
Abstract
Knowledge about the dynamic metabolism and function of cerebrospinal fluid (CSF) physiology has rapidly progressed in recent decades. It has traditionally been suggested that CSF is produced by the choroid plexus and drains to the arachnoid villi. However, recent findings have revealed that the brain parenchyma produces a large portion of CSF and drains through the perivascular glymphatic system and meningeal lymphatic vessels into the blood. The primary function of CSF is not limited to maintaining physiological CNS homeostasis but also participates in clearing waste products resulting from neurodegenerative diseases and acute brain injury. Aneurysmal subarachnoid hemorrhage (SAH), a disastrous subtype of acute brain injury, is associated with high mortality and morbidity. Post-SAH complications contribute to the poor outcomes associated with SAH. Recently, abnormal CSF flow was suggested to play an essential role in the post-SAH pathophysiological changes, such as increased intracerebral pressure, brain edema formation, hydrocephalus, and delayed blood clearance. An in-depth understanding of CSF dynamics in post-SAH events would shed light on potential development of SAH treatment options. This review summarizes and updates the latest physiological characteristics of CSF dynamics and discusses potential pathophysiological changes and therapeutic targets after SAH.
Keywords
Introduction
Cerebrospinal fluid (CSF) is a clear, colorless fluid that fills the space around/within the brain and spine. It was first revealed in the 19th century that the CSF was secreted by the choroid plexus within the ventricles, collected in the arachnoid villi, and absorbed by the superior sagittal sinus. 1 Since then, the CSF was believed to primarily function as a physical buffer and waste sink for the central nervous system (CNS).1,2 Ongoing advancements have broadened our understanding of the physiological role of CSF in brain metabolism and function. The CSF is reportedly produced by the choroid plexus and the brain parenchyma.2,3 Arachnoid villi may not be the only pathway of CSF drainage due to the newly identified meningeal lymphatic drainage 4 and glymphatic system.5,6 CSF circulation participates in maintaining CNS homeostasis, which stabilizes intracerebral pressure (ICP), cerebral blood perfusion, solute (e.g. glucose, ions, and proteins) concentration, and pH levels.7,8 Conversely, CSF is responsible for transporting nutrients and hormones, as well as waste protein (e.g. beta-amyloid and tau) clearance via its dynamic fluid circulation. 9 Given the critical physiological function of CSF, emerging evidence has shown that CSF circulation was impaired in both chronic neurodegenerative disorders and acute brain injury, which may contribute to the pathology of disease progression and poor outcome.3,5,9–11
Subarachnoid hemorrhage (SAH), often caused by ruptured cerebral aneurysm, is a life-threatening subtype of stroke that results from bleeding into the subarachnoid space. The mortality rate of SAH patients ranges from 8.3%–66.7%. 12 SAH survivors often have a decreased quality of life due to the high incidence (nearly 45%) of neurological and cognitive impairment. 13 Neurological deficits following SAH primarily result from the initial injury and secondary pathological response.9,14 The initial injury, also known as the first part of early brain injury (EBI), occurs in the first 72 h after SAH and is characterized by increased ICP, as well as reduced cerebral perfusion pressure (CPP) and cerebral blood flow (CBF), inevitably leading to global ischemia. 15 This ischemia-induced hypoxic state further results in mitochondrial failure in neurons and glial cells, and initiates cytotoxic edema formation and cell death. 16 In addition, ischemia also leads to astrocytic and microglial activation and endothelial cell death, further triggering an inflammatory response and blood-brain barrier (BBB) dysfunction. 17 Ionic homeostasis disruption, excitotoxicity, and oxidative stress occur simultaneously after initial ischemia.17–19 In this process, toxic products of subarachnoid blood (e.g. hemoglobin, heme, Fe2+) and EBI metabolites (e.g. Damage-associated molecular patterns (DAMPs), cytokines) are transported by CSF movement, and further aggravate the environment within and around the brain. This contributes to the secondary injury after SAH, which is considered the second part of EBI. The secondary injury reportedly lasts for 2–4 weeks after SAH, and leads to delayed neuronal death, neuroinflammation, cerebral vasospasm, and chronic hydrocephalus. 20
Undoubtedly, ischemia and toxic products of subarachnoid blood are responsible for the poor outcomes following SAH. Abnormal CSF flow is suggested to be an important participant of both short- and long-term pathophysiological sequelae of SAH, such as increased ICP, decreased CPP, brain edema formation, as well as acute and chronic hydrocephalus. 17 Meanwhile, the clearance of blood and its lysis products by the CSF circulation is considered an important mechanism of self-repair in the brain. 21 While surgical interventions, such as lumbar drainage and external ventricular drainage, are widely used to accelerate SAH blood clearance in the clinical setting, the mortality and morbidity remain high. 22 An in-depth understanding of the role that CSF dynamics have in SAH pathological process would shed light for SAH treatment. In this review, we summarize and update the physiological characteristics of CSF dynamics, and discuss potential pathophysiological changes and therapeutic targets after SAH.
Physiology of CSF dynamics in normal brain
CSF formation
Currently, there are two known sources of CSF production within the brain: the choroid plexus system and the brain parenchymal system.2,9,11,23 The choroid plexus system, considered the main source of CSF production, is reportedly responsible for secreting nearly 80% of CSF.2,9,24 Recently, the bulk fluid exchange, which occurs between the brain interstitial fluid (ISF) and the capillaries within the brain parenchyma, has also been proposed to participate in CSF formation. 25 In the following section, we will briefly introduce the roles of these two systems in CSF formation (Figure 1).
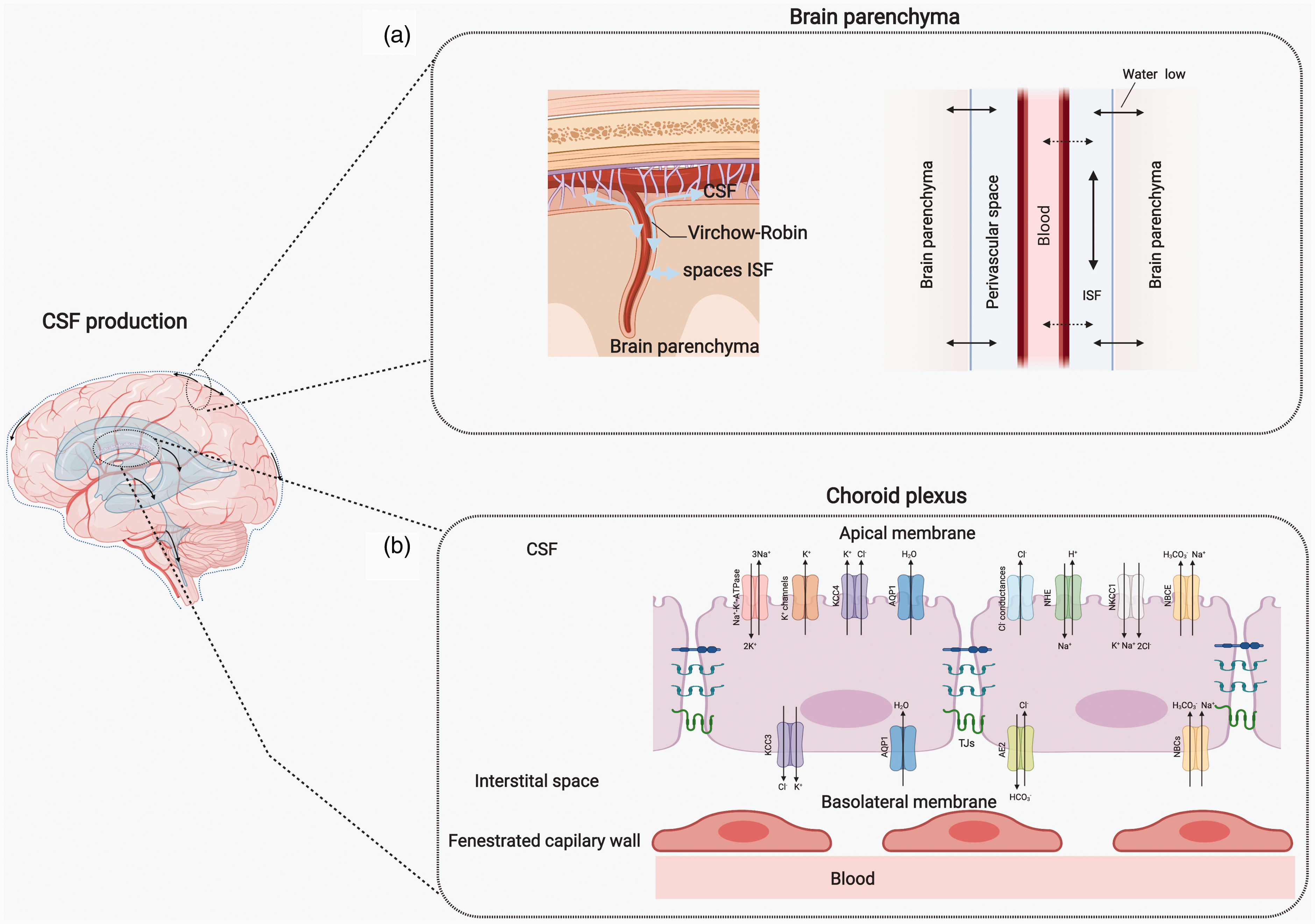
CSF production. a) The BBB and brain parenchyma are currently considered critical components of CSF formation. A large amount of fluid is exchanged bi-directionally among blood, CSF, and interstitial fluid within the perivascular space and the glymphatic system contributes to the CSF production. b) Traditionally, CSF is primarily produced by the choroid plexus in cerebral lateral ventricles, comprising cuboidal epithelium and fenestrated capillary endothelium. A variety of transporters on the basolateral (plasma side) and apical (CSF side) membranes of choroid plexus epithelium facilitate CSF formation. The apical membrane is occupied by the Na+-K+-ATPase, K+ channels (Kv1.3; Kv1.1 and Kir7.1), Cl- conductances (VRAC and Clir), potassium cotransporters NKCC1 (Na+-K+-2Cl−) and KCC4 (K+-Cl−), Na+/H+ exchanger (NHE1), Na+-HCO3− cotransporters NBCE, and water channel aquaporin (AQP)1. Transporters on the basolateral membrane of the choroid plexus epithelium are the K+-Cl− cotransporter, KCC3, Na+-HCO3− cotransporters (NBCn and NCBE), Cl−/HCO3− exchanger AE2, glucose transporter-1 (GLUT1), and AQP1.
Choroid plexus system
Choroid plexuses are located in the cerebral ventricular system, which remain the relatively well-known anatomical structure responsible for CSF production. 23 The choroid plexuses are an epithelial-endothelial unit consisting of central capillaries with fenestrated endothelium and cuboidal epithelium. 24 The cuboidal epithelial cells on ventricular site stay tightly connected with the assistance of the tight junction (TJ) proteins, which controls the diffusion of excess fluid and plasma contents. The main TJ proteins include the occludins, claudins, and Zonula occludens (ZO) proteins. 24
Due to different hydrostatic pressure and solutes (ions, pH, and proteins) found between CSF and plasma, 9 hydrostatic pressure and osmotic gradients between the blood, choroid plexus epithelial cells, and CSF are the proposed potential drivers of CSF secretion. 3 However, CSF formation is found relative insensitive to the alteration of ICP or plasma osmolarity. 2 This insensitivity is considered may be caused by the existence of many ion and water transporters found on the choroid plexus epithelial cells. 24 These transporters are divided into basolateral (plasma side) and apical (CSF side) transporters according to their facing side.23,24 The apical membrane is occupied by Na+-K+-ATPase, K+ channels (Kv1.3; Kv1.1, and Kir7.1), Cl– conductances (VRAC and Clir), potassium cotransporters (NKCC1 (Na+-K+-2Cl−) and KCC4 (K+-Cl−)), Na+/H+ exchanger (NHE1), Na+-HCO3− cotransporters NBCE, and water channel aquaporin AQP1. Conversely, fewer transporters are found on the basolateral membrane of the choroid plexus epithelium, including the K+-Cl− cotransporter (KCC3), Na+-HCO3− cotransporters (NBCn and NCBE), Cl−/HCO3− exchanger (AE2), glucose transporter-1 (GLUT1), and AQP1. 24
As these transporters are easily affected by the pathological changes that occur after acute brain injury, we will briefly introduce mechanisms of transporter-mediated CSF formation in choroid plexuses. Accordingly, the Na+, Cl–, HCO3–, and Cl– movements initiate CSF formation. The uptake of Na+ on the choroid plexus epithelium of the basolateral side is driven by the Na+ gradient, and contributes to the primary step of CSF formation.3,26 Na+-HCO3− cotransporters, such as NBCn and NCBE, facilitate intracellular accumulation of Na+ and HCO3–, which accompany H+ and Cl– outflow.26,27 Carbonic anhydrases within the choroid plexus epithelium provide intracellular HCO3– and H+ from H2O and CO2, which has been proposed to be directly associated with HCO3– transporters (e.g. AE2 and NBCE) on two sides to maintain CSF pH. 28 Cl– uptake from plasma is also dependent on the basolateral transporter, AE2, 29 which is further transported by Cl– conductances and Cl–-related cotransporters into CSF. 30 The apical Na+-K+-ATPase drives the Na+ efflux and K+ influx in the choroid plexus epithelium, which provides the gradient for Na+ influx from plasma and secondary K+ transport. 31 Meanwhile, the luminal NKCC1 and NHE also participate in the choroid plexus-contributed Na+ in CSF. 32 The high cellular K+ is transported outward, which is driven by the transmembrane chemical K+ gradient through the KCC3 and KCC4. 33 The luminal K+ channels also facilitate the recycling of CSF K+ from the choroid plexus. 34
Using the osmotically driven force between plasma and CSF formed by abundant ion transporters, AQP1 facilitates transcellular water transfer. 35 Besides, the potential water transport on the basolateral membrane may be accompanied by ion movement with transporters, such as NKCCs and KCCs. The AQP7 also reportedly participates in transcellular water transfer. 23 Additionally, it should be noted that the water and cations are reportedly transported by a paracellular pathway due to the stranding connection of TJs. 30 This form of paracellular claudin connection leads to pore formation, which allows water and cations to cross. 36
Brain parenchyma system
The brain parenchymal system has been proposed as a source of CSF production, as the CSF produced from the trans-endothelium fluid exchange and glymphatic CSF-ISF fluid exchange in the brain parenchyma. 2 Traditionally, the BBB was considered an endothelial cell-based barrier responsible for mediating the exchange of ions, nutrients, and cells between the blood and the brain parenchyma. 37 However, the detailed mechanisms involved in substance exchange at the cellular and molecular levels remain ambiguously described. It was believed that the capillary endothelium has high electrical resistance and low permeability to polar solutes due to its tight connection. 38 Upon the discovery of ion channels and transporters on the brain side, its function was further extended to include substance transportation, such as ions, water, larger molecules, and even cells located between blood and CSF. 37
Recently, the BBB was revealed to be more than an independent unit of endothelial cells. As the “neurovascular unit”, the BBB includes pericytes, astrocytes, smooth muscle cells, microglia, and neurons.16,39,40 The capillary-astrocyte complex of the neurovascular unit has been proposed as the site of CSF and brain ISF production and exchange. ISF fills the extracellular space between neurons and glia in the brain parenchyma. The CSF-ISF fluid exchanges in the perivascular space are driven by the osmotic forces (i.e. ion and protein gradients) between the blood, perivascular space, and brain parenchyma.25,41 While the water has been proposed to be filtered from the high hydrostatic arterial vessels to the ISF and CSF, this phenomenon is reversed when the ISF and CSF reaches the low hydrostatic pressure of capillaries and venules. 6 This blood-CSF-ISF fluid exchange system is known as the “cerebral glymphatic system”.6,10 We will discuss its detailed mechanism in the following sections.
CSF absorption
Both CSF formation and absorption play important roles in regulating CSF volume. Similar to the divergent presumptions on CSF formation, the current view on CSF absorption also remains under debate. According to the anatomical location, features, and functional capabilities, several hypotheses were proposed, including arachnoid villi drainage, meningeal lymphatic drainage, and glymphatic drainage.2,3,9,42,43 (Figure 2(a))
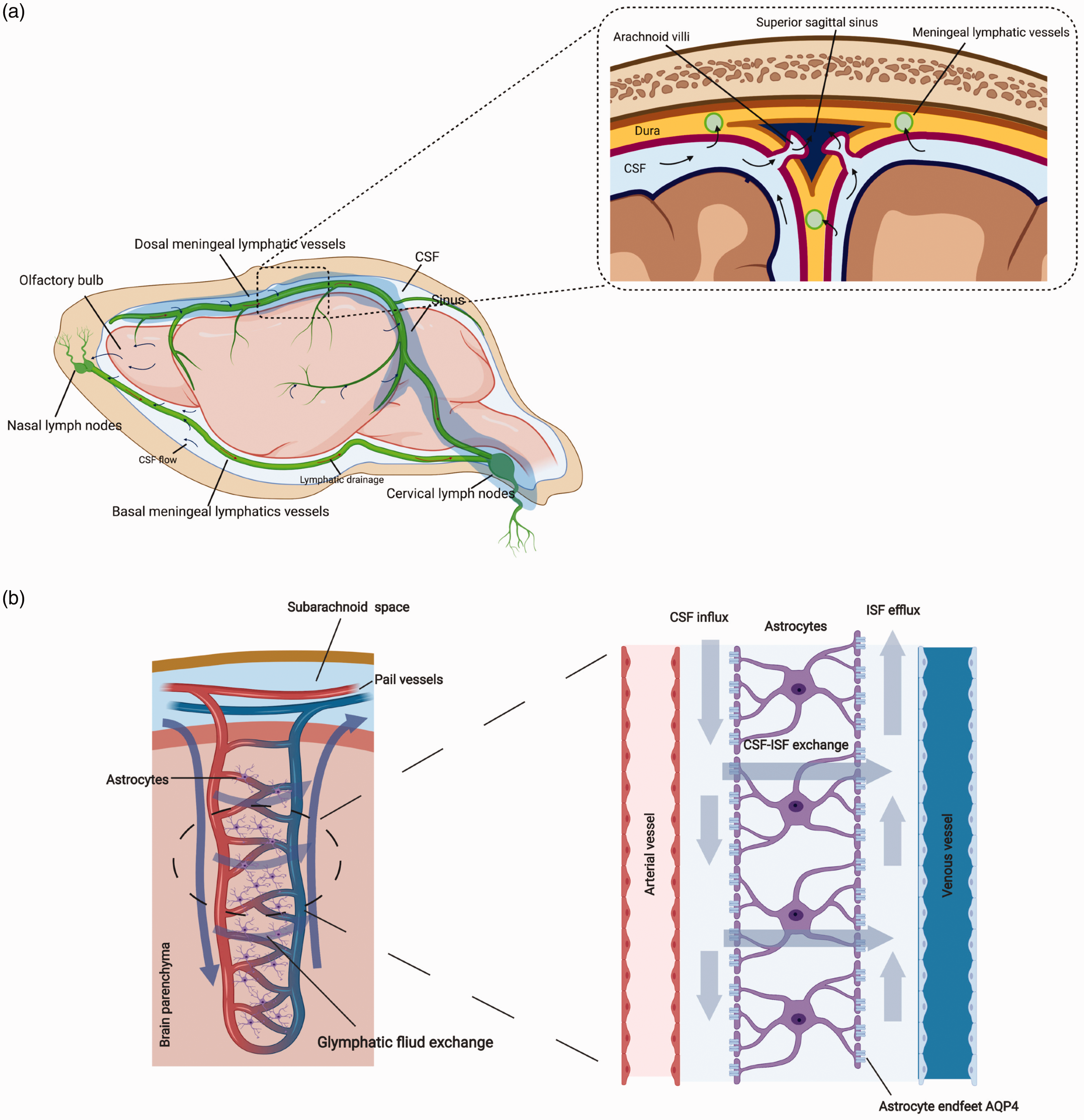
CSF absorption and glymphatic system. a. CSF absorption. The majority of CSF directly drains into the arachnoid villi along the superior sagittal sinus into the blood. Some CSF drains along the paraneural sheaths of olfactory nerves across the cribriform plate and nasal submucosa to the nasal lymph nodes. The meningeal lymphatic vessels distributed on the dorsal and basal dura (green vessels) also facilitate CSF drainage into cervical lymph nodes. b. Glymphatic system. The subarachnoid CSF communicates with parenchymal ISF in the perivascular space around the penetrating pial vessels. The hydrostatic gradient between the arterial and venous perivascular space drives continuous bulk CSF-ISF exchange from the arterial side to the venous side. Polarized AQP4 water channels located on the astrocytic endfeet around arteries and veins facilitate CSF movement from periarterial spaces into the brain parenchyma, and from the brain parenchyma into perivenous spaces.
Arachnoid villi drainage
Arachnoid villi (i.e. arachnoid granulations) are small protrusions that extend from the arachnoid mater into the dural venous sinuses located on the outer membrane of the dura mater. The discovery of arachnoid villi drainage resulted from an experiment conducted by Quincke, who injected cinnabar into the CSF of animals and found accumulation around arachnoid granulations. 44 Colored tracers injected into CSF consistently pass the arachnoid villi to the sagittal sinus, and eventually travel to internal jugular veins. 45 Arachnoid villi are considered as an anatomical structure with “one-way valve”, depending on the hydrostatic pressure gradient between CSF and dural venous blood.3,44 Particles up to 7.5 μm in size were found to pass through the arachnoid villi and enter blood in monkeys. 46 The potential transmembrane transport may rely on endothelial pinocytosis and vacuolization, as well as extracellular cistern. 46 It is necessary to ensure that there is a 3–5 mmHg hydrostatic pressure gradient between CSF and dural venous blood to maintain CSF drainage from arachnoid villi.8,43 However, the exact changes related to hydrostatic pressure dynamics under physiological and pathological conditions remain unclear due to the difficulties of replicating the dynamic changes of ICP in an in vitro model. 47
Lymphatic drainage
Numerous studies have corroborated that CSF injection of tracer could be found not only in cervical lymphatics,41,48–50 but also in spinal extradural lymphatic vessels 51 in mammals. The reported percentage of CSF-lymphatic drainage ranges from 5–50%48–50 across several preclinical studies. The discrepancy may result from differences in molecular weights of the tracers, access time after injection, and experimental methods. The routes that mediate lymphatic drainage include the perineural sheaths of cranial and spinal nerves, 52 as well as the meningeal lymphatic vessels. 53
Experimental tracers have been shown to move along the olfactory nerve (considered the major nerve drainage pathway 54 ) with CSF, and appear sequentially in the cribriform plate, 55 nasal submucosa, and the nasal lymph nodes. 56 This movement was present during the inpiration phase, but stopped in expiration phase of respiratory. 57 The potential explanation may be that the altered ICP and venous pressure was associated with respiration. However, current research regarding the driving force between perineural subarachnoid space and lymphatics remains limited, but further exploration of anatomy and physiology in the cribriform plexus and the perineural pathway may promote the understanding of this area.
Recently, numerous studies have revealed that lymphatic vessels exist within the dura meninges in rodents, humans, and non-human primates.4,58–62 Fluid, immune cells, macromolecules, and antigens in the CSF could be transported via lymphatic vessels of the meninges toward the deep cervical lymph nodes.4,61,62 These intracranial lymphatic networks express all of the molecular hallmarks of lymphatic endothelial cells, such as homeobox protein 1 (PROX1), vascular endothelial growth factor receptor 3 (VEGFR3), lymphatic endothelial hyaluronan receptor (LYVE)-1, podoplanin, and C-C motif chemokine ligand (CCL) 21.4,58 Similar to peripheral lymphatics, there is a high correlation between meningeal lymphangiogenesis and the expression of VEGFR3 and vascular endothelial growth factor C (VEGFC), the growth factors secreted by blood vessel smooth muscle cells.42,63 Impairment of meningeal lymphatic drainage is shown to impede the diffusion of CSF tracer into the perivascular spaces and the brain parenchyma, as well as the clearance of intraparenchymal tracer. 60
Interestingly, the meningeal lymphatic vessels display distinct junctional organization on the basal and dorsal sides. 42 The dorsal meningeal lymphatic vessels shared similar characterization with peripheral lymphatic vessels, and are proposed to be responsible for the transport of macromolecules and immune cell in brain.4,58,64,65 In contrast, the basal meningeal lymphatic vessels move on the skull base, and are proposed to maintain unidirectional lymph flow toward the deep cervical lymph nodes.53,58,63 Due to its proximity to the basal subarachnoid space, the basal lymphatic network is believed to contribute more in CSF clearance from brain parenchyma and ventricular system.53,58 Additionally, some studies have suggested that the CSF collected from olfactory nerves is also transported by the basal lymphatic network to the deep cervical lymph nodes.4,42,52 However, it remains unclear the molecular mechanisms underlying CSF-meningeal lymphatic exchange and the connective anatomy of the meningeal-extracranial lymphatic network.
Glymphatic drainage
Fluid exchange exists among blood, CSF, and ISF. According to the newly-defined glymphatic system, CSF communicates with ISF in the brain parenchyma through the perivascular space (will be discussed in the following section). 66 Water and solutes are proposed to move across the endothelium in a bidirectional manner driven by the osmotic and hydrostatic forces. Because of the unavailable methodology in monitoring such fluid exchange, as well as the difficulty in quantification, it remains unclear if, or how much, CSF is drained by the glymphatic system. Overall, this theory is akin to a theoretical hypothesis of CSF drainage, 9 which warrants further exploration.
CSF movement and glymphatic system
Traditionally, CSF movement is believed to originate from the choroid plexus and epithelium in the lateral ventricles, which then travels through the third and fourth ventricle, passes into the subarachnoid space, and is then absorbed in the arachnoid villi.7,9 This unidirectional pattern of CSF flow is now being challenged by the glymphatic CSF-ISF fluid exchange system, in which CSF flow is bidirectional.10,11 The glymphatic system is named due to its functional similarity to the peripheral lymphatic system and the importance of astrocytic AQP4 in this pathway.66,67 Using either two-photon imaging or light sheet fluorescence microscopy for visualizing the small fluorescent tracer movement in subarachnoid space, it was revealed that the bulk of the CSF in the subarachnoid space enters the brain parenchyma along perivascular spaces around pial-perforating arteries, and flows out of the brain parenchyma through the perivenous space (the perivascular space also named Virchow-Robin space).67,68 The AQP4 channels located on the perivascular astrocytic endfeet facilitated this flux in fluid exchange 17 (Figure 2(b)). The CSF movement in the glymphatic system is suggested to be driven by the hydrostatic gradient between the arterial and venous perivascular spaces, and is affected by arterial pulsatility and respiration, as well state of consciousness and head position.69–73
Of interest, the perivascular fluid flux highly correlates with the molecular weight and size of the substances. Intracisternally infused small single-domain antibodies (17 kDa; 4.5 nm diameter of hydration) accessed perivascular basement membranes more readily than large full-length immunoglobulin G antibodies (150 kDa; 10 nm diameter of hydration). 74 The lower molecular weight and diameter of hydration tracers allowed them to enter the brain parenchyma much more easily, as demonstrated by the contrast-enhanced MRI. 75 This may potentially be due to the tight connection between astrocytic endfeet around the vessel basement membranes, in which only 20-nm clefts between overlapping processes can provide direct communication with the interstitium. 76
Accumulating studies report that the glymphatic system participates in the maintenance of metabolism 77 and clearance of waste products, such as beta-amyloid 64 and ɑ-Synuclein 78 in brain parenchyma. However, the research pertaining to the glymphatic system is still in its infancy. The questions that remain unanswered include: 1) whether ISF drainage can directly cross the venules, the dural lymphatics, or arachnoid villi; 2) which methods can distinguish the perivascular space around the venules and arterioles, as well as the fluid exchange form; 3) whether fluid exchange stays identical between substances with different molecular weights; 4) and what are the other fluid exchange molecular mechanisms besides AQP4 participation.
CSF dynamics after SAH
SAH is characteristically described as blood that influxes into the subarachnoid space, mixes with the CSF throughout the entire circulation system of brain CSF, and diffuses throughout the brain. In humans, the first three days after SAH are the most critical and life-threatening. 79 Cognitive deficits develop in the clinical setting 6–14 days after SAH. 80 Current knowledge regarding pathological changes after SAH is mainly derived from pre-clinical studies. The brain injury is divided into four phases: acute phase (0–24 hours post-hemorrhage), sub-acute phase (1–3d post-hemorrhage), delayed phase (3–14d post-hemorrhage), and chronic phase (>14d post-hemorrhage). 20 In the acute phase, early vasospasm, acute hydrocephalus, increased ICP, and decreased CBF account for the initial damage. 15 When SAH progresses to the sub-acute phase, the blood and its degradation products within the subarachnoid space are detrimental, not only in the tissue proximal to the vasculature, but also in the parenchyma. 20 In the delayed and chronic phases, the blood clot in the subarachnoid space begins to be visibly cleared. While brain water content, CBP, and ICP return to baseline, 81 delayed vasospasm, neuronal death, secondary inflammatory response, and hydrocephalus appear in this period. 20 Besides, both acute and chronic pathological changes after SAH are associated with the CSF dynamic disorder that involves CSF production, movement, and drainage. A plethora of studies report pathological impairment found in the choroid plexus, 82 BBB, 83 arachnoid granulations, 84 lymphatic, 85 and glymphatic system, 17 which may be related to post-SAH blood clearance. (Figure 3)
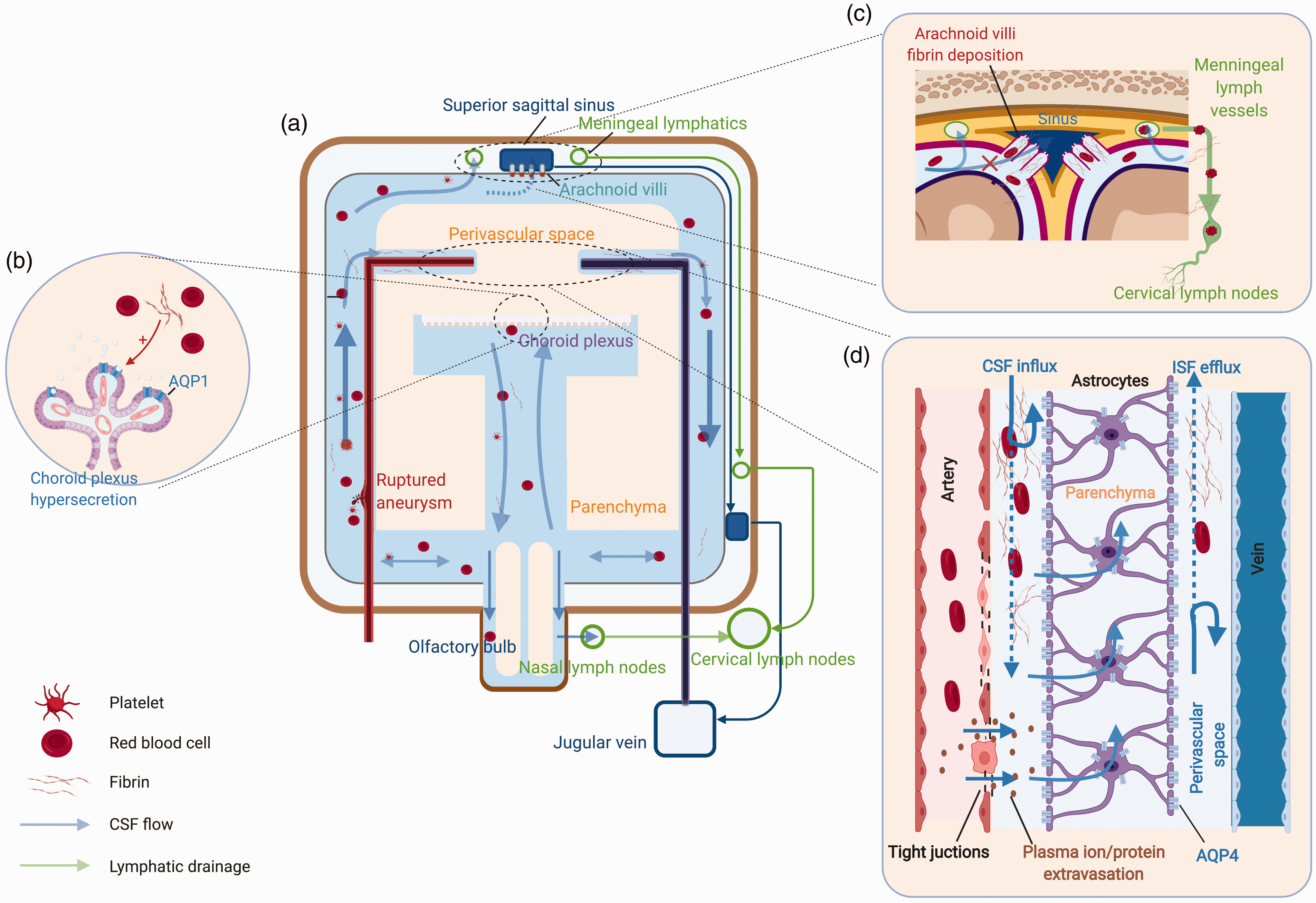
Pathological CSF circulation after SAH. a. Blood, with its lysis products, fulfill the subarachnoid space and ventricular system with CSF circulation after SAH. b. Toxic products of blood stimulates choroid plexus epithelium facilitating AQP1 expression and CSF hypersecretion. c. Fibrin/fibrinogen fill in the arachnoid villi and block the arachnoid villi CSF drainage. Erythrocytes were found to drain with CSF to the cervical lymph nodes. d. Blood clot with fibrin occupying the perivascular space inhibits CSF/ISF exchange. Dysfunctional endothelium results in plasma ions and protein extravasation, which promotes water influx to the perivascular space. Dislocated astrocytic AQP4 further leads to parenchymal water accumulation.
CSF production responses to SAH
Choroid plexus system responses to SAH
SAH is associated with a 30-50% incidence rate of visualized intraventricular hemorrhage (IVH).86,87 Blood, with its products of degradation that are toxic to the choroid plexus and epithelial cells, are inevitably carried by the CSF flowing from the bleeding site to the ventricular system in both the acute and chronic phases. 88
Several clinical studies have reported ependymal damage and loss in the fetal or neonatal post-hemorrhagic periventricular areas.89,90 Preclinical research further demonstrated the acute choroid plexus cell death and ultrastructural changes that occur within 3 days after IVH in preterm rabbits and hemoglobin/heme-treated in vitro model. 91 Moreover, acute choroid plexus and ependymal death were also consistently found in the adult rat model of IVH 92,93 and the rabbit model of SAH. 94 The inflammatory responses and caspases activation, as well as intracellular inclusions (IgG), accompanied cell death after hemorrhagic insults in neonatal and adult animal models.91–94 In addition, the choroid plexus atrophy and ependymal loss commonly exists in the chronic phase (1 week to 3 months) after IVH and SAH in various animal models, such as pigs 95 and rabbits.96,97 The post-hemorrhage fibrin deposition, which induces thrombosis and inflammatory changes in the vessel wall, 95 as well as choroidal artery vasospasm 96 are considered the primary contributors of these pathological changes.
IVH is a risk factor in the development of hydrocephalus and poor prognosis after SAH 98 Hydrocephalus developed in 52-62% of SAH patients with IVH.98,99 Using the findings of various preclinical SAH models, 24 a potential mechanism underlying hydrocephalus is suggested to be the CSF hypersecretion from the choroid plexus. In the rabbit cisternal blood injection SAH model, Aydin et al. 100 revealed a large number of water-filled vesicles within the choroid plexus at 2–14 days after SAH, supporting the theory of choroid plexus hypersecretion. They also proposed that the increased number of cytoplasmic water vesicles is probably induced by the stimulation of the glossopharyngeal and vagus nerves, which innervate the choroid-related arteries. The second contributor may be the dysfunction of channels and transporters on choroid plexus epithelium and ependyma after SAH. The expression of water channels, AQP1 and AQP5, on the apical and basolateral membranes were increased 1–7 days at the protein and mRNA levels after IVH in the preterm rabbit, 101 whereas the AQP4 found on the ependymal cells was increased at 1–3 days after SAH in rats. 102 It should be mentioned that both AQP1 and AQP4 levels highly correlated with the hydrocephalus occurrence. 102 Besides, the apical membrane potassium cotransporter, NKCC1, was also reportedly upregulated 1–7 days after IVH in rats, and was accompanied with the CSF secretion. 103 Thirdly, inflammatory responses and choroid plexus damage were linked. The epithelia can increase their rate of fluid secretion in response to proinflammatory stimuli, such as fibrin, DAMPs, hemoglobin, and heme. 103 IVH reportedly activates Toll-like receptor 4 (TLR4)- and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB)-dependent inflammatory response in the choroid plexus epithelium and the downstream Ste20-type stress kinase (SPAK), which stimulates NKCC1 upregulation. 103 The peripheral immune cells are reportedly trafficked into the choroid plexus after SAH stimulation and are accompanied by increased levels of chemoattractant molecules CCL2, tumor necrosis factor-α (TNF-α), and interleukins. 104 The increased ICP and alteration of TJ proteins at the choroid plexus after SAH are also believed to contribute to the acute choroid plexus damage-induced CSF hypersecretion.103–105
Several questions exist in the current study of choroid plexus pathophysiological changes after SAH. 1) Long-term choroid plexus degeneration was found to occur after SAH. However, its effect on actual CSF production remains questionable, as well as whether it is related to the chronic hydrocephalus; 97 2) The choroid plexus epithelium forms the blood-CSF barrier and maintains the osmotic and hydrostatic gradients, but current knowledge pertaining to the acute and chronic changes after SAH on such barriers and gradients remains limited. 3) the specific liquid flow mechanism still requires further investigation, which should include both bilateral channels/transporters and TJ proteins that were mentioned previously in the section of physiological CSF dynamics.
Brain parenchymal system responses to SAH
Endothelium impairment initiates immediately after aneurysm rupture, which disrupts the local equilibrium existing between blood and CSF/ISF. The blood content flowing into the CSF/ISF increases the osmotic concentration, further driving water influx to the parenchymal side, and contributing to acute cerebral edema formation. 16 Such dysfunction is associated with increased ICP and expansion of brain volume within 24 h after SAH in humans. 106
During the sub-acute and delayed phases (2–14d) after SAH, there is a decrease in TJ proteins, ion/water transporter dysfunction, matrix metalloproteinase (MMPs)-induced basal lamina degradation, neuroinflammation, and endothelial cell death. 16 The brain suffers a global ischemia after the spread of blood from the local site, which makes the second phase of endothelium dysfunction partially similar to the ischemia-induced endothelial cell injury. The increased endothelial permeability peaked at 72 h after SAH in the rat endovascular perforation model. 107 Endothelial cell-related ionic and vasogenic edema successively contribute to the increased CSF production and global cerebral edema formation in this period after SAH. 17 The dysfunction of ion and water transporters on astrocytes resulted in Na+ influx into the intracellular space with low osmotic concentrations in the interstitial space. The changes of the Na+ gradient between the vascular compartment and interstitial compartment drives the subsequent influx of ionic edema fluid from the vascular compartment to the interstitial compartment. 108 This transmembrane ion (Na+) transport relies on endothelial channels and transporters, such as NHE, 109 NKCC1, 110 and sulfonylurea receptor (SUR). 111 The Cl– and water crosses the endothelium accompanied with Na+, and further improves CSF production. Furthermore, with the disruption of TJs and with the death of endothelial cells, the brain capillaries form the permeability pores, which allow plasma proteins, such as albumin and IgG, to diffuse into the brain interstitial compartment. 112 Hydrostatic and osmotic pressure gradients directly prompt further water influx. 113
CSF absorption responses to SAH
Arachnoid villi responses to SAH
Blockage of arachnoid villi CSF drainage is initiated during the acute phase, and is sustained until the late stage after SAH. A brain autopsy revealed that the accumulation of blood clots in arachnoid granulations inhibited CSF drainage, raising the ICP after SAH in patients. 114 This granulation blockage could still be observed under the microscope at 4 weeks after SAH. 114 In addition to the blood clot, collagen and fibrin deposition were also found to block the arachnoid granulation-mediated CSF drainage in the autopsy.84,115 Proliferation of leptomeningeal cells with increased collagen synthesis enzyme (prolyl 4-hydroxylase) prompts CSF collagen production in response to the circulating toxic products of blood, which further block the arachnoid granulations.84,116 After SAH, the physiological structure of arachnoid granulations is destroyed by the increased ICP and by the chemical effects of digestive enzymes released from inflammatory cells, 84 which consequently stimulates the fibro-cells to facilitate granulation fibrosis. 115 Fibrin/fibrinogen derived from the impaired BBB could be an additional source of granulation fibrin deposition. 117 Knowledge regarding post-SAH pathophysiological changes of arachnoid villi has stagnated, as most studies were performed 20 years ago. A recent in vitro study demonstrated the significant decrease in small molecule permeability through arachnoid membranes when treated with whole and lysed blood. 118 Future studies should focus on other potential underlying mechanisms involving arachnoid granulation blockage, and they should uncover how different ICPs affect erythrocyte clearance by the arachnoid granulation after SAH.
Lymphatic drainage responses to SAH
The lymphatic drainage plays an important role in post-SAH CSF drainage. Emerging animal studies show that ligation of cervical lymphatic drainage worsens early brain injury (brain edema, oxidative stress, and neuronal apoptosis) within 72 hours after SAH.119–121 Since the discovery of meningeal lymphatic vessels, it has been widely accepted that they are involved in clearing amyloid-β in mouse (both aged and young) models of Alzheimer’s disease.60,122 The finding that the meningeal lymphatic drainage clears macromolecule waste in the CNS raises the question as to whether it also participates in post-SAH blood clearance. Interestingly, a recent study revealed that the erythrocytes in CSF drained into the deep cervical lymph nodes via meningeal lymphatic vessels in mice 4 hours after SAH was induced by cisterna magna autologous blood injection. 85 Ablation of meningeal lymphatic vessels blocked erythrocytic drainage into the deep cervical lymph nodes, and aggravated neuroinflammation and short-term neurological deficits. 85 Meningeal lymphatic flow was augmented for 7 days after SAH without lymphangiogenesis or expansion. 85 The augmented meningeal lymphatic flow may be in response to the increased CSF production and neuroinflammation after SAH, similar to the functionality of the peripheral lymphatic system.123,124 The inhibition of lymphatic growth factor, VEGFR3, exacerbated the neuroinflammation and neurobehavioral impairments 30 days after SAH, 85 suggesting the presence of slow lymphangiogenesis in the chronic phase.
Future studies are necessary to elucidate whether erythrocytes and their products of degradation enter meningeal lymphatics vessel through glymphatic pathways, and to determine how meningeal lymphatics regulate the immune response to SAH. An improved understanding of the mechanisms and regulation of lymphatic drainage in both the acute and chronic phases following SAH would be meaningful.
CSF movement and glymphatic system responses to SAH
The ventricular CSF movement is significantly blocked after the blood coagulation response is initiated in the subarachnoid space and ventricular system after SAH. And it would be life-threatening when the blood clot blocks the third and fourth ventricles, which may lead to severe hydrocephalus and herniation. 125
Meanwhile, decreased glymphatic fluid exchanges were recently observed in rodents and non-human primates after SAH.17,125–129 Gaberel et al. 125 first demonstrated the impaired glymphatic system after SAH by using MRI in the rat SAH model of chiasmatic cistern blood injection. The diffusion of contrast agent, DOTA-Gd (557.6 Da), from the cisterna magna into parenchymal and olfactory bulbs was significantly restricted in the acute phase (24 h) after SAH. 125 Further research found that this impairment started as soon as SAH was induced in non-human primates. 127 In addition, the impaired distribution of both low molecular tracer fluorescent microspheres (0.02 μm) and large molecular tracers, such as Evans Blue (961 Da) and fluorophore Alex594 (758 Da) from cisterna magna to perivascular space and parenchyma, were also blocked 24 hours after SAH in rodents.17,129 Consistently, we demonstrated that the clearance speeds of Evans blue from the cisterna magna to deep cervical lymph nodes and blood were simultaneously decreased at 24 hours after arterial perforation-induced SAH in rats. 17
The blockage of glymphatic fluid exchange is attributed to several pathological changes after SAH. The primary cause is considered the occlusion and stimulation of the perivascular space by blood components. 5 Blood rapidly flows into the perivascular space following SAH and diffuses into the perivascular parenchyma, resulting in a sequelae of pathological processes, including cerebral vasospasm, cytotoxic edema, blood coagulation in CSF, and other early brain injury.17,126 Cerebral vasospasm enlarges the perivascular spaces and doubles glymphatic inflow speeds, facilitating immediate cytotoxic edema. 130 With the initiation of CSF blood coagulation, accumulating fibrin/fibrinogen deposits occupy the perivascular space and inhibit CSF-ISF exchange.125,129 The fibrin/fibrinogen was found to exist in CSF for 7 days, even in the absence of a visible blood clot.117,128,129 Hypoxia, cytokine infiltration, and blood toxicity prompts AQP4 depolarization on astrocytes in the first 24 hours after SAH, which is considered the mechanism of cytotoxic edema formation.17,128 However, AQP4 knockout did not improve brain water content or neurological deficits.126,131 Generally, AQP4 on the astrocytic endfeet facilitate CSF/ISF fluid exchange from the arterial side to the venous side. After brain injury, dislocated AQP4 impaired glymphatic function, resulting in water accumulation in astrocytes and brain parenchyma. 5 The potential mechanism of AQP4 depolarization was attributed to the activation of SUR1/TRPM4 on the membranes of astrocyte bodies, which recruit AQP4 to form a heteromultimeric complex and cytotoxic edema. 111 Additionally, the expression of other transporters (e.g. NKCC1, excitatory amino acid transporter, glucose transporter) were increased in astrocytic cell somas, and contributed to the hypoxia-induced cytotoxic edema, which could be the potential cause of AQP4 depolarization. 108 SUR1 is only expressed during ATP deficiency, which may not last to the late stage of SAH. 111 However, the AQP4 depolarization reportedly lasts from days to a month after cerebral hemorrhage in rodents.126,132 The long-term glymphatic changes warrant further exploration. Other transporters (e.g. NKCC1, excitatory amino acid transporter, and glucose transporter) increased expression on the astrocytic cell soma and contributed to the cytotoxic edema after hypoxia induction, which could also be the potential cause of AQP4 depolarization. 108
Therapeutic targets of CSF dynamic modulation after SAH
Surgical interventions, such as lumbar cistern drainage, external ventricular drainage, ventriculoperitoneal shunt, and endoscopic third ventriculostomy, are widely used in the clinical setting to accelerate blood CSF clearance and improve clinical outcome in SAH patients. 14 Of note, pharmacological interventions may also be a potential treatment to assist in post-SAH clearance of blood CSF. Here, we focus on the molecular therapeutic targets of CSF dynamic modulation after SAH.
Intracisternal fibrinolysis is considered a promising treatment in favor of reducing subarachnoid clot and fibrin deposition in CSF for an extended period after SAH. Several randomized trials have shown that intracisternal administration of tissue plasminogen activator (tPA) and urokinase improved neurological outcomes in SAH patients, including the prevention of cerebral vasospasm, 133 delayed cerebral ischemia, 134 and hydrocephalus. 135 As mentioned earlier, subarachnoid clot and fibrin deposition could block CSF/ISF exchange and drainage via arachnoid villi. A recent pre-clinical study demonstrated that intraventricular injection of tPA cleared the perivascular space fibrin deposition and promoted glymphatic function after SAH in mice.125,129 Fibrin deposition in the subarachnoid space mainly results from the activation of the extrinsic coagulation cascades mediated by glycoprotein tissue factor (TF) on the cell membrane surface.129,136 The brain expresses high levels of TF on astrocytes. 137 One study showed that neutralized TF in CSF could also reduce fibrin deposition and improve glymphatic function. 129 However, the causes of post-SAH brain TF activation remain unclear. TF was a link between inflammation and coagulation in the peripheral system. 138 Thus, TF may serve as a promising target of anti-fibrin deposition and anti-neuroinflammation after SAH.
Water/ion transporters and channels are another potential molecular target for modulating CSF dynamics after SAH. Since AQP1 and NKCC1 have an important role in choroid plexus injury-induced CSF hypersecretion and hydrocephalus after SAH,102,103 it is rational to evaluate the efficacy of their inhibitors in reducing CSF production. A variety of endothelial water/ion transporters and channels mediate endothelium (BBB)-derived CSF hypersecretion. The inhibition of SUR1/TRPM4 by glibenclamide alleviated BBB disruption and brain edema. 139 Additionally, selective metabotropic glutamate receptor 1 (mGluR1), a negative allosteric modulator, prevented BBB disruption and edema formation after SAH. 140 We recently demonstrated that pituitary adenylate cyclase-activating peptide (PACAP) provided potent BBB and glymphatic protection by down-regulating SUR1 expression on endothelial cells and astrocytes, attenuating the post-SAH astrocytic AQP4 translocation to the soma, promoting CSF movement in the subarachnoid and perivascular spaces, and accelerating CSF clearance to the deep cervical lymph nodes and blood. 17 Thus, a better understanding of the function of other transporters and channels (e.g. NKCCs, NHE) located on endothelial cells (BBB), astrocytes (glymphatic function), and the choroid plexus after SAH would further facilitate the development of targeted pharmacological therapeutics for SAH patients.
Furthermore, the inflammatory response and oxidative stress may also play critical roles in dysregulating CSF dynamics after SAH. 141 Inhibition of oxidative stress and inflammatory responses in the acute phase after SAH could prevent CSF hypersecretion and dysfunction of endothelial cells and choroid plexus dysfunction.142,143 A recent study reported that lipopolysaccharide (LPS)-induced inflammation markedly limited perivascular CSF tracer flow and penetration into the parenchyma of mice. However, no AQP4 depolarization was found in their study. They attributed this glymphatic system dysfunction to TLR4 activation-induced choroid plexus hypersecretion. 144 It is insufficient evidence to support the proposal that chronic plexus hypersecretion may cause glymphatic system dysfunction. LPS is considered a potent pyroptosis inducer for immune cells, including astrocytes. 145 Astrocytes have large amount TF and fibrinogen in brain, 137 and we propose that LPS may induce astrocytic pyropotsis and TF release and fibrin formation, which blocks the perivascular space. However, it is known that the expression of glymphatic system key channel protein, AQP4, is widely affected by cytokines and reactive oxygen species, 146 but the effects on the AQP4 polarization remains unknown. In addition, a recent study revealed that meningeal and perivascular macrophages are involved in erythrocyte uptake at 2–5 days after SAH in mice, which highlights the importance of the glymphatic-lymphatic-mediated immune response to the post-SAH outcome. 147 The interventions that target the inflammatory response and oxidative stress, with benefits to post-SAH CSF circulation in the glymphatic-lymphatic system, are worthy of further investigation.
Conclusion
The new findings of the glymphatic system and meningeal lymphatics reshape our traditional knowledge that the choroid plexus–ventricle-arachnoid villi system is the sole pathway of CSF circulation. Undoubtedly, both systems work as an entire network to regulate CSF dynamics. However, due to a lack of real-time, high-quality methodology for monitoring fluid exchange between blood, CSF, ISF, meningeal lymphatics and extracranial lymphatic network, the concept of a newly proposed glymphatic- meningeal lymphatics system remains debatable. Besides, the proportions by which each system is responsible for CSF production and drainage remains unclear. It also remains unknown how specific proteins (e.g. ions/water transporters and channels) and what mechanisms (e.g. hydrostatic and osmotic driven) regulate the movement of ions, water, and immune cells in CSF. Overall, uncovering the participation of the glymphatic-meningeal lymphatic system in SAH will introduce a new modality for using CSF modulation as treatment for SAH. However, current research of CSF dynamics in the setting of SAH remains limited to investigations of visible CSF movement changes and blood metabolites after SAH insults, rather than molecular and cellular mechanism. Altogether, a better and deeper understanding of the dynamics of CSF physiology could provide a solid foundation for identifying the pathophysiological changes of post-SAH CSF dynamics. The functionality clarification of specific proteins that participate in fluid exchange in the CSF circulatory network at molecular and cellular levels would further assist in identifying therapeutic targets against blood clearance, cerebral edema formation, and hydrocephalus after SAH. Moreover, future studies are also needed to explore the relationship between other risk factors (e.g. hypoxia, oxidative stress, and inflammation) and pathophysiological dynamics of CSF circulation.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This review was supported by grants from the National Institutes of Health (NS081740 and NS082184) of John H. Zhang, and the National Natural Science Foundation of China (82071287 and 81870916) of Jianmin Zhang, (81971107) of Sheng Chen
Acknowledgements
All figures were created using Biorender.com.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.