
Abstract
Immunohistochemistry for haptoglobin (Hp) in the postischemic hippocampus demonstrated an immunoreactivity visible one day after reperfusion and continuing to increase until 14 days after ischemia. The immunoreactivity was most prominent in CA1 and the dentate hilar region, especially in cells with astroglial morphology. Double immunofluorescence histochemistry confirmed colocalization of the Hp and glial fibrillary acidic protein. Furthermore, a reverse transcription-polymerase chain reaction study confirmed an elevated Hp mRNA level in the postischemic hippocampus. The Hp gene expression was also upregulated in C6 and A-172 glioblastoma cell lines after H2O2 treatment. These findings suggest that Hp is synthesized in reactive astrocytes in response to ischemia-reperfusion injury.
Haptoglobin (Hp) is a major acute-phase plasma protein that forms a stable complex with hemoglobin (Hb) (Bowman, 1993). Because of its strong Hb-binding capacity, Hp can minimize Hb-stimulated lipid peroxidation and protect DNA and tissues against oxidative damage during hemolytic injury (Miller et al., 1997; Lim et al., 2000). The physiologic function of Hp has, therefore, been considered to be an antioxidant. In addition, several reports have suggested new roles for Hp such as an angiogenic factor (Cid et al., 1993) or an immunomodulator (Oh et al., 1990; Berkova et al., 1999). Although these biologic activities of Hp have been confirmed, little is known about gene expression and pathophysiologic significance of the Hp in the injured brain.
Although it is well known that Hp is mainly synthesized in the liver, several investigators have reported tissue-specific Hp expression in extrahepatic tissues (Yang et al., 1995; Friedrichs et al., 1995; Kim et al., 2001). Recently, Hp transcripts were detected in human glioblastoma cell lines (Sanchez et al., 2001) and increased Hp was observed in cerebrospinal fluid from patients with central nervous system disorders (Johnson et al., 1992). Therefore, the possibility is raised that Hp is locally synthesized in brain cells to play a role in modulating the pathophysiologic reactions of brain insult such as cerebral ischemia.
In the present study, to determine whether the Hp is locally expressed in brain cells, we investigated the expression pattern and cellular localization of Hp in the adult rat hippocampus after transient forebrain ischemia. Our results indicate that Hp is synthesized in reactive astrocytes of the hippocampus after ischemia-reperfusion. To further confirm the Hp gene expression in astrocytes, in vitro studies using glioblastoma cell lines were also performed.
MATERIALS AND METHODS
Animal preparation
Adult male Sprague-Dawley rats (250 to 300 g) were used in this study. All experimental procedures performed on the animals were conducted with the approval of the Catholic Ethics Committee of the Catholic University of Korea, and were in accordance with the NIH Guide for the Care and Use of Laboratory Animals. Transient forebrain ischemia was induced by four-vessel occlusion and reperfusion, as previously described by Pulsinelli and Brierley (1979) with minor modifications (Lee et al., 2000). Briefly, the vertebral arteries were electrocauterized and cut completely to abolish circulation. After 24 hours, both common carotid arteries were occluded for 10 minutes with miniature aneurysmal clips. Only animals with complete EEG flattening on vascular occlusion were classified as ischemic and used for the study (Lee et al., 2000). Rectal temperature was maintained at 37.5 ± 0.3°C with a heating lamp during and after ischemia. Sham-operated rats with cauterized vertebral arteries and ligatures placed around carotid arteries were used as controls. No animal convulsed or died after reperfusion or sham operation. Animals were maintained for 1, 3, 7, or 14 days after reperfusion. For each time point, three rats were treated for immunohistochemistry and two rats for reverse transcription-polymerase chain reaction (RT-PCR). Sham-operated animals were treated on the same schedule as the ischemic-reperfused animals. At each time point after reperfusion, animals were killed by transcardial perfusion with a fixative containing 4% paraformaldehyde in 0.1 mol/L phosphate buffer, pH 7.4, or by transcardial perfusion with saline followed by decapitation and dissection of the hippocampi, which were quickly frozen in liquid nitrogen.
Immunohistochemistry
Coronal sections, 25-μm thick, were cut from the septal pole of the hippocampus. Free-floating sections were processed for Hp. After blocking with 10% (v/v) normal goat serum for 1 hour, the sections were incubated with a rabbit polyclonal antibody against Hp (Sigma, St Louis, MO, U.S.A.; diluted at 1:20) or a mouse monoclonal antibody to Hp (Sigma; diluted at 1:20) overnight at 4°C. Primary antibody binding was visualized using peroxidase-labeled goat antirabbit antibody (Jackson Laboratories, West Grove, PA, U.S.A.; diluted at 1:200), and 0.05% 3,3'-diaminobenzidine tetrahydrochloride and 0.01% H2O2 as substrate. The specificity of Hp immunoreactivity was confirmed by the absence of immunohistochemical reaction in sections from which the primary antibody was omitted, or in which it was substituted with nonspecific rabbit immunoglobulin G (IgG).
Double immunofluorescence histochemistry
Sections were incubated in a combination of a mouse monoclonal antibody to Hp (Sigma; diluted 1:20) and a rabbit polyclonal antibody against glial fibrillary acidic protein (GFAP; Sigma; diluted 1:400) overnight at 4°C. After washing in phosphate-buffered saline, the sections were incubated with a mixture of FITC-conjugated goat antimouse IgG (Jackson Laboratories, West Grove, PA, U.S.A.; diluted at 1:100) and Cy3-conjugated goat antirabbit IgG (Jackson; diluted at 1:100) for 2 hours at room temperature, respectively. Control sections were prepared as described above. Slides were viewed using a confocal microscope (MRC-1024, BioRad, Hercules, CA, U.S.A.). Images were converted to TIFF format, and contrast levels of images were adjusted using Adobe Photoshop.
Cell culture and treatment
The C6 and A-172 glioblastoma cell line, originating from rat and human respectively, were purchased from the American Type Culture Collection (Rockville, MD, U.S.A.), and maintained in RPMI 1640 medium (Sigma) supplemented with 10% (v/v) fetal bovine serum (HyClone, Logan, UT, U.S.A.). To induce Hp gene expression, the cells were plated at a density of 1 × 10 /mL. After overnight incubation, the cells were treated with 100 to 300 μmol/L of H2O2 (Sigma) for the indicated periods.
RNA preparation and reverse transcription-polymerase chain reaction
Total cellular RNAs were extracted from H2O2-treated C6 and A-172 cells, and ischemic hippocampi using RNAzol B solution (Tel-test Inc., Friendswood, TX, U.S.A.), based on the modified single-step guanidinium isothiocyanate lysis method. Two micrograms of total RNA from each sample was reverse-transcribed for 30 minutes at 42°C in 50 μL of PCR buffer containing 5 mmol/L MgCl2, 1 mmol/L dNTPs, RNase inhibitor, random hexamer, and 0.1 U/μL avian myeloblastosis virus reverse transcriptase. A 2-μL aliquot of the RT product was amplified in a total volume of 50 μL. The amplification was performed for 30 cycles under the following conditions; denaturation at 95°C for 1 minute, annealing at 55°C for 1 minute, and extension at 72°C for 1 minute. A 10-μL aliquot of the PCR product was then analyzed electrophoretically on a 1.5% (w/v) agarose gel. The sequences of oligonucleotide primers used in this study are as follows: human Hp primers, 5'-CG-GATCCTAGGTGGACACCTTGAT-3' (forward) and 5'-GCATTAGTTCTCAGCTATGGTCTT-3' (reverse); rat Hp primers, 5'-CATTGAAGATGACAGCTGCCC-3' (forward) and 5'-GGAGTTACAGCGCCTTTCTTC-3' (reverse); β-actin primers, 5'-ATCTGGCACCACACCTTCTACAATGAGC-TGCG-3' (forward) and 5'-CGTCATACTCCTGCTTGCT-GATCCACATCTGC-3' (reverse).
RESULTS
Upregulation of haptoglobin in the hippocampus in response to ischemia-reperfusion
To study the induction and cellular localization of Hp in the postischemic hippocampus, immunohistochemistry was performed both in control hippocampus and in experimental rats at different time points after ischemic insults. Incubation of adjacent brain sections with monoclonal and polyclonal antibodies against Hp, respectively, resulted in similar immunohistochemical labeling patterns (data not shown). Hp levels in the hippocampus of control rats were too low to be detected by immuno-labeling (Figs. 1A and A1). After transient global ischemia, reproducible changes in both staining intensity and the distribution of Hp immunoreactivity were observed. Immunoreactivity became visible one day after reperfusion and the signal intensity further increased until 14 days after ischemia. At one day after reperfusion, Hp immunoreactivity was distributed homogeneously over the hippocampus proper and the dentate gyrus (Fig. 1B). As shown in the higher magnification of the CA1 sector in Fig. 1B (B1), the immunoreactivity was localized in cells with glial morphology of the stratum radiatum, but was absent or low in the pyramidal cell layer. At three days of reperfusion, Hp immunoreactivity had increased preferentially in the CA1 sector and the hilar region of the dentate gyrus (Figs. 1C and C1). Labeling intensity further increased between three and seven days after ischemia. During the second week after ischemia, the labeling pattern in the hippocampus remained similar to that at seven days, but labeled cells displayed a more hypertrophic appearance. The distribution pattern and morphology of Hp immunoreactive cells suggested that they might represent reactive astrocytes (Figs. 1D and D1). Colocalization of Hp and GFAP confirmed the Hp expression in reactive astrocytes (Figs. 1E–G).
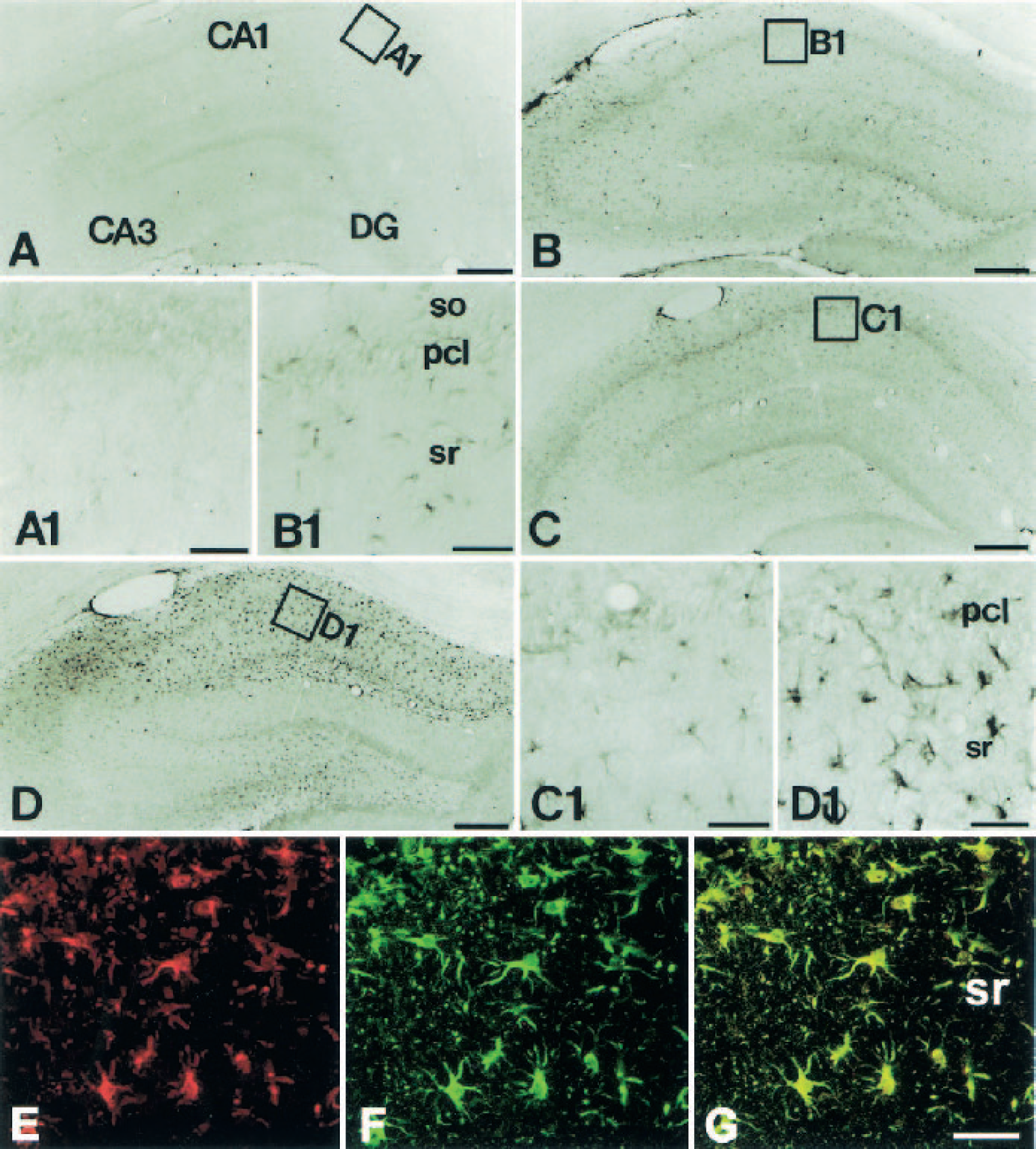
(
Because Hp protein was detected in the postischemic hippocampus, we examined whether the Hp mRNA was also enhanced after ischemic injury. In contrast to the absence of Hp mRNA in sham control hippocampus, the transcript could be detected seven days after ischemia by RT-PCR (Fig. 2A).
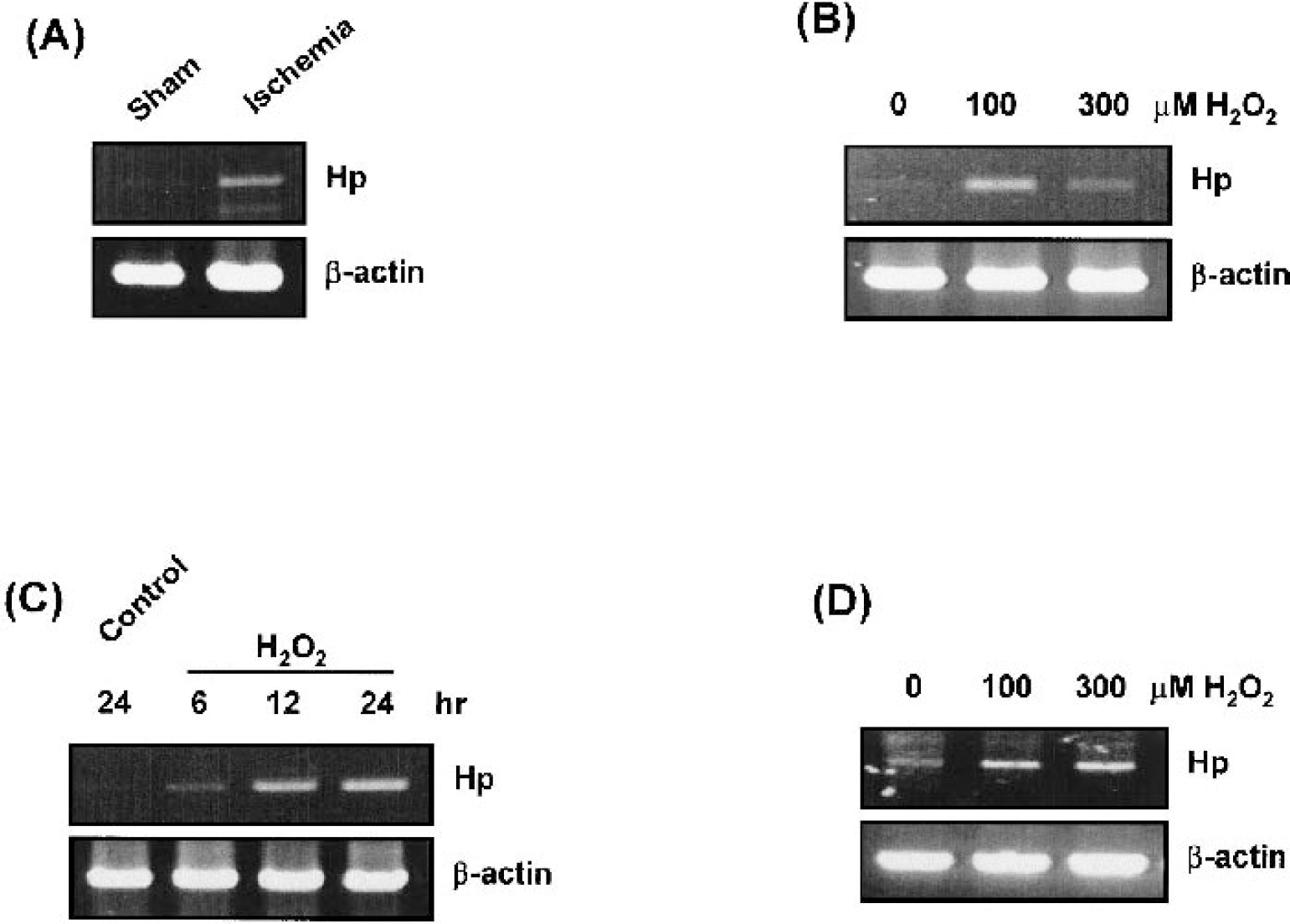
Induction of haptoglobin (Hp) gene expression in ischemic hippocampus and H2O2-treated glioblastoma cells. Total cellular RNAs were extracted and then reverse transcription-polymerase chain reaction was performed to detect gene transcript of Hp using the same procedure as described in Materials and Methods.
Haptoglobin gene expression in H2O2-treated glioblastoma cells
To further confirm the upregulation of Hp gene expression in astrocytes in response to ischemia, the level of Hp transcripts was analyzed in H2O2-treated glioblastoma cells, where the cells were caused an oxidative stress damages. Reverse transcription-polymerase chain reaction experiments showed that 100 μmol/L of H2O2 increased the Hp mRNA level in C6 rat glioblastoma cells (Fig. 2B) and the induction was time dependent until 24 hours (Fig. 2C). Treatment of H2O2 also enhanced Hp expression dose-dependently in A-172 human glioblastoma cells (Fig. 2D). However, Hp protein was not detected in these cell lines by western blot analysis, although it was well detected in damaged hippocampus by immunohistochemistry (Fig. 1). It is likely that some differences exist in signaling for translational activation between glioblastoma cell lines (C6 and A-172) and astrocytes in normal rat hippocampus. This possibility is supported by other studies (Sanchez et al., 2001), reporting that Hp protein could not be detected by western blot in human glioblastoma cell lines (U-87MG and U-138MG), although the presence of Hp mRNA was evidenced by RT-PCR. In that report, Sanchez et al. (2001) demonstrated the presence of Hp mRNA in untreated U-87MG and U-138MG cells. However, in our study, Hp mRNA was barely detected in untreated glioblastoma cells, and only induced by H2O2 treatment (Fig. 2).
DISCUSSION
Our study provides the first evidence that the reactive astrocytes in the hippocampus locally synthesize the Hp after transient forebrain ischemia. However, the possibility that the increased Hp level in reactive astrocytes may be due to intracellular uptake of plasma Hp must be considered, because plasma Hp could escape into the brain parenchyma after ischemic insults and be passively absorbed by astrocytes. But we think this possibility can be ruled out for the following reasons. First, the immunoreactivity of Hp in astrocytes did not peak at an early phase, but at the late phase (7 to 14 days) after reperfusion, where remodeling occurs (du Bois et al., 1985; Kirino et al., 1985). Second, this aberrant expression was not global, but was restricted mainly to the hippocampal CA1 and hilar region, in which neuronal death has also been observed after a four-vessel occlusion insult of the same period (Pulsinelli et al., 1982). Third, all of the cells that are immunoreactive for Hp also showed GFAP immunoreactivity in double immunofluorescent staining, indicating that the expression of Hp was astrocyte specific. Finally, the level of Hp mRNA in the postischemic hippocampus was significantly increased compared with that of sham-operated animals. Taken together, our present findings suggest that Hp expression is temporally upregulated in reactive astrocytes within the vulnerable region of the postischemic hippocampus. Furthermore, RT-PCR study using human and rat glioblastoma cells treated with H2O2 supports that reactive astrocytes can synthesize Hp in response to oxidative stress in the brain.
The mechanisms of the induction of Hp in reactive astrocytes are of significant interest. It is well known that the proinflammatory cytokines such as interleukin (IL)-6, IL-1β and tumor necrosis factor (TNF)-α are principal Hp-inducers and that CCAAT/enhancer binding protein β and δ (C/EBP-β and C/EBP-δ) and signal transducer and activator of transcription-3 (STAT-3) are important transcription factors for Hp gene expression (Baumann and Gauldie, 1994). Orzylowska et al. (1999) demonstrated that IL-6, IL-1β and TNF-α were induced in rat hippocampus after ischemic injury. Immunoreactivities against these cytokines were detected in astrocytes especially in the CA1 area. In addition, the staining intensity was maximal 14 days after ischemia. These results are concomitant with the Hp induction in the present study, in terms of cell type, localization and induction time. Furthermore, previous reports indicated that proinflammatory cytokines enhanced C/EBP-β, C/EBP-δ (Cardinaux et al., 2000) and STAT-3 activity in the reactive astrocytes after brain injury (Justicia et al., 2000). Considered together, the expression of Hp in reactive astrocytes could be induced, at least partly, via the signal pathway including C/EBP-β, C/EBP-δ, or STAT-3 activated by proinflammatory cytokines produced during ischemic injury.
Another issue that should be addressed is the function of Hp produced in astrocytes after ischemia-reperfusion. Cerebral ischemia elicits glial and inflammatory reactions in the brain, and the glial and inflammatory environment may determine the survival of neurons after ischemia (Kato and Walz, 2000). A strong antiinflammatory environment, therefore, is needed after brain insults such as ischemia-reperfusion. Considering the accumulative evidence suggesting that Hp is increased during inflammation and exerts for antiinflammatory actions in tissues (Gabay and Kushner, 1999), it might be postulated that Hp plays an important role in defending against oxidative damages and other harmful inflammatory reactions in the injured brain. In addition, with the recent finding that Hp possesses angiogenic activity (Cid et al., 1993), its role in repairing damaged capillaries in the tissue during inflammation is also an interesting possibility.
In summary, our present findings, for the first time, demonstrate that reactive astrocytes upregulate Hp production in response to ischemic insult in an in vivo model of brain ischemia. Further experiments are needed to clarify the functional significance of Hp expression in reactive astrocytes after ischemic insult.