
Abstract
The energy cost of information processing is thought to be chiefly neuronal, with a minor fraction attributed to glial cells. However, there is compelling evidence that astrocytes capture synaptic K+ using their Na+/K+ ATPase, and not solely through Kir4.1 channels as was once thought. When this active buffering is taken into account, the cost of astrocytes rises by >200%. Gram-per-gram, astrocytes turn out to be as expensive as neurons. This conclusion is supported by 3D reconstruction of the neuropil showing similar mitochondrial densities in neurons and astrocytes, by cell-specific transcriptomics and proteomics, and by the rates of the tricarboxylic acid cycle. Possible consequences for reactive astrogliosis and brain disease are discussed.
Introduction
The notion that neurons are the major energy sink in brain tissue appears solid enough. Neurons spend considerable energy on the recovery of cation gradients challenged by excitatory postsynaptic potentials (EPSPs) and action potentials, while the neuronal signaling suppression by deep anesthesia or by pharmacological inhibition of the Na+/K+ ATPase pump (NKA) reduces brain metabolism by 50–60%. 1 Accordingly, a seminal bottom-up analysis based on the microscopic properties of ion channels, transporters and pumps concluded that 95% of the energy expenditure of gray matter signaling is neuronal and 5% is glial. 2 When action potentials were later found to be more efficient than anticipated, 3 the budget was modified, with 70% ascribed to dendrites, 15% to axons, and 7% to astrocytes. 4 In line with the energetic prominence of dendrites, glucose consumption in gray matter is several times higher than that in white matter. 5
There is however evidence that astrocytes use the NKA to capture the K+ that is released by neurons during neurotransmission. 6 Here I discuss why this active process, together with fresh insights on astrocytic oxidative metabolism, prompts a revision of the energy budget of gray matter.
K+ buffering is chiefly mediated by the astrocytic NKA
The EPSP is the transient dendritic depolarization caused by Na+ and Ca2+ entry through AMPA and NMDA receptors. Less widely appreciated is that these ionotropic glutamate receptors are permeable to K+, and that during neurotransmission the inward movement of cations is mirrored by the release of K+.2,7 The journey of Na+ in the aftermath of the EPSP is straightforward. It diffuses along the dendritic shaft to be pumped out later on by the NKA. 8 The EPSP lasts for a few milliseconds, whereas the extrusion of Na+ by the sluggish NKA takes seconds to minutes. 8 Most of the Ca2+ load is transformed by the Na+/Ca2+ exchanger NCX into a Na+ load, which is then dealt with by the NKA. Hence, the energy burden of the Na+ and Ca2+ load is carried entirely by the postsynaptic neuron. The fate of the K+ is more convoluted. After leaving the dendrite, K+ enters an intricate space bounded by many different cells, mostly neurons and astrocytes. Assuming a square law for the escalation of cell surface and a typical diffusional distance of 100 micrometers,8,9 it can be calculated that <0.01% of the surface screened by the K+ belongs to the neuron that released it. In fact, it is well established that most of the K+ released by synaptic activity is not cleared by neurons, but by astrocytes, a phenomenon known as K+ buffering.6,10
The molecular mechanism responsible for K+ buffering is paramount to this article’s main point. Over the years, three candidates were considered: the Na+-K+-2Cl− co-transporter NKCC, the K+ channel Kir4.1 and the NKA. The NKCC hypothesis, based on culture experiments, lost impetus when it was realized that the NKCC is not significantly expressed in adult gray matter, although the NKCC may play a role in development, in white matter oligodendrocytes and in glioma cells.6,11 The second candidate, Kir4.1, mediates a form of K+ buffering termed spatial buffering. According to this hypothesis, K+ can be captured by astrocytes via constitutively open Kir4.1 channels and at the same time an identical amount of K+ leaves distant astrocytes, also via Kir4.1. This long-range electrotonic phenomenon requires gap junction coupling between astrocytes. 10 Because permeation across ion channels does not involve ATP hydrolysis, spatial buffering is costless, which explains the minor role attributed to astrocytes in various energy budgets.2,4,12
However, spatial buffering and Kir4.1 channels are no longer regarded as the main mechanisms of K+ buffering, a conclusion reached from anatomical and physiological considerations and the outcome of pharmacological and genetic manipulation of gap junctions and Kir4.1 9,.11,13–18 The participation of Kir4.1 in K+ buffering has been analized in depth by Nanna MacAulay. 6 Revised roles for Kir4.1 in astrocytes and oligodendrocytes are in setting the membrane potential, limiting the amplitude of the K+ rise at high frequency stimulation, and mediating the delayed release of astrocytic K+, which is then captured by neurons to complete the cycle. Thus, Kir4.1 remains an integral element of the K+ cycle. Spatial buffering is still thought to be dominant in the highly polarized retina. 19
The lion’s share of K+ buffering in brain tissue is nowadays attributed to the astrocytic NKA. 6 The NKA is an oligomeric enzyme consisting of one catalytic subunit α (four isoforms) and one regulatory subunit β (three isoforms). The preferential role of astrocytes over neurons in K+ buffering is explained by NKA subtype allocation.16,20–22 At 3 mM resting K+, the neuronal NKA (α3β1) is saturated with K+ and therefore primed to respond to variations in intracellular Na+, for which it has low affinity. The neuronal NKA behaves as a true Na+ pump, that is, driven by Na+, with K+ playing a permissive role. Conversely, the major astrocytic NKA (α2β2) is primed by intracellular Na+ and ready to respond to extracellular K+, for which it has low affinity. Moreover, α2β2 is dormant at resting membrane potential and is activated by depolarization, which is not the case for the neuronal pump.16,22 Because of parallel sodium and chloride conductances, the membrane potential of neurons is four times less sensitive to [K+]e than the astrocytic membrane potential. 23 The compounded effect of NKA sensitivity to K+, NKA sensitivity to membrane potential, and membrane potential sensitivity to K+, is that a physiological rise in K+ from 3 to 6 mM will stimulate the astrocytic NKA by 107% but the neuronal NKA only by 6% (Figure 1), an 18-fold difference. Driven by intracellular Na+, the NKA of the stimulated dendrite would be more efficient at buffering K+ than the non-stimulated neurons, but this uptake occurs in delayed fashion and far from the active zone. 8
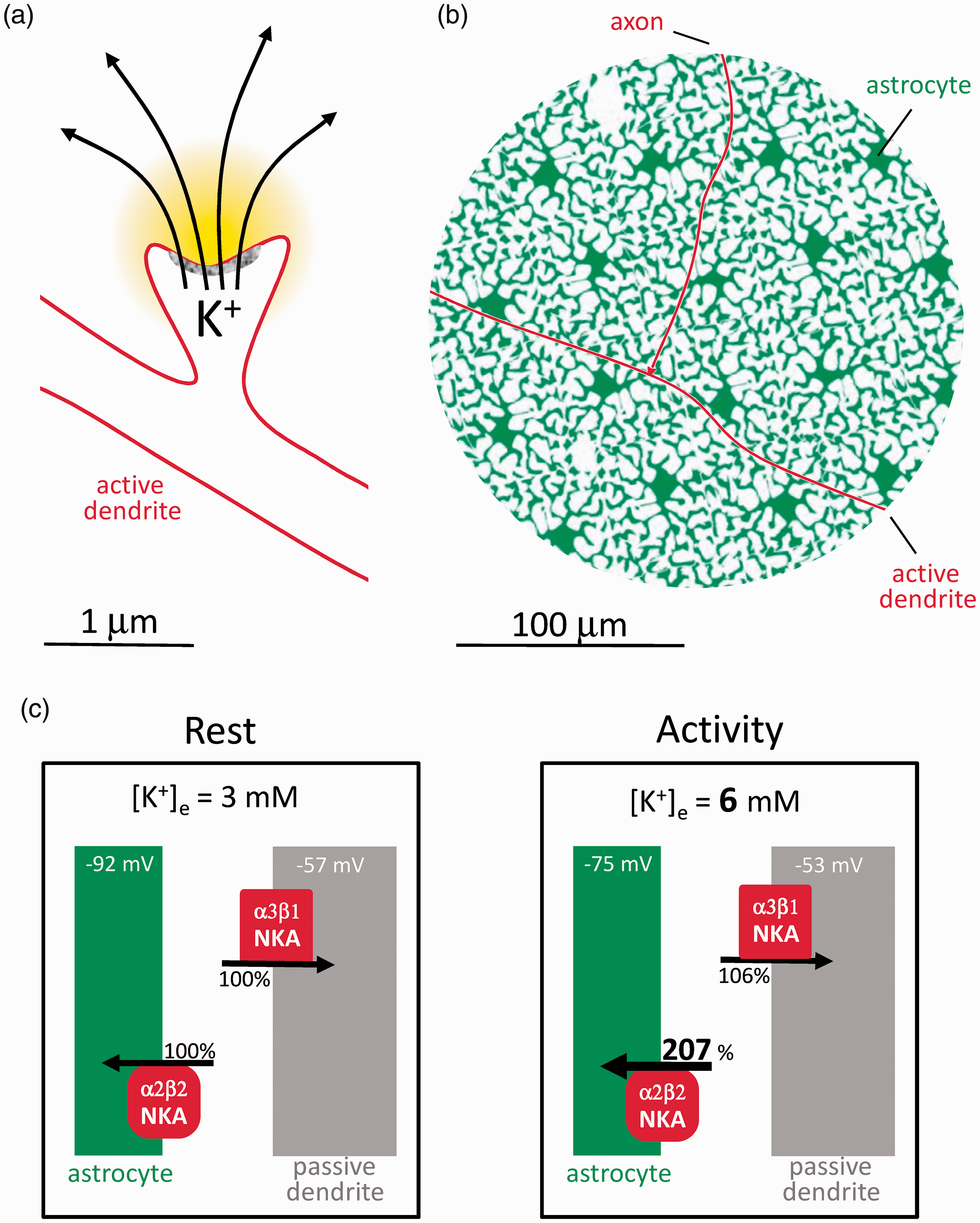
Astrocytes are poised to capture K+ while neurons are not. (a) The postsynaptic neuron releases K+ during excitatory neurotransmission. (b) Compared to astrocytes, the active postsynaptic neuron represents a minor fraction of the cellular surface to be screened by K+. Passive neurons are omitted for simplicity. (c) According to the Goldman-Hodgkin-Katz equation, membrane potential Vm = 59 x log(PK x [K+]e + PNa x [Na+]e + PCl x [Cl−]I/PK x [K+]I + PNa x [Na+]I + PCl x [Cl−]e) where PK, PNa and PCl (permeabilities) are assumed to be 1.0, 0.04 and 0.45 for neurons and 1.0, 0 and 0 for astrocytes, 23 with resting concentrations (mM; e, extracellular; i. intracellular) of [K+]e = 3, [Na+]e = 140, [Cl−]I = 20 mM, [K+]I = 110 mM, [Na+]I =10 mM and [Cl−]e = 110 mM. Under these conditions, a [K+]e rise from 3 to 6 mM will depolarize astrocytes by 17 mV (−92 mV to −75 mV) and neurons by 4 mV (- 57 mV to −53 mV). These depolarizations will double the K+ affinity of α2β2 without affecting α3β1 . 16 With the compounded effects of higher K+ and higher affinity, α2β2 occupancy rises from 35 to 73% (a 107% increase), whereas α3β1 occupancy rises only from 88 to 94% (a 6% increase).
The astrocytic NKA should perhaps be best understood as a K+ pump, whose permissive co-factor is intracellular Na+. The NKA exchanges three Na+ ions per two K+ ions and therefore leads to cell shrinkage. Yet, synaptic activity is accompanied by astrocytic swelling, 24 which is partly explained by parallel influx of Na+ and bicarbonate via the NBCe1, and partly by metabolic stimulation.25,26 The Na+/glutamate cotransport is deemed a minor contributor, as only 3 Na+ ions are imported per glutamate, compared with the 150 Na+ ions per glutamate exported by the NKA. Additional Na+ influx is provided by voltage-sensitive Na+ channels, TRP channels and the Na+/Ca2+ exchanger.11,27–29
Astrocytes as major energy spenders
How much energy is consumed by astrocytes? According to the current budget, most K+ is passively buffered via Kir4.1, and astrocytes use just 7% of the signaling budget, for glutamate recycling and membrane potential generation (Figure 2(a)). At the opposite end of the possibility spectrum, if all synaptic K+ were cleared by the NKA, astrocytes would spend 37% of the ATP (Figure 2(b)). With the NKA and Kir4.1 contributing equally, a conservative estimate, astrocytes would use 25% of the ATP (Figure 2(c)), plus a minor contribution from the astrocytic Ca2+ pumps. 30 In light of the experimental evidence cited above showing that the NKA surpasses Kir4.1 in the buffering of K+, it seems safe to state that astrocytes account for at least 25% of the ATP spent in gray matter signaling, i.e. more than 200% higher than previously thought. In this hybrid model, the NKA would possibly cater for low and moderate activity and be supplemented by Kir4.1-mediated K+ buffering during high frequency stimulation, 6 thus limiting maximum energy demand. Brain tissue must also spend energy for protein and lipid turnover, membrane trafficking, amino acid and nucleotide metabolism, pH regulation, cytoskeleton remodeling, etc, collectively estimated at 20–30% of the total tissue ATP turnover.2,4,30 Several of these housekeeping functions have been evolutionary outsourced from neurons to astrocytes, including control of interstitial fluid composition, glycogen storage and mobilization, supply of energy substrates and precursors for biosynthesis, and the recycling of neurotransmitters, oxidized lipids and scavengers, and other waste products.31–33 The astrocytic share of the housekeeping cost might be even larger than its share of the signaling cost, further supporting the case for the expensive astrocyte.
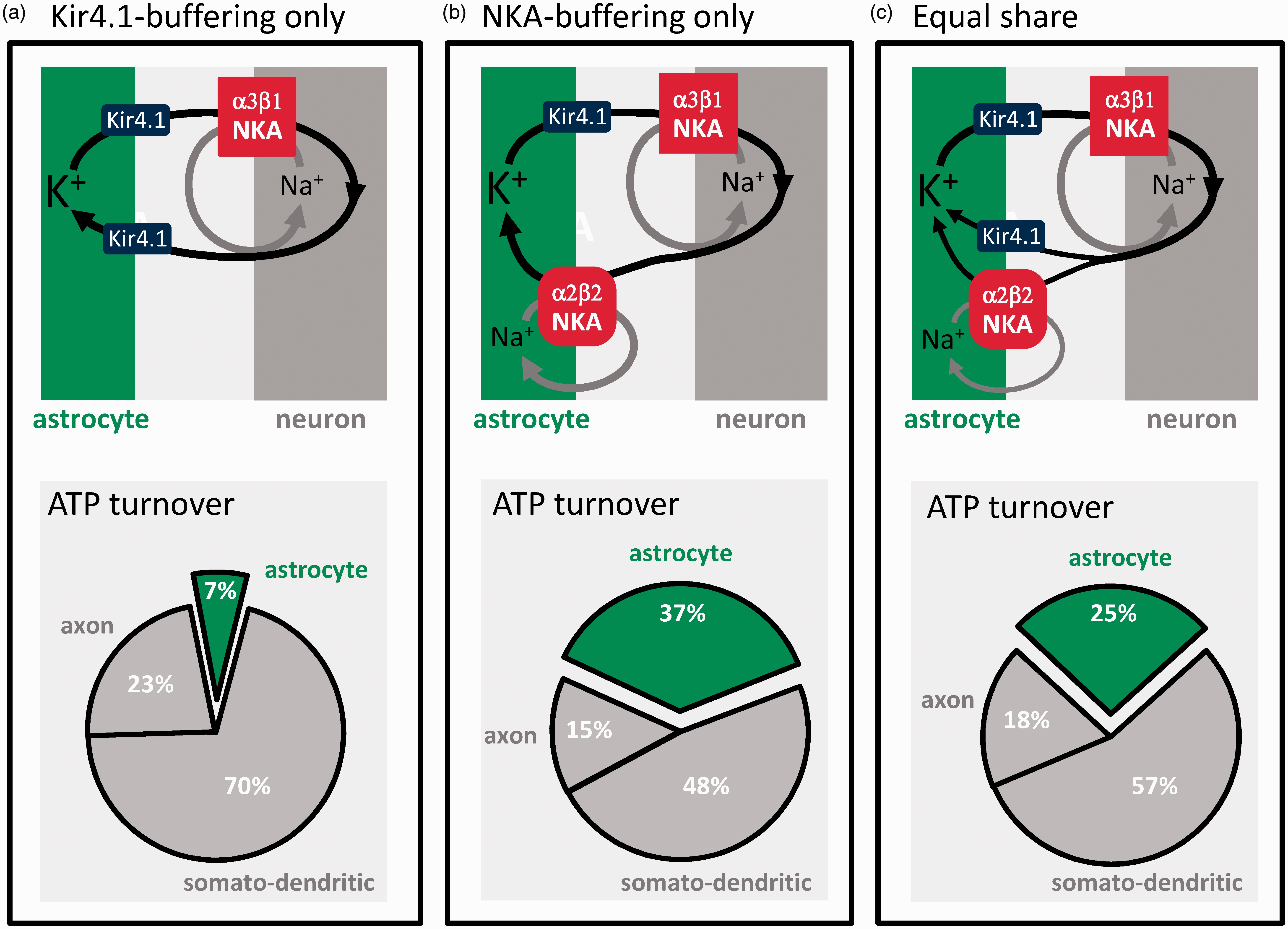
The active buffering of K+ makes astrocytes energetically costly. Postsynaptic neurons release approx. 100 K+ ions for exocytosed glutamate. 2 The current energy budget of gray matter signaling 2 assumes that these ions are passively recycled via Kir4.1 channels without energy expenditure. In such case, astrocytes would use only 7% of the energy budget (a). If the contribution of Kir4.1 were negligible and all synaptic K+ were captured by the NKA, astrocytes would use 37% of the budget (b). In an intermediate scenario, K+ buffering is equally shared between Kir4.1 and the NKA, and astrocytes use 25% of the signaling budget (c).
Mitochondrial function and distribution also point to expensive astrocytes
Lack of immunological and histochemical detection of cytochrome oxidase led to the pervasive idea that astrocytes do not possess significant oxidative phosphorylation, OXPHOS, 34 a conclusion supported by the apparent absence of mitochondria in the astrocytic processes that surround synapses. 35 In those days, astrocytes were even thought to produce 100% of their ATP in glycolysis. 36 Later work has shown otherwise. Even in culture, a condition that promotes glycolysis via the Warburg effect, 75% of the astrocytic ATP is generated by mitochondria. 37 In vivo determinations have shown that 20–33% of the brain TCA cycle flux is glial.30,38–41 When these figures are fed into mathematical models, once again, over 75% of the astrocytic ATP is shown to be mitochondrial.12,42 Noticing that, relative to deep anesthesia, oxidative glial ATP synthesis in awake animals is well in excess of that required by glutamate recycling, Gulin Oz, Rolf Gruetter and colleagues advanced that “glial oxidative ATP synthesis sustains additional metabolic reactions in the astrocyte that are related to overall brain activity and not only those that maintain glutamatergic action”. 39 K+ buffering fits the bill. And the anatomy has also been revised. Refinements of electron microscopy and targeted fluorescent markers have detected abundant mitochondria in astrocytes.43–47 In fact, at 7–10%, the fraction of the cellular volume occupied by mitochondria is similar in astrocytes and neurons.48,49 Transcriptomics and proteomics concurred by showing similar expression of TCA cycle and OXPHOS enzymes in astrocytes and neurons.43,50,51 On top of that is the ATP production of glycolysis, for which astrocytes are better equipped.52–54
There are however qualitative differences in mitochondrial metabolism between the two cell types. In neurons, OXPHOS complex I is predominantly assembled into supercomplexes, whereas in astrocytes the abundance of free complex I is higher, associated with several-fold higher reactive oxygen species (ROS) production compared with neurons. 55 Neurons degenerate in response to partial genetic inhibition of OXPHOS, whereas astrocytes upregulate glycolysis and survive indefinitely, without ostensible functional deficit.56,57 Astrocytes express PDK4, a kinase that inhibits pyruvate dehydrogenase, and the anaplerotic enzyme pyruvate carboxylase,50,58 suggesting tonic deviation of pyruvate away from oxidation. Instead, astrocytic mitochondria are better endowed than those of neurons for fatty acid oxidation.59–61 Fatty acids can be transferred from neurons to astrocytes in an activity-dependent fashion32,62,63 and enter the TCA cycle beyond the PDH blockage, as do ketone bodies, fuels of pathophysiological and therapeutic relevance 64,65 that have a powerful inhibitory effect on astrocytic glucose metabolism. 66 Determining the relative weight of glucose, neuronal fatty acids, blood-borne fatty acids, and glutamate in fueling astrocytic respiration is a pending task.67–69 Another emerging aspect of astrocytic respiration is that it appears to be highly regulated. In vitro experiments have shown that astrocytic respiration is subject to short-term modulation by physiological levels of extracellular K+, NH4+ and NO.70–72 These signals, which are released by neurons and endothelial cells during local activity and reactive hyperemia, are proposed to generate glycolytic lactate and spare tissue oxygen for the benefit of active neurons. 73
In summary, there is a good correlation between the energy demand imposed by K+ buffering on astrocytes, and the mitochondrial endowment and speed of respiration of these cells. The volume occupied by astrocytes in the neuropil is less than half that occupied by neurons,12,42,49,74 which means that gram-per-gram, astrocytes are as expensive as neurons. This conclusion is not in conflict with the metabolic effects of anesthesia and NKA inhibition 1 nor with the faster metabolic rate of gray matter versus white matter. 5
Expensive astrocytes and disease
A common pathogenic factor in stroke, traumatic brain injury and other acute diseases of the brain is a local energy deficit, which has been assumed to be neuronal. With astrocytes promoted to non-trivial energy spenders, their metabolism becomes more interesting. Neurons are more fragile than astrocytes. Brain disease, acute and chronic, causes the preferential death of neurons, while astrocytes survive and often thrive, a phenomenon termed reactive astrogliosis. Reactive astrocytes are pleiotropic and may differ from normal astrocytes in morphology, cytoskeleton, transcription factors, signaling, chaperones, secreted proteins, etc. 75 A common feature of these cells is a reduced ability to cater for the needs of neurons, i.e. they become “selfish”. 31 For example, astrocytes located near human tumors showed a 60-70% reduction in the mRNA expression of the three transport proteins that link the buffering of K+ to energy metabolism (α2β2 NKA, Kir4.1 and NBCe1). 76 Glial K+ buffering is also downregulated in a Drosophila seizure model, 77 whereas its reactivation by genetic means suppressed neuronal hyperactivity and seizures, and increased the life span of the fly. 77 The role of K+ buffering and associated energy demand in neuroprotection is complex. Reactive astrocytes are predicted to spend less energy and glucose during neural activity,78,79 contributing to the well-being of neurons, which use glucose for antioxidation; 52 but they would also fail to remove extracellular K+ and reduce their own oxygen consumption on demand,70,73 to the detriment of neurons. Supporting the latter, in a mouse model of Huntington’s disease Kir4.1 was found to be diminished in astrocytes, leading to K+ accumulation and neuronal hyperexcitability. Viral delivery of Kir4.1 to these astrocytes normalized extracellular K+, ameliorated the neuronal dysfunction and prolonged neuronal survival. 80 Huntington´s astrocytes switch from glucose to fatty acid metabolism, a phenomenon associated with reactive oxygen species-induced damage in the striatum but not in the cerebellum, a clue to the baffling regional damage of this genetic disease. 81 And in a mouse model of Alzheimer´s disease glycolysis was impaired in the early stages, leading to reduced production of serine and altered synaptic plasticity and memory, which was reverted by dietary serine. 33 In summary, reactive astrocytes may be protective or detrimental to neurons according to brain region, pathology, disease stage and experimental model.75,82 More research focusing on astrocytes, both normal and reactive, is needed to clarify the role of energy metabolism in brain disease.
Footnotes
Funding
The author disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was partially funded by Fondecyt Grant 1200029 and by ANID-BMBF Grant 180045.
Acknowledgements
My thanks to all members of my laboratory and to our collaborators for generous and fruitful discussions. I also thank Karen Everett for critical reading of the manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.