
Abstract
Glial cells constitute a large percentage of cells in the nervous system. During recent years, a large number of studies have critically attributed to glia a new role which no longer reflects the long-held view that glia constitute solely a silent and passive supportive scaffolding for brain cells. Indeed, it has been hypothesized that glia, partnering neurons, have a much more actively participating role in brain function. Alteration of intraglial ionic homeostasis in response to ischemic injury has a crucial role in inducing and maintaining glial responses in the ischemic brain. Therefore, glial transporters as potential candidates in stroke intervention are becoming promising targets to enhance an effective and additional therapy for brain ischemia. In this review, we will describe in detail the role played by ionic transporters in influencing astrocyte, microglia, and oligodendrocyte activity and the implications that these transporters have in the progression of ischemic lesion.
INTRODUCTION
Glial cells constitute a large percentage of cells in the nervous system. During recent years, a large number of studies have attributed to glia a new role that no longer reflects the long-held view that glia constitute solely a silent and passive supportive scaffolding for brain cells. Indeed, it has been hypothesized that glia, partnering neurons, have a much more actively participating role in brain function.
Indeed, glial cells are involved in almost every type of neurodegenerative diseases. However, conversely, alteration of intracellular ionic homeostasis in response to ischemic injury has a crucial role in inducing and maintaining glial responses in the injured brain.1-3 For instance, neuronal injury may elicit a series of glial reactions that are of critical importance for the progress of neural pathology.
Astrocytes in Brain ischemia
Astrocytes perform several functions that are essential for normal neuronal activity, including glutamate uptake, glutamate release, K+ and H+ buffering, and water transport. Accordingly, they can influence neuronal survival during ischemia. Furthermore, they also influence neurite outgrowth and other processes that contribute to brain recovery.
Astrocytes are divided into four major types.4,5
Protoplasmic astrocytes with numerous highly branched short processes.
Interlaminar astrocytes extending striking long, frequently unbranched processes throughout the layers of the cortex.
Polarized astrocytes residing in the deep layers of the cortex and extending one or two long unbranched processes away from the white matter.
Fibrous astrocytes displaying more stellate shapes, with smooth, long less-branched processes.
Astrocytes also include other cells such as velate astrocytes of the cerebellum, tanycytes (found in the periventricular organs, the hypophysis, and the raphe part of the spinal cord), pituicytes in the neuro-hypophysis, and perivascular and marginal astrocytes. 4
After brain ischemia, a massive and extensive astrocyte death in the core of the lesion occurs.6,7 In the surrounding penumbra region, astrocytes become reactive and subsequently form the glial scar.6,7 Several investigations suggest that astrocytes are better preserved than neurons in animal models of stroke inside the core.6,7 Indeed, although in the ischemic core neuronal markers are decreased as soon as 1 hour after middle cerebral artery occlusion (MCAO), GFAP expression, a common marker of fibrous astrocytes, is preserved over the first 3 hours of reperfusion.
3
At later reperfusion periods, GFAP increases in the periinfarct area that later develops into the glial scar (Figure 1). In contrast, Liu
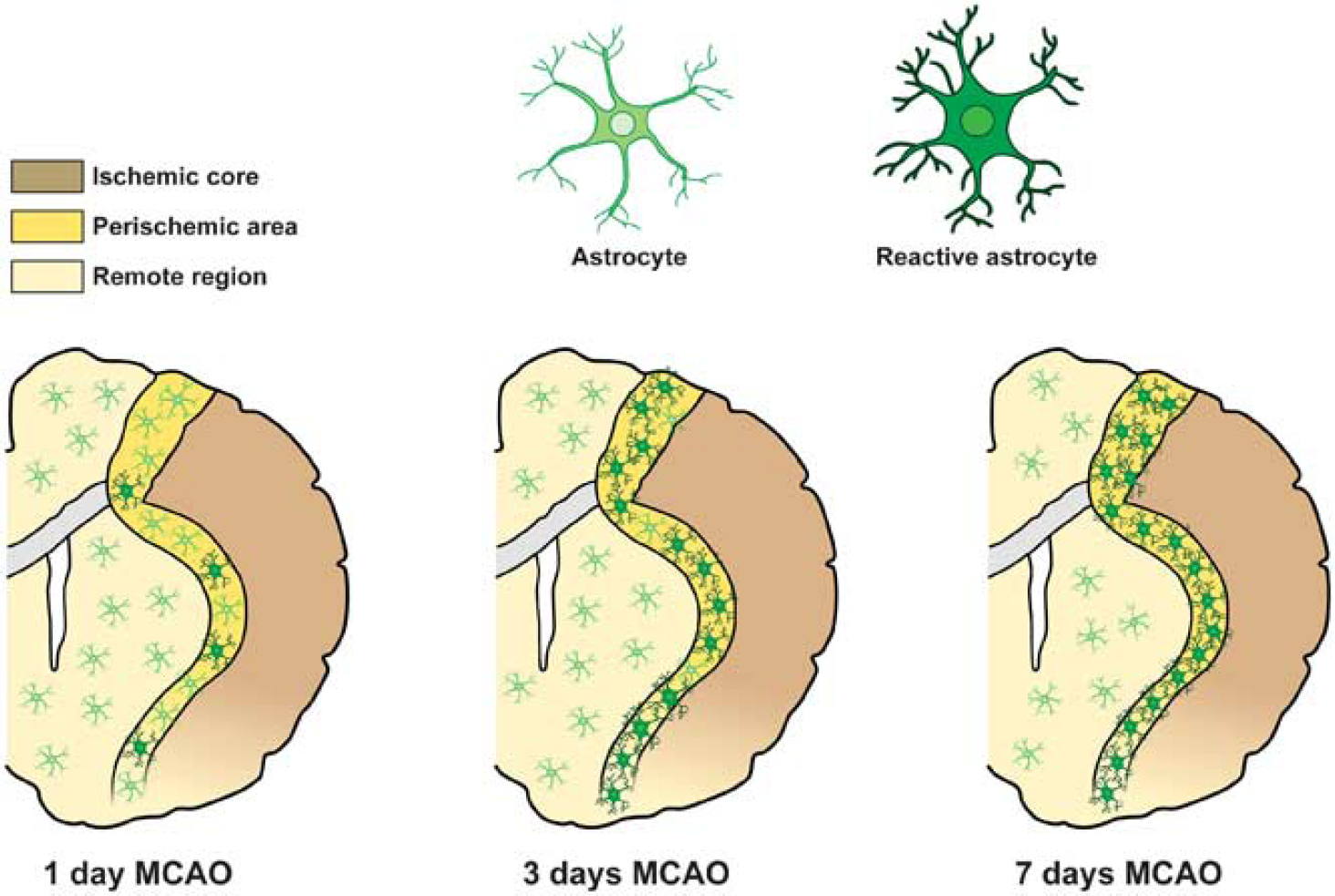
Schematic anatomic diagram describing distribution of the different astrocyte phenotypes in the postischemic brain. MCAO, middle cerebral artery occlusion.
The effects of astrocytes on brain injury are divided into those (I) with immediate influence on cell survival and those (II) with long-term or delayed influence, eventually affecting later recovery. Astrocytes react to stroke by triggering the complex phenomenon known as reactive astrogliosis. 7 Reactive astrocytes undergo dramatic morphological changes 9 and alterations in gene expression. 7 It has been long debated whether reactive astrocytes are harmful or beneficial. In the past few years, both types of effects have been observed10,11 and raise the question of whether there might be different subtypes of reactive astrocytes, elicited depending on the nature and localization of the injury, that differ in their functions.
An expanding and intriguing research direction aimed at unraveling mechanisms by which astrocytes may be more or less vulnerable than neurons to brain ischemia is the study of ionic homeostasis in astrocytes during cerebral injury. Indeed, there is now strong evidence that astrocytes by maintaining ionic homeostasis have a critical role in the processes related to cell death and survival of other brain cells. In fact, glial cells are essential for the maintenance of (1) cellular pH in a range compatible with central nervous system (CNS) cell survival and (2) of Na+, Ca2 +, and K+ homeostasis.
In gray matter, astrocytes are connected by an extensive network of gap junctions 12 permeable to K+. In white matter, astrocytes are much less abundantly coupled. 13 Nevertheless, ionic redistributions in interconnected cells of the CNS are of fundamental importance.
Thus, astrocytes form a syncytium for rapid redistribution of K+ from regions with high neuronal activity to perivascular areas. This astrocytic-mediated involvement in K+ redistribution is primarily mediated by inwardly rectifying K+ channels. 14
Other mechanisms, such as the exchange between K+ and Na+, catalyzed by the astrocytic Na+, K+ -ATPase, contribute to extracellular K+ homeostasis. Active uptake of K+ into astrocytes may also be mediated by the Na+ -K+ -Cl_ cotransport. 15 Particularly relevant is the function of all those transporters, suchas Na+ /H+ exchanger (NHE) and Na+ -HCO3− cotransporter, that by controlling the concentration of H+ and HCO3− ions regulate cellular pH.16,17 In addition, other transporters, including Na+ /Ca2+ exchanger (NCX) and Na+ -K+-Cl_ cotransport, by controlling Ca2+ homeostasis influence gliotransmitter release and cerebral blood flow.18,19 In addition to generally acknowledged Ca2+ excitability of astroglia, recent studies have demonstrated that neuronal activity triggers transient increases in the cytosolic Na + concentration ([Na+] i) in perisynaptic astrocytes. These [Na+]i transients are controlled by multiple Na+ -permeable channels and Na+ -dependent transporters; spatiotemporally organized [Na+] i dynamics in turn regulate diverse astroglial homeostatic responses, such as metabolic/signaling utilization of lactate and glutamate, transmembrane transport of neurotransmitters and K+ buffering. In particular, near-membrane [Na+] i transients determine the rate and the direction of the transmembrane transport of GABA and Ca2 + .20
Microglia in Brain Ischemia
Microglia are the immunocompetent cells of the CNS and account for 10% of the total glial cell population in the brain. 2 During embryonic development, microglia differentiate in the bone marrow from hematopoietic stem cells to monocytes and then travel to the brain, where they settle and further differentiate into microglia. 2
In the brain, microglial cells may appear in resting and multiple activated states, including ameboid and phagocytic. Under physiological conditions, microglial cells are in the resting state, characterized by a small cell body with fine, highly branched processes. In the developing brain, these cells support neuronal differentiation and clearance of cells deemed for elimination through programed cell death. In the mature brain, resting microglia serve the role of immune surveillance and host defence. Any disturbance or loss of brain homeostasis, as it occurs during an ischemic insult, evokes a rapid transformation of resting microglia either into an activated or into a phagocytic state. During this transition, resting microglia retract their processes, increase the size of cell body, modify the expression of enzymes, receptors, and immune response molecules. Microglial cells become motile, and using ameboid-like movements, migrate rapidly to the injury site along the chemokine gradients and also in response to chemoattractants released after the injury. However, if the damage persists and the CNS cells die, microglial cells undergo further transformation and become phagocytes. In fact, besides morphological changes and surface molecule upregulation, activated microglia secrete a wide array of soluble factors. Microglia response to pathological tissue alterations in the postischemic brain may be beneficial when neurotrophic factors, such as IGF-1, NGF, NT-3, NT-4, and BDNF, are released or become harmful when neurotoxic and inflammatory molecules such as free radicals, reactive oxygen species, nitric oxide (NO), superoxide, and fatty acid metabolites molecules are secreted. In addition, they produce a wide range of immunocompetent molecules, which include the proinflammatory cytokines interleukin (IL)1-β, IL-6; IL-3, and the tumor necrosis factor-alpha. All these molecules regulate the inflammatory processes and control the immune response. 2
The signals triggering microglial activation are not completely known. It is recognized that both withdrawal of molecules normally released under physiological conditions, ‘off signals', or release or upregulation of molecules, ‘on signals', might be involved in microglial activation. Neurotransmitters such as ATP and neuropeptides are recognized as ‘on signals'. Microglia are clearly susceptible to cell death induced by severe hypoxia, particularly when combined with aglycemia. 21 The temporal sequence of microglial activation during an ischemic event has been well characterized using animal models of focal cerebral ischemia.22,23 In particular, microglia response begins in the ischemic penumbra within few hours after reperfusion. Its activation is protracted in the periinfarct zone and becomes striking by 3 days and peaks at 7 days. After 14 days, the number of activated microglia in the periischemic area is significantly lower than that after 3 and 7 days. In the ischemic core, the majority of microglial cells degenerate by 12 hours after reperfusion. Later, 24 hours after the insult, several round microglia/macrophage cells invade the ischemic core and dramatically increase within this region throughout the first week after MCAO. At this time, they can no longer be microscopically or immunohistochemically differentiated from invading blood-borne brain macrophages. At later time points, after 2 weeks, the number of round cells decreases. However, the innate microglia, rather than the blood-borne immune cells, are the predominant immunocompetent cells in the brain for the first 3-4 days after ischemia (Figure 2). 24
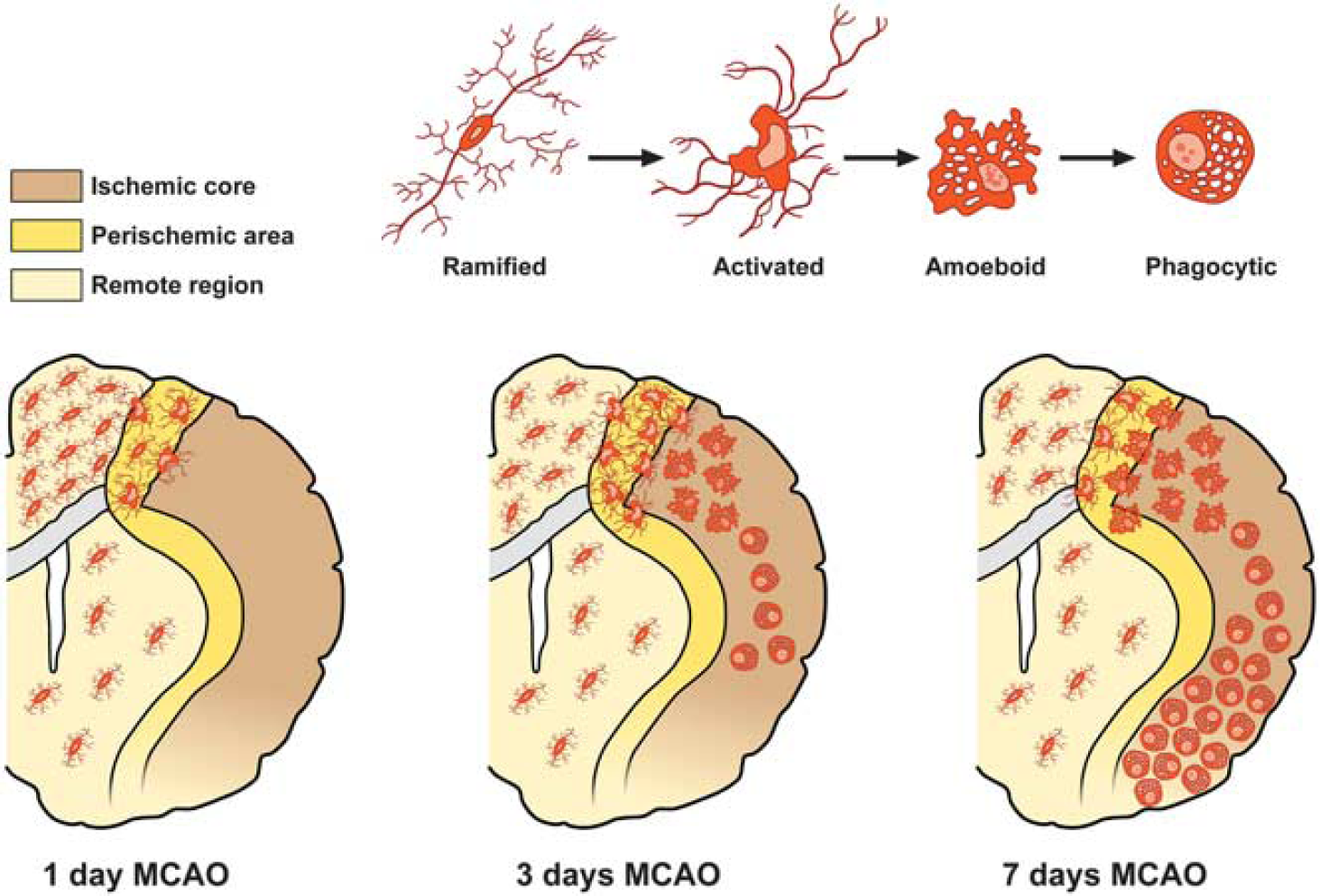
Schematic anatomic diagrams describing the distribution of the different microglia phenotypes in the postischemic brain. MCAO, middle cerebral artery occlusion.
During the last two decades, a large number of papers have been published describing both the detrimental and beneficial roles of microglia in various brain disorders, including stroke. However, the specific conditions that induce microglia to take beneficial phenotypes remain unknown. Furthermore, although in the ischemic brain microglia appears to be a major cellular contributor of postischemic inflammation, there is also growing evidence that some aspects of the inflammatory response are important for tissue repair. 25 Indeed, microglia has been shown to be neurosupportive by removing cell debris, by engulfing polymorphonuclear neutrophiles, 26 and by promoting adult neurogenesis. 27 The benefits of microglial activation have been further demonstrated by several findings: (1) the exacerbation of neuropathology in inducible mouse models lacking microglia, 28 (2) the finding of protective microglia in cases of cerebral ischemia, 26 and (3) the improvement, after CNS injury, of neurite growth and functional recovery after microglia transplantation. 29
Oligodendrocytes in Brain Ischemia
The term oligodendroglia was introduced by Rio Hortega in 1921 to describe those neuroglial cells that show few processes, hence the prefix ‘olig'. 30
In the adult CNS, oligodendrocytes (OLGs) can be classified as follows: (a) OLG precursor cells (OPCs), (b) myelin-forming cells, and (c) non-myelinating OLGs.
Oligodendrocyte Precursor cells
Oligodendrocytes arise from OPCs that colonize both the gray and white brain matter during development. 31 Oligodendrocytes develop from OPCs through distinct phenotypic stages, which can be identified by the sequential expression of specific markers characteristic of progenitors, including A2B5, NG2, PDGFalpha receptor, O4, or of mature and myelinating OLGs, identified by selective markers such as CNPase, myelin-associated glycoprotein, and myelin basic protein. Although many OPCs differentiate into mature myelinating OLGs during the early and much later stages of human brain development, a considerable number of them do persist in the adult brain at the pro-OLG stage, and may provide a source of new OLGs, protoplasmic astrocytes, and neurons.32,33 Because of their apparent stem-cell-like characteristics, adult OPCs have recently gained much attention for their potential reservoir of cells capable of self-renewal, differentiation, and remyelination after CNS injury. 34
Myelin-forming Oligodendrocytes
Myelinating OLGs are classified in four categories, according to the morphology, size, and thickness of the myelin sheath they form. Type I and type II are small cells supporting the short, thin myelin sheaths of 15 to 30 small-diameter axons. Types III OLGs have larger cell bodies and myelinate up to five thick axons. Finally, type IV OLGs are the largest cells forming long, thick myelin sheaths of 1 to 3 large-diameter axons.
The main function of myelinating OLGs is the production of myelin, which insulates axons and facilitates the fast saltatory conduction of action potential. In addition to myelination, these glial cells, by producing and releasing several neurotrophins, such as NGF, BDNF, and NT-3, can provide trophic support on nearby neurons. 35 Furthermore, myelinating OLGs use the lactate transporter MCT1 to provide metabolic support to neurons. 36
Non-myelinating Oligodendrocytes
A population of non-myelinating OLGs attached to large neurons are present in the gray matter and are known as ‘perineuronal’ or ‘satellite’ OLGs. Satellite OLGs have generally received little attention and their precise function is still unknown. Previous
Among the different glial cell types, gray and white matter OPCs and OLGs are the most vulnerable to perturbations of ionic homeostasis, as it occurs in stroke. 39 Their vulnerability to ischemia, comparable to that occurring in neurons, is owing to their high sensitivity to energy deprivation, oxidative stress, and hypoxia. Damage to OLGs during cerebral ischemia causes demyelination, white matter dysfunction, and axonal impairment (Wallerian degeneration). Actually, demyelination is a pathological process in which myelin sheaths are lost around axons with consequent alteration of conduction and impairment of sensation, movement, cognition, or other functions depending on the nerves involved. White matter ischemia that is usually more severe than gray matter ischemia because of the lower blood flow, and little collateral blood supply 39 is a clinically important part of stroke. Indeed, white matter lesions are often observed in stroke patients and have been thought to contribute mainly to cognitive impairment. 40
Glutamate-mediated excitotoxicity is implicated in the loss of myelin and OLGs occurring in stroke.41,42 OPCs and OLGs located in the infarct core undergo anoxic depolarization rapidly. Loss of ion homeostasis triggered by ischemia causes acute axonal depolarization and elevation in axoplasmic [Na]i+; these changes result in reversal of the Na+ /glutamate transporter and massive release of glutamate. Glutamate, in turn, triggers Ca2+ entry into OLGs mainly through AMPA receptors. Oligodendrocytes do not possess the GluR2 AMPA subunit rendering their AMPA receptors permeable to Ca2+ ions. Oligodendrocyte precursor cells, which express high levels of AMPA/Kainate receptors, appear to be the most vulnerable to glutamate toxicity. Depending on the intensity of the insult, Ca2+ overload in OLGs triggers oxidative stress and mitochondrial damage, with subsequent induction of necrosis or apoptosis.
Although a rapid decrease in the number of OLGs, OPCs, and myelin density occurs in the infarct core 1-2 days after tMCAO, the periinfarct area, including white matter regions bordering the lesion, exhibits a steady increase in the number of OPCs 3 and 7 days after ischemia onset and a gradual recovery of OLGs 2 weeks after MCAO.43,44 Periinfarct OLGs progressively increase the expression of myelin marker PLP and MBP from 24 hours to maximal levels at day 7 after tMCAO (Figure 3). 45 Interestingly, in the periischemic ischemic lesion, NG2 cells display multipotent differentiation after focal ischemia. 44
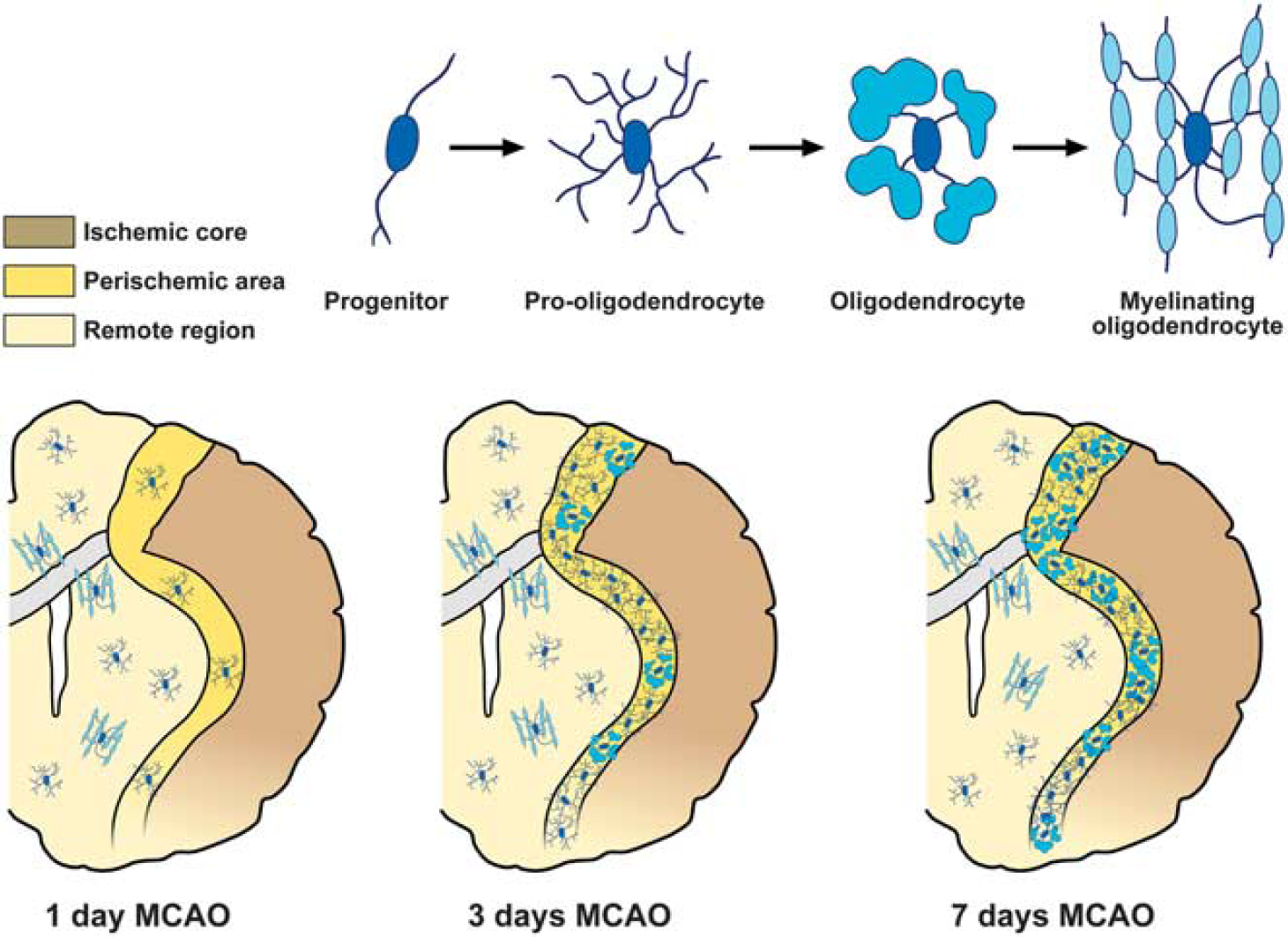
Schematic anatomic diagrams describing the distribution of the different adult oligodendrocyte (OLG) progenitor cells (OPCs) and OLG phenotypes in the postischemic brain. MCAO, middle cerebral artery occlusion.
Alterations in ionic homeostasis may have a crucial role in OPC response during demyelination and remyelination processes. In fact, changes in ionic homeostasis, in particular the increase in [Ca2+]i levels, not only influence the developmental processes that accompany the transition of OPCs into mature myelinating OLGs, but also intervene in the initiation of myelination and remyelination processes. 46 Furthermore, the increase in [Ca2+] i levels influences the developmental processes that accompany the transition of human OPCs into mature myelinating OLGs, including OPC migration, lineage progression, and differentiation.47,48
Finally, as the ionic fluxes through plasmamembrane transporters in OLGs or OPCs are involved in several functions regulating OLGs development, cell death, or repair processes, it would be valuable to further elucidate the role of selective transporters expressed on these cells (Figure 4).
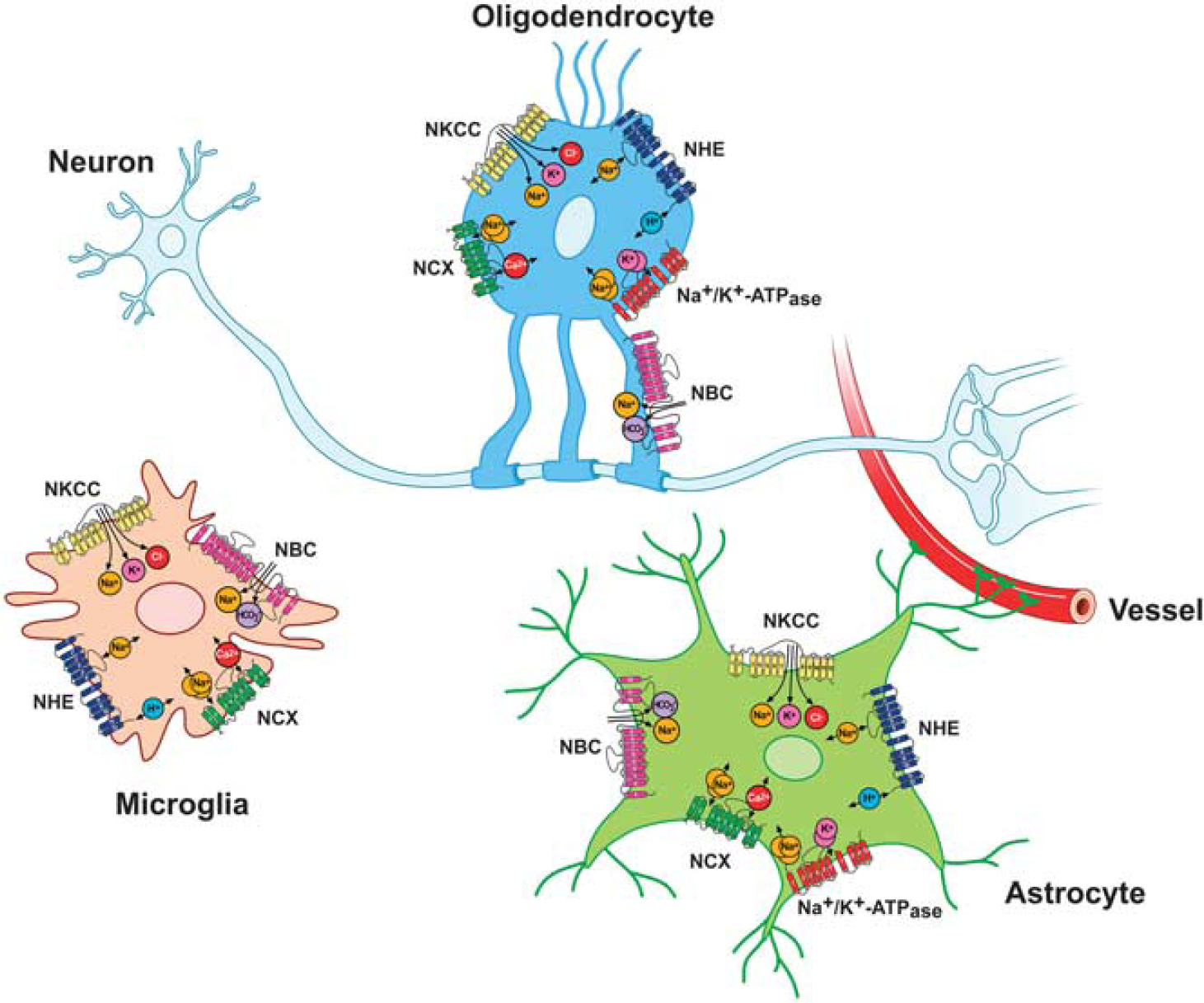
Major ionic transporters expressed in microglia, astrocytes, and oligodendrocytes (OLGs). NBC, Na+ /HCO3− cotransporter; NCX, Na+ /Ca2+ exchanger; NHE, Na+ /H+ exchanger; NKCC, Na+ -K+-Cl− cotransporter.
EXPRESSION AND FUNCTION OF IONIC TRANSPORTERS IN GLIAL CELLS DURING PHYSIOLOGICAL CONDITIONS AND IN BRAIN ISCHEMIA
Na + /Ca2 + Exchanger
The NCX is an ionic transporter that exchanges three Na+ ions for one Ca2 + ion.49,50 When [Ca2+] i rise, this exchanger couples the uphill extrusion of Ca2+ ions to the entry of Na+ ions into the cells. This mode of operation, defined as forward mode, keeps the 10 4 -fold difference in [Ca2+]i across the cell membrane. In other physiological or pathophysiological conditions when the [Na+]i rise, NCX reverses its mode of operation and mediates the extrusion of Na+ and the entry of Ca2+ ions. This mode of operation is defined as reverse mode. Na+ /Ca2 + exchanger is composed by nine transmembrane segments (TMSs) 51 that can be divided into an N-terminal hydrophobic domain, composed of the first five TMS1-5, and into a C-terminal hydrophobic domain, composed of the last four TMS6-9, separated through a large intracellular loop named f loop (Figure 5). 51 The f loop is responsible for the regulation of NCX activity elicited by several transductional mechanisms, such as H+, Ca2+, and Na+ ions, NO, phosphatidylinositol, PKC, PKA, and ATP. 49
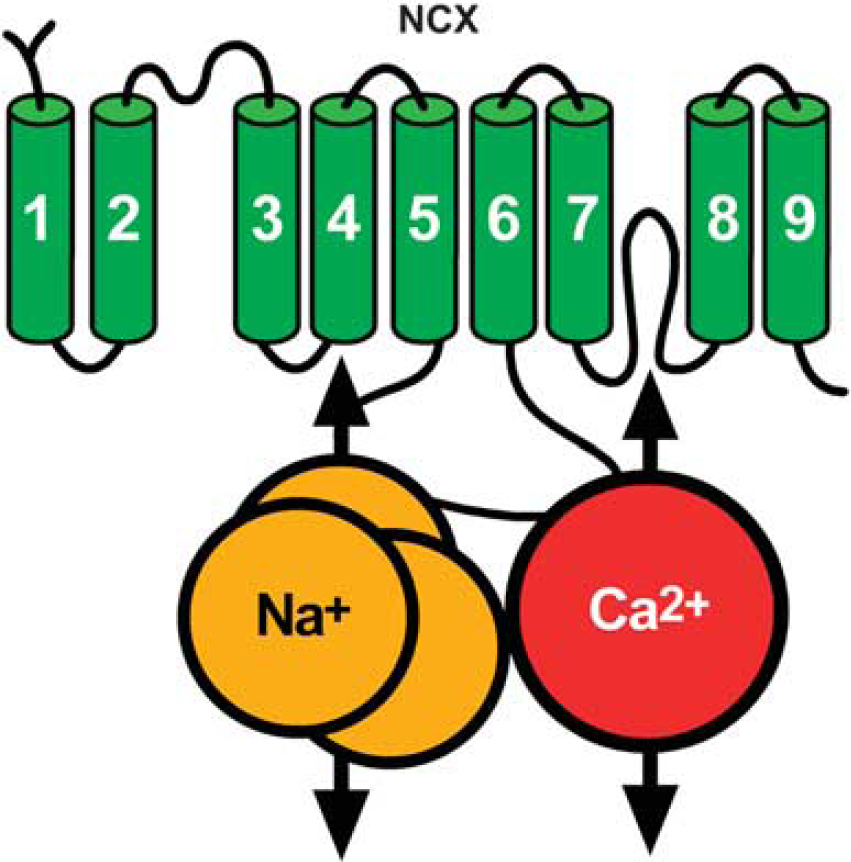
Putative topology of Na+ /Ca2+ exchanger (NCX).
Three genes coding for the three different NCX1-352-54 proteins have been identified. Na+/Ca2+ exchanger 1 displays an ubiquitous expression, whereas NCX2 and NCX3 are present exclusively in neuronal and skeletal muscle tissues. 55 Notably, these NCX isoforms have a fundamental role in the pathophysiology of stroke damage. In particular, NCX1 and NCX3 downregulation or genetic ablation worsens ischemic damage in mice and rats,56-58 whereas its pharmacological activation determines a reduction in the brain infarct volume. 59
The most important role played by NCX1 in astrocytes is related to the regulation of gliotransmitter release. 18 Hence, mild depolarization of astrocytes forces NCX to operate in the reverse mode and generates cytosolic Ca2+ increases leading to release of gliotransmitters, such as glutamate.62,63
Only few studies have investigated the pathophysiological role of astrocytic NCX in ischemic astrocytes. In
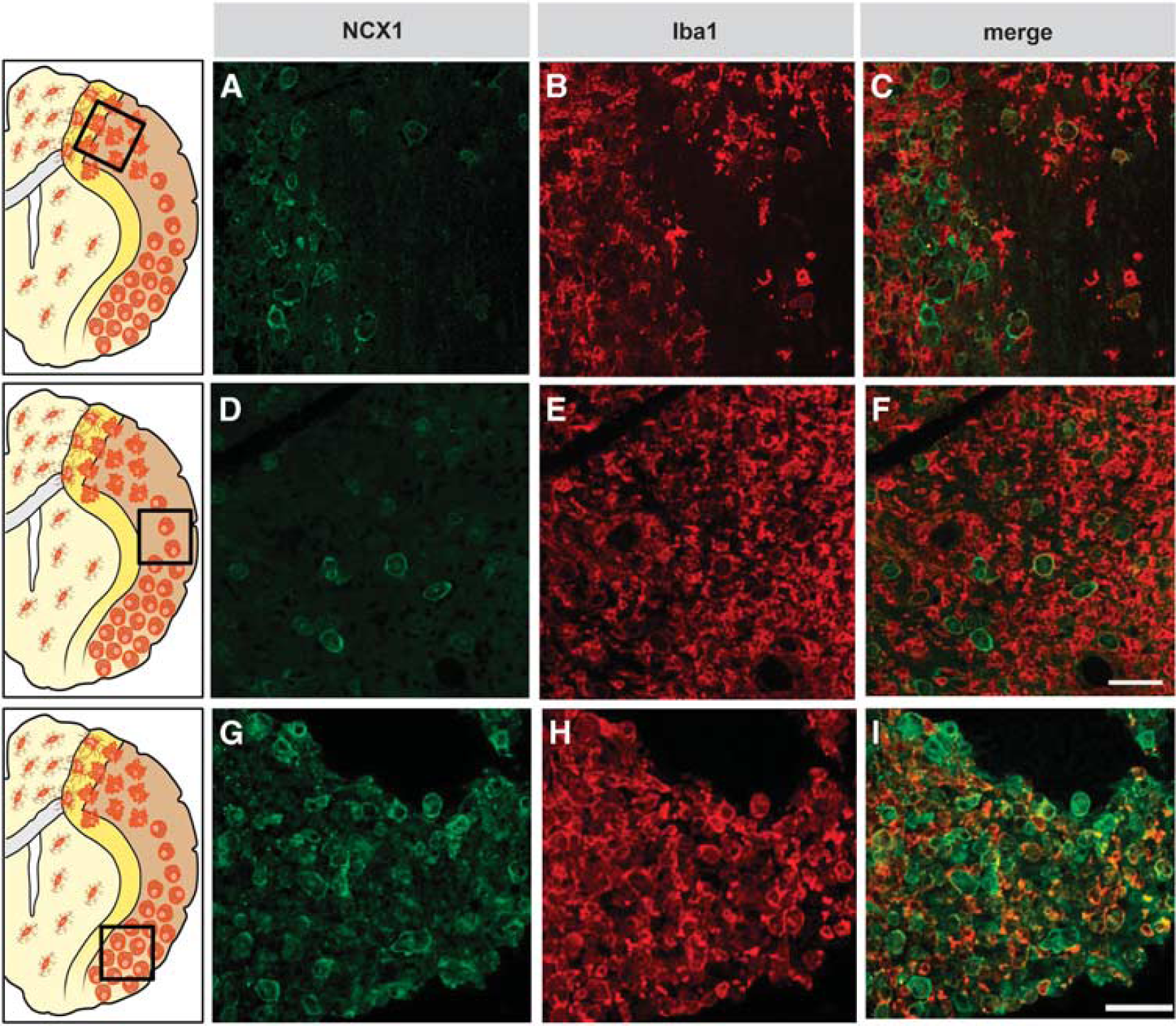
(
To further explore NCX1 expression and NCX activity in microglial cells of the infarct core, Boscia
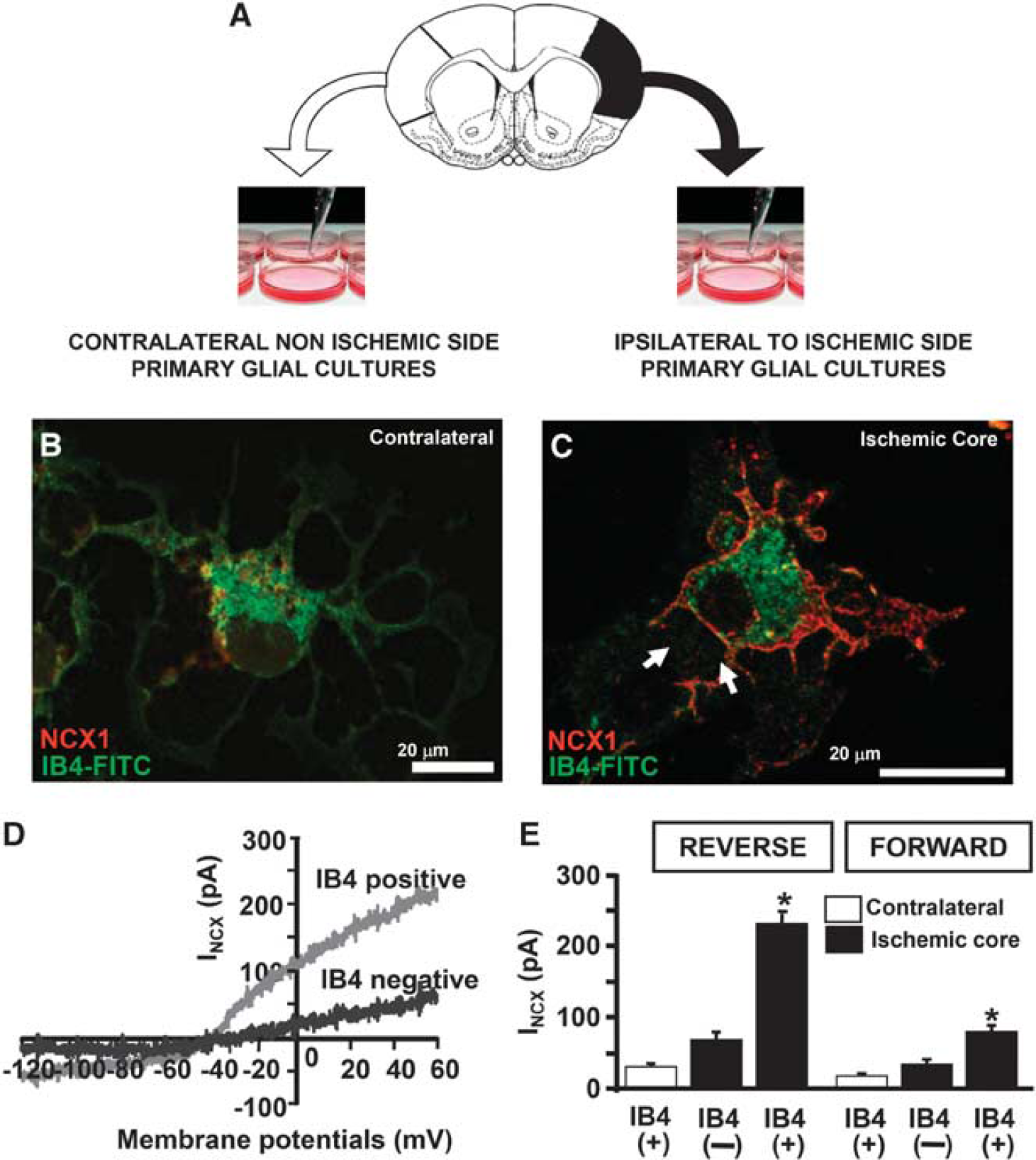
(
When NCX1-silenced BV2 cells are exposed to OGD plus 6 hours of reoxygenation, the increase in [Ca2+]i is completely prevented. This finding, together with the results showing that the protein expression of the other NCX isoforms—NCX2 and NCX3—significantly decreases after OGD, demonstrates the relevant role played by the NCX1 isoform in round phagocytic microglia during hypoxic conditions.
Previous studies have demonstrated a central role of NCX in anoxic and ischemic injury of both central and peripheral myelinated axons. Indeed, NCX reverse mode of operation has been implicated in axonal damage during spinal cord anoxia, 74 stretch injury of axons, 75 and experimental allergic encephalomyelitis. 76 In OLGs, NCX1 and NCX3 are co-localized with Na+ -K+-Cl− cotransporter 1 (NKCC1). The reverse-mode operation of NCX and NKCC1 has been also implicated in OLG cell death induced by AMPA-mediated excitotoxicity. Activation of AMPA receptors leads to NKCC1 phosphorylation that, in turn, enhances NKCC1-mediated Na+ influx. The latter triggers NCX in the reverse mode of operation with consequent Ca2+ overload, thereby compromising mitochondrial function and cellular viability. 77
Na+ /H+ Exchanger
The mammalian NHE family is a group of integral membrane transport proteins that mediates an electroneutral 1:1 exchange of intracellular H+ for extracellular Na+ and in doing so regulates pHi homeostasis and cell volume. 78 All members of NHE family are composed of 12 transmembrane domains and contain two functional domains: the N-terminal transmembrane domain, which is necessary and sufficient to catalyze ion translocation, and the C-terminal cytoplasmic domain, which is crucial for modulating NHE activity (Figure 8). 78 To date, nine isoforms have been cloned. These isoforms differ in their tissue expression, subcellular distribution, kinetic properties, inhibitor sensitivity, and physiological functions. Na+/H+ exchanger 1 is ubiquitously expressed and is considered to be a ‘housekeeping’ isoform. Na+ /H+ exchanger 2-3 are highly expressed in the apical epithelia of the kidney and intestine. Na+ /H+ exchanger 4 is mainly present in the stomach and basolateral epithelia of the kidney. Na+ /H+ exchanger 5 predominantly resides in the brain. Na+ /H+ exchanger 6-9 are targeted to the membranes of intracellular organelles. Na+ /H+ exchanger 1 is by far the most extensively studied isoform and is associated with many physiological and pathophysiological conditions. 79
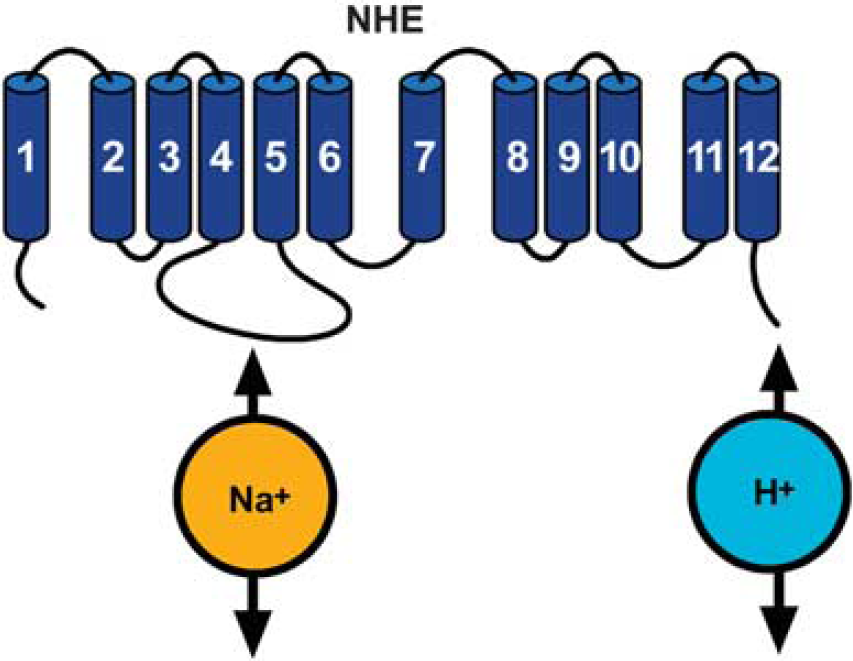
Putative topology of Na+ /H+ exchanger (NHE).
As NHE is involved in numerous essential cell functions, its activity is crucial for the correct cell functioning even under pathophysiological conditions. Indeed, the pharmacological blockade of NHE-1 activity reduces infarct volume in animal models of brain ischemia. 80 In addition, ischemia/reperfusion determines an increase in NHE-1 expression/activity. 78
Na+ /H+ exchanger-1 is the major protein involved in the regulation of pHi in activated microglia, whereas bicarbonate transporters (see below section Na+ /HCO3− cotransporter) have only a minor role. Blockade of NHE-1 function with the more potent and selective NHE inhibitor cariporide (HOE642) significantly acidifies primary microglia and abolishes the H+ extrusion. Liu
Thus, given that NHE regulates OLG pHi, it can be hypothesized that during ischemic damage the modulation of NHE in OLG lineage may give rise to a neuroprotective effect.
Potassium-chloride Cotransporters
Four distinct but related genes encoding potassium-chloride cotransporter (KCC) have been cloned, KCC1-4, all of which have 70% sequence identity to each other.
86
They share the same topology with 12 putative membrane-spanning domains, a large extracellular loop between fifth and sixth transmembrane domains, a short cytoplasmic amino terminus with a long carboxy terminus.
86
All
Potassium-Chloride Cotransporter in Astrocytes
The expression pattern of KCC isoforms in astrocytes reveals strong expression levels for KCC1, KCC2, and KCC3. 89 , 90
Maintenance and regulation of cell volume is a major function of KCC as demonstrated for red blood cells and many other cell types.89,90 Furthermore, a similar role for KCC has been recently reported also in astrocytes. 90 As KCC is involved in brain cell volume regulation, it may also have a role during the development of ischemic or traumatic brain edema, pathological conditions where cell volume regulation is disturbed.
Potassium-Chloride Cotransporter in Microglia
All four KCC subtypes are constitutively expressed in primary microglial cells and BV2 microglia cell line. 91 Potassium-chloride cotransporter cotransporters participate in cellular mechanisms that promote the transformation of microglial cells from ramified into ameboid morphology. Indeed, changes in cell phenotype after lysopho-sphatidylcholine-induced microglia activation are prevented by the simultaneous inhibition of cation channels and KCC with Gd3+ and the potent KCC blocker DIOA, respectively. 92 More recently, the involvement of KCC in the formation of lamellipodial protrusion during microglial cell migration has been demonstrated. 91 Ameboid microglia form broad lamellipodia, required for cell migration and phagocytosis of cell debris. After ischemia, the increase in extracellular K+ concentration reverses flux direction of KCC with a consequent increase in intracellular osmolarity. Such increase evokes volume changes that are essential for the sprouting of a lamellipodium. When BV-2 cells or primary microglial cells are exposed to a high KCl (70-135 mmol/L) or hypoosmotic saline solution they do not show an equal swelling in each direction but develop a delicate extension at one pole of the cell, which then proceeds rapidly into a vigorously moving lamellipodium within seconds. After the formation of a lamellipodium, the cell starts to migrate. Blockade of chloride influx with DIOA significantly prevents the formation of lamellipodia. 91
Potassium-Chloride Cotransporter in Oligodendrocytes
In OLGs and Schwann cells, KCC may participate in the regulation of cell volume under physiological and pathological conditions. Potassium-chloride cotransporter 2 signal is detected within the cell bodies of OLGs, whereas KCC3 expression is abundantly observed in white matter-rich regions of the brain, including the myelinated tracts of the internal capsule, corpus callosum, and spinal cord.93,94 Loss-of-function mutations of the
Oligodendrocyte damage under anoxia/ischemia may be attributable more to volume alterations in myelin sheath than to cytoskeletal dissolution as it occurs in axons. 97 As a consequence, altered conduction along white matter tracts will be impaired. Potassium-chloride cotransporter may also contribute to such Cl− -dependent volume alterations under ischemic conditions. In fact, combined inhibition of Na+ influx and K+ and Cl− efflux, obtained through the blockade of KCC, protects white matter against anoxia better than Na+ -channel blockers alone. 98
Na+ -K+-Cl− Cotransporter
The electroneutral NKCC mediates the influx of Na+, K+, and Cl− with a stoichiometry of 1Na+:1K+:2Cl− . 99
To date, only two distinct isoforms, NKCC1 and NKCC2, have been identified. Na+ -K+ -Cl− cotransporter 1 has a broad tissue distribution, whereas NKCC2 is only found in vertebrate kidney. Na+ -K+-Cl− cotransporter serves multiple functions, including ion and fluid movements in secreting or reabsorbing epithelia and cell volume regulation. 99 The structure of NKCC1 consists of 12 putative transmembrane domains, flanked by large cytoplasmic amino and carboxyl termini, which contain regulatory phosphorylation sites (Figure 9). 99
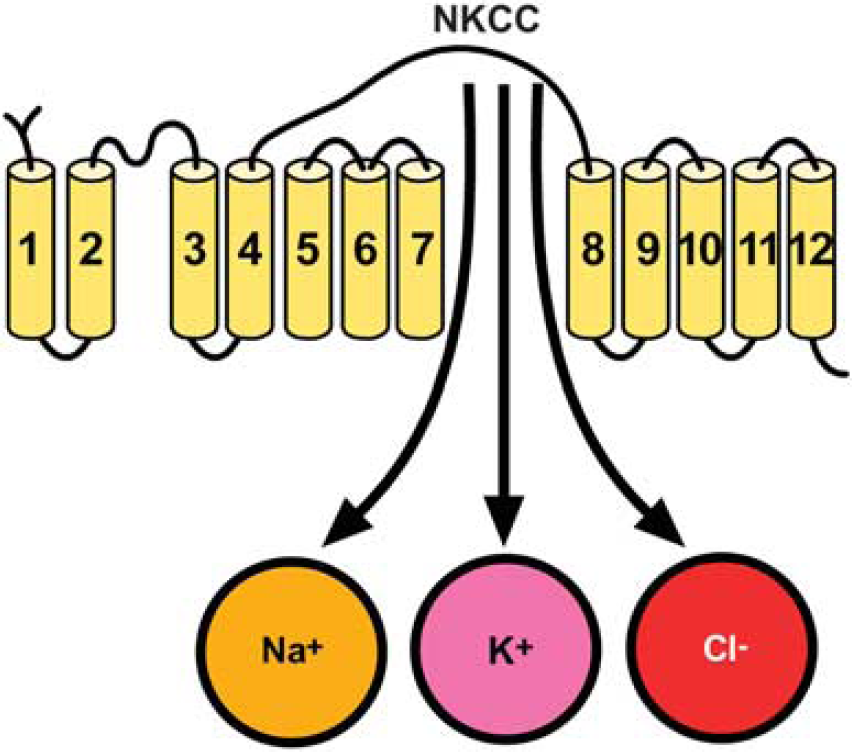
Putative topology of Na+ -K+ -Cl− cotransporter (NKCC).
Na+ -K+ -Cl− cotransporter 1 protein is expressed in neurons throughout the brain where it contributes to the maintenance of [Cl−]i. Thus, NKCC1 may affect neuronal excitability by regulating [Cl−]i. Expression of NKCC1 protein has also been found in astrocytes and OLGs. 100 The pharmacological inhibition of NKCC1 with bumetanide 101 or its transgenic ablation significantly attenuates infarction and swelling after tMCAO. 99 Therefore, NKCC1 represents an attractive target for novel therapeutic strategies against stroke.
Another aspect that establishes a direct relationship between astrocytic NKCC and progression of ischemic damage is the influence that NKCC exerts on glutamate concentration. Indeed, astrocytic NKCC1 stimulation takes part to excitotoxic injury by increasing swelling-induced glutamate release or decreasing glutamate clearance from the extracellular space.104,105,109
Another key element promoting NKCC1-dependent cell damage in astrocytes during ischemia is represented by the loss of the plasmamembrane Na+ gradient.
110
In this regards, the activation of several Na+ entry pathways, including NKCC1, also contributes to ischemia-induced loss of Na+ homeostasis. In fact, OGD in astrocytes results in both an increase in NKCC1 phosphorylation and activity.
107
This stimulation of NKCC1 activity is accompanied by an accumulation of Cl− and Na+ in astrocytes.
111
Na+ -K+ -Cl− cotransporter 1-mediated increases in [Na+]i after
Na+ /HCO3− Cotransporter
Na+ /HCO3− cotransporter, 12 transmembrane domains containing an intracellular C and N terminus (Figure 10), represents, together with NHE, one of the main cellular systems involved in the regulation of ionic and pH homeostasis in the CNS 117 where it can transport either two or three bicarbonate ions per one sodium ion. 117 Several evidence demonstrate that exposure to acute and chronic hypoxia or ischemia produces changes in intracellular pH in neurons and glia, 118 and more severe acidosis correlates with more severe injury, 118 therefore NBC modulation may profoundly influence ischemic pathophysiology.
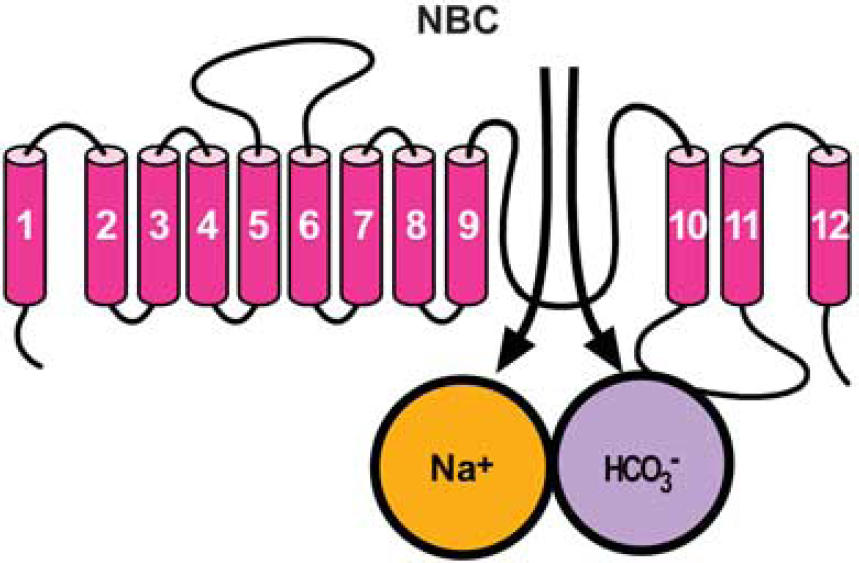
Putative topology of Na+ /HCO3− cotransporter (NBC).
Several evidence reported that NBC in astrocytes may modulate neuronal excitability through pH changes. In fact, when a neuron fires an action potential, there is an increase in extracellular K+ that depolarizes neighboring astrocytes. This depolarization leads to stimulation of NBC activity in astrocytes and transport of Na+, HCO3− and net-negative charge into the cells. The ensuing decrease in pHo tends to dampen further neuronal activity by inhibiting many pH-sensitive voltage- and ligand-gated channels. This negative-feedback model is predicted to be neuroprotective under pathophysiological conditions associated with ischemia.
Jung
Na+ /K+-ATPase
The Na+/K+-ATPase is an important protein complex ubiquitously expressed in the human body128-130 that mediates the active exchange of cytosolic sodium for extracellular potassium in a 3:2 ratio, a process required for the maintenance of transmembrane ionic gradients for all mammalian cells, which in turn is essential for setting the cellular resting potential, regulating osmolarity, and powering the secondary transport of other important solutes, such as calcium and protons.131,132 As the brain is the primary consumer of ATP, the sodium-potassium pump is particularly vulnerable to ATP depletion, a condition characterizing ischemic stroke. This vulnerability suggests that pharmacological inhibition of the Na+/K+ -ATPase could further compromise ATP-depleted neurons, nevertheless there is accumulating evidence that inhibiting the sodium-potassium pump can actually provide neuroprotection in the context of ischemia.131,133 The debate on the neuroprotective or neurodetrimental role of Na+/K+-ATPase in brain ischemia is still open and several research groups are carrying out their research in the attempt to solve this question.
The Na+/K+-ATPase consists of a heterodimeric core of a and β subunits that may be accompanied by a third γ subunit.131,132 The expression of different α and β isoforms is regulated in both a developmental and tissue-specific manner. 134 The α1, α2, and α3 isoforms are expressed in the brain. 135 In particular, the α2 isoform is widely expressed in neurons during development 136 but becomes confined primarily to glia and few types of neurons in the adult brain;134,137 in contrast, the α3 isoform shows neuronal expression in the adult.134,137 Similar to the a subunit, the three b subunit isoforms are expressed to varying degrees in the brain, both in neurons and glia.134,138,139
In effect, there is a substantial heterogeneity among reactive astrocytes, with some close to the ischemic lesion showing decreased buffering capacity and those in the penumbra region showing a higher ionic buffering ability. 142
IONIC TRANSPORTERS AS POTENTIAL TARGET TO MODULATE THE ACTIVITY OF ASTROCYTES, MICROGLIA, AND OLIGODENDROCYTES IN BRAIN ISCHEMIA
In the last few years, the development of neuroprotective strategies for brain ischemia has been primarily focused on targeting neuronal cell death. However, considering the role played by the different glial cells in neuronal survival, in debris removal and in functional remodeling after stroke, these cells are becoming a promising target to enhance an effective and additional therapy for brain ischemia. Thus, maintenance of supportive glia function or limitation of pathological processes needs to be considered when designing future stroke neurotherapeutics.
Although few studies have specifically targeted glial cells for their protective role in stroke, there is some evidence indicating that glial cells can actually represent a successful target to setting on effective strategies against stroke.
Indeed, recent results indicate that induction of BDNF in astrocytes by galectin-1 reduces neuronal apoptosis in ischemic boundary zone and improves functional recovery. 149 In addition, enhancing astrocyte resistance to ischemic injury by overexpressing protective proteins or antioxidant enzyme results in improved survival of CA1 neurons after forebrain ischemia. 150
In addition, recent work suggests that the maintenance of ionic homeostasis, intracellular pH control, and cell swelling in glial cells may contribute to the modulation of brain damage after stroke.
Concerning the modulation of ionic transporters expressed in glial cells, preliminary studies indicate NHE as a potential druggable target in stroke interventions. Indeed, the genetic ablation of NHE-1 reduces the OGD-mediated rise in [Na+]i and swelling in cortical astrocytes, suggesting that the inhibition of NHE-1 activity reduces ischemic injury through an effect mainly mediated by astrocytes.
79
Another important target could be the plasmamembrane exchanger NCX;in fact, Kintner
DISCLOSURE/CONFLICT OF INTEREST
The authors declares no conflict of interest.