoxia-induced dysfunction of OPCs, which was consistent with the results that metformin administration alleviated the white matter damage after BCAS. Therefore, these results strongly suggest that metformin alleviates white matter damage and cognitive decline through promoting oligodendroglial proliferation and differentiation under hypoxia.
Chronic hypoperfusion provoked hypoxia in white matter, and HIF-1α is an important regulator of tissue and cell responses to hypoxia. 32 It has been reported that hypoxia-induced hypomyelination and maturation arrest requires intact HIF function within OPCs. 43 HIF-1α was upregulated in white matter lesions of BCAS mice and mediated the blockage of OPCs differentiation. 44 Moreover, by using a model of chronic HIF-1α accumulation in pluripotent stem cell-derived OPCs, HIF-1α was demonstrated to interact with OLIG2 to inhibit Sox10, thereby blocking the differentiation of OPCs. 45 Consistently, our results showed that the density of Sox10+ cells and the proportion of CC1+ cells in Sox10+ cells decreased after BCAS. Moreover, we found that CoCl2 induced HIF-1α elevation and inhibited OPCs proliferation and differentiation in vitro. Importantly, metformin was shown to inhibit HIF-1α expression and restore the functions of OPCs under hypoxic conditions. Metformin has been reported to rejuvenate aging OPCs, which are mainly AMPK-dependent. 34 In addition, metformin has been shown to promote the repopulation of mature oligodendrocytes via AMPK activation in acute or chronic demyelination animal models.18,46,47 In our study, metformin abrogated hypoxia-induced pAMPK decrease and restored OPCs functions, which were inhibited by dorsomorphin, suggesting that metformin recovery of OPCs dysfunctions under hypoxia is achieved through AMPK activation. Previous reports also demonstrated that the AMPK pathway participated in the degradation of HIF-1α by PHDs in cancer-associated fibroblasts and colon cancer cells.48,49 In the present study, we showed that PHD2 (a primary molecule degrading HIF-1α) was upregulated by metformin treatment under hypoxia, resulting in the reduction of HIF-1α in OPCs. Moreover, inhibiting AMPK activation with dorsomorphin would reduce the PHD2 level, leading to the accumulation of HIF-1α. Notably, when metformin-treated BCAS mice were administered intraperitoneally with DMOG or dorsomorphin for 60 days, OPCs proliferation and differentiation abilities were correspondingly inhibited. Therefore, these results suggested that metformin restores the OPCs functions under hypoxia most likely through the AMPK-PHD-HIF-1α pathway.
It is important to note that the effects of neuroinflammation and vascular injury on white matter and neurological function cannot be ignored after chronic cerebral hypoperfusion. Several studies show interactions between blood vessels and OPCs, for example, endothelial Cav-1 damaged oligodendrogenesis and myelin regeneration under ischemic conditions. 50 Chronic CBF dysregulation and BBB impairment may change Aβ metabolism and result in a cognitive decline apart from typical vascular dementia. 51 In addition, reparative angiogenesis can reduce white matter lesions and irreversible neuronal injury under chronic hypoperfusion. 52 Previous studies indicate that the occurrence of blood-brain barrier (BBB) leakage is earlier than demyelination, and vascular abnormality may compromise oligodendrogenesis and myelin regeneration in BCAS.53,54 Considering that metformin has been reported to improve endothelial dysfunction, 55 the possibility that metformin against white matter injury via preventing vascular dysfunction deserves further investigation. Matrix metalloproteinases (MMPs) which are secreted in an attempt to remodel the blood vessel wall, have the undesired consequences of opening the BBB and attacking myelinated fibres. 56 MMP9, the main form of cerebral MMPs, has been reported to mediate the BBB disruption at the early stage after BCAS. 53 Consistently, we found that the expression of MMP9 by endothelial cells gradually increased with the time of BCAS (Figure S1A-C). However, metformin treatment did not reduce MMP9 expression in endothelial cells after BCAS (Figure S1B, C). The neurovascular unit is crucial for the maintenance of the BBB, which encompasses endothelial cells, a basement membrane, pericytes, and astrocyte endfeet, and damage to any of these components may cause BBB leakage. 57 Therefore, the direct protective effect of metformin on neurovascular unit components in white matter injury needs to be studied in depth in the future. Moreover, inflammation and oxidative stress may be one of the underlying causes of oligodendrocyte injury after chronic hypoperfusion, as there is evidence that inhibition of inflammation and oxidative stress enhances OPCs proliferation and improves the disruption of white matter integrity. 58 We observed that neuroinflammation occured at 60 days post-BCAS, as shown by increased numbers of microglia and astrocytes in the white matter, which was not alleviated by metformin. However, the above results are not sufficient to assess the effect of the ischemic-hypoxic microenvironment on the role of OPCs, and we need more detailed studies in this regard.
Notably, we found that metformin alleviates cognitive impairment in BCAS mice in association with white matter repair. Numerous studies have been conducted to describe the link between cognitive impairment and white matter damage, particularly myelin damage. Imaging data showed that WMH burden may cause cognitive decline, especially impairment of executive functions. 59 The BCAS model exhibited working memory deficits due to myelin loss in the corpus callosum and other white matter regions, which is consistent with the pathological and clinical findings in human patients with WMH of vascular origin. 29 The hippocampus is important for both spatial memory and recognition memory. 60 Many studies have shown impaired hippocampal function after BCAS, as evidenced by increased neuronal apoptosis and hippocampal atrophy,61,62 although it has been shown that the white matter including the corpus callosum was consistently found to be rarefied without clear ischemic lesions in the hippocampus after BCAS. 38 The different manifestations of the hippocampus are likely to be related to the timing of the ischemic-hypoxic injury. We examined the hippocampus at 60 days post-BCAS. We found a significant reduction in Ki67+ cells which represent neurogenesis in dentate gyrus (DG) region of hippocampus after BCAS, and metformin could alleviate this reduction (Figure S2A, C). But no reduction of neurons in DG region was found after BCAS (Figure S2B, C). In addition, we found that synapses were affected in the DG region due to BCAS, as shown by a decrease in the number of Synapsin1+ puncta, yet metformin did not seem to improve synaptic damage (Figure S2D, E). In summary, our results showed that part of hippocampal function was affected by ischemia and hypoxia, but metformin appeared to have a limited effect on restoration of hippocampal function, which required further investigation.
As the population ages, the number of dementia patients is expected to reach 115 million by 2050. 63 Vascular dementia is the second leading cause of dementia, and up to 75% of dementia patients have evidence of vascular pathology. 1 The epidemiologic data strongly suggest the need for early intervention for VCID. Current prevention and treatment principally focus on the control of risk factors such as elevated blood pressure and blood lipids, platelet aggregation, diabetes mellitus, overweight, and smoking.1,64 However, there is currently no effective therapy for VCID. Therefore, exploring novel therapeutic strategies based on the pathology of VCID is greatly needed. In the present study, we demonstrated that metformin alleviated the white matter damage and improve cognitive functioning in BSCA mice, supporting the idea of a clinical trial with metformin to treat VCID with chronic cerebral ischemia. Beyond its current clinical application to treat Type 2 diabetes, an accumulation of pre-clinical and clinical data has stimulated interest in re-purposing metformin for anti-aging, anti-cardiovascular disease, and anti-cancer. 12 Recently, a clinical study in a cohort of 234 patients with Type 2 diabetes revealed that long-term use of metformin was associated with reduced cognitive impairment and lower CSVD burden. 15 Importantly, a proportion of the relation between long-term use of metformin and cognitive impairment was attributable to the alleviation of the CSVD burden. Therefore, it would be encouraged to carry out a clinical trial with metformin to treat VCID in multi-center with relatively large sample sizes.
In summary, this study shows that metformin alleviates white matter lesions and cognitive impairment after BCAS. The underlying mechanism for the anti-hypoxia effect of metformin involves AMPK-dependent PHD-mediated HIF-1α inhibition in OPCs. The findings provide a potential strategy for re-purposing metformin against cognitive dysfunction and white matter injury of VCID.
Supplemental Material
sj-pdf-1-jcb-10.1177_0271678X231175189 - Supplemental material for Metformin attenuates white matter injury and cognitive impairment induced by chronic cerebral hypoperfusion
Supplemental material, sj-pdf-1-jcb-10.1177_0271678X231175189 for Metformin attenuates white matter injury and cognitive impairment induced by chronic cerebral hypoperfusion by Yixi He, Zhenghao Li, Xiaoyu Shi, Jing Ding and Xin Wang in Journal of Cerebral Blood Flow & Metabolism
Supplemental Material
sj-pdf-2-jcb-10.1177_0271678X231175189 - Supplemental material for Metformin attenuates white matter injury and cognitive impairment induced by chronic cerebral hypoperfusion
Supplemental material, sj-pdf-2-jcb-10.1177_0271678X231175189 for Metformin attenuates white matter injury and cognitive impairment induced by chronic cerebral hypoperfusion by Yixi He, Zhenghao Li, Xiaoyu Shi, Jing Ding and Xin Wang in Journal of Cerebral Blood Flow & Metabolism
Supplemental Material
sj-pdf-3-jcb-10.1177_0271678X231175189 - Supplemental material for Metformin attenuates white matter injury and cognitive impairment induced by chronic cerebral hypoperfusion
Supplemental material, sj-pdf-3-jcb-10.1177_0271678X231175189 for Metformin attenuates white matter injury and cognitive impairment induced by chronic cerebral hypoperfusion by Yixi He, Zhenghao Li, Xiaoyu Shi, Jing Ding and Xin Wang in Journal of Cerebral Blood Flow & Metabolism
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the National Natural Science Foundation of China (32100798) and the China Postdoctoral Science Foundation (2021M700821).
Acknowledgements
The authors thank Prof. Z.D. Su from NMU for critical reading of the manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
Y.H., Z.L., J.D. and X.W. conceived and designed the experiments. Y.H., Z.L. and X.S. performed the experiments. Y.H., Z.L., J.D. and X.W. analyzed the data and prepared the manuscript. All authors reviewed and approved the manuscript.
Supplementary material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.