
Abstract
The aetiology of congenital hydrocephalus (cHC) has yet to be resolved. cHC manifests late in rodent gestation, and by 18–22 weeks in human fetuses, coinciding with the start of the major phase of cerebral cortex development. Previously we found that cerebrospinal fluid (CSF) accumulation is associated with compositional changes, folate metabolic impairment and consequential arrest in cortical development. Here, we report a proteomics study on hydrocephalic and normal rat CSF using LC-MSMS and a metabolic pathway analysis to determine the major changes in metabolic and signalling pathways. Non-targeted analysis revealed a proteome transformation across embryonic days 17–20, with the largest changes between day 19 and 20. This provides evidence for a physiological shift in CSF composition and identifies some of the molecular mechanisms unleashed during the onset of cHC. Top molecular regulators that may control the shift in the CSF metabolic signature are also predicted, with potential key biomarkers proposed for early detection of these changes that might be used to develop targeted early therapies for this condition. This study confirms previous findings of a folate metabolic imbalance as well as providing more in depth metabolic analysis and understanding of cHC CSF.
Introduction
Congenital hydrocephalus (cHC) is a multifactorial condition, resulting from combinations of environmental insults and genetic predispositions. In experimental animal models, pregnancies exposed to x ray radiation, nutritional deficiencies, alcohol consumption and other teratogenic chemicals can all result in cHC, revealing environmental toxicity and nutritional abnormalities as risk factors.1–4 However, the mechanisms leading to, and underlying cHC remain matters of great debate and research, while early diagnosis and treatment of cHC is crucial to the outcome of the new-born. Although prenatal ventriculomegaly (enlarged brain ventricles) can be identified by ultrasound imaging between 18–20 weeks of gestation, only certain aetiologies of cHC can be identified in utero. 5 Other aetiologies may thus pass undiagnosed with severe consequences for the fetal brain. 1 , 6 , 7 Elucidation of the complex pathophysiology of cHC and a better understanding of metabolic changes involved should aid the development of diagnostic tools and, potentially, novel treatments to replace current surgical treatments that can only be carried out postnatally. 8 With a prevalence of 4.65 per 10,000 births, cHC is particularly difficult to treat, with poor neurological outcome or common terminations of affected fetuses. 5 , 8 , 9 CSF composition changes, triggered by fluid drainage insufficiency and abnormal fluid accumulation, result in deficient cortical development. 7 CSF flow obstruction commonly occurs in the aqueduct of Silvius, an Arnold Chiari malformation in the subarachnoid space, or by brain tumours. 10 , 11
Past proteomic analyses have described changes in CSF composition across a broad spectrum of neurological disorders, and importantly, have served as a useful tool for biomarker discovery. 12 , 13 Thus, we decided to analyse cHC CSF in the hydrocephalic Texas (HTx) rat 14 using a proteomic and metabolic profiling approach to assess changes in normal and hydrocephalic fetal brain metabolism during the critical period of cortical development. High volume CSF secretion into the ventricles commences at gestational days 17–18, as evidenced by the initial stages of ventricular dilation, which is equivalent to the third-fourth month of pregnancy (week 12–16) in humans. 15 , 16 We compared CSF from HTx rats and normal Sprague-Dawley rats using liquid chromatography with tandem mass spectrometry (LCMS)17–20 protein identification and metabolic pathway analysis by Ingenuity Pathway Analysis (IPA) software (Qiagen).
Material and methods
Animals and CSF collection
All experiments were sanctioned by The Home Office, Animals (Scientific Procedures) Act 1986 through project and personal licence and by The University of Manchester Animal Procedures Ethical Review Committee. ARRIVE guidelines have been followed in execution and reporting of animal experiments. 21 Hydrocephalic Texas (HTx) and Sprague Dawley rats were kept as previously described 7 in 12 hour light-dark cycles at constant temperature, low light levels and with unlimited access to food and water. Timed mating provided fetal tissue at gestational ages from day 17 to 20. Pregnant dams were killed by overdose of anaesthetic and fetuses were collected onto wet ice. CSF from six controls and six HTx rat fetuses were used for each gestational day. In total, eighteen controls and eighteen HTx fetuses were used. The rationale behind the sample size chosen was the fact that in previous LCMS analysis between three and six animals were utilized to get successful results when comparing proteomes from body fluid samples to achieve repeatability and reproducibility as reported in the literature. 21 – 23 CSF was collected by insertion of a fine glass pipette into the cisterna magna and lateral ventricles. 50 µl CSF per sample were centrifuged at 6500 rpm for 10 min to remove cellular debri. The supernatant was stored at −80°C until use.
LCMS protein analysis
A non-targeted proteomic analysis of normal and abnormal CSF was performed as described by Nabiuni. 24 Briefly, CSF was subjected to trypsin enzymatic digestion for protein fragmentation. Because CSF contains around 2000 proteins and the LCMS can reliably detect only around 200, isoelectric focussing was used to produce 20 fractions which were each put through physical separation of proteins/peptides by liquid chromatography (LC) with detection of mass and charge by mass spectrometry (MS) analysis. The resulting LCMS data was entered into the MASCOT database to identify proteins according to trypsin fragment molecular weights and amino acid sequences. The result was lists of proteins for each sample with assigned accession numbers (Uniprot) and corresponding chemical names. Lists of proteins, imported into Microsoft Excel, were contrasted and their datasets categorized to investigate what proteins and associated metabolic pathways were either common, missing, or present in each of the CSF categories. Each protein was assigned a specific numerical value based on its place in the list that also gives information about its relative abundance since MASCOT software lists proteins in order of abundance of trypsin fragments. Missing proteins were assigned a “−1” value, which means “error”. This is achieved by using the “if error” formula. This formula allows the error to be tracked, and a “−1” number will be displayed in the cell. Three lists of proteins were obtained using the corresponding “autofilters”: proteins unique to normal CSF, proteins unique to abnormal CSF and common proteins present in both.
Qiagen ingenuity pathway analysis (IPA)
IPA was used for protein-gene matching (mapping) and core analysis to predict what proteins/genes were associated with specific canonical pathways (www.targetexploreringenuity.com). Protein expression profiling lists produced by LCMS and categorized in Microsoft Excel were uploaded to the IPA server and then analysed to predict molecular interactions between proteins and involvement and association with specific metabolic functions. A global canonical pathway analysis (GCP) was carried out on Excel categories: common proteome, normal unique proteome and abnormal unique proteome. P-value measurements obtained from GCP analysis were equivalent to the relationships found between related genes in the Excel categories and their associated metabolic pathways. IPA uses the Fisher’s Exact Test analysis for hypothesis testing to assess metabolic pathway enrichment (−log (p-value)), which is based on the specific number of genes found in our dataset, and the reference total set of genes retrieved from the IPA database. P-values are a measure of the probability that the IPA relationships between LCMS focus genes and a given metabolic pathway or biological process is due only to random chance. Only significantly predicted pathways (–log (p-value) = 1.3; p < 0.005) were considered in this study.
Results
Percentage intervariability and the variation coefficients (CV) between the six individual controls and the six hydrocephalic CSF samples were calculated and represented in Figure 1(a) to (f) for each of the three gestational days analysed (E17-20). Data for normal sample 1 (NS1) represent comparisons between proteome/sample 1 and proteome/samples 2, 3, 4, 5 and 6. Data for normal sample 2 (NS2) represent comparison of proteome/sample 2 with sample 1, 3, 4, 5, and 6 and etc. Abnormal samples (AS) are presented in the same manner. None of the individual proteome/samples across the study showed CVs >10, which confirms the similarity between samples and thus, the reliability of our results.
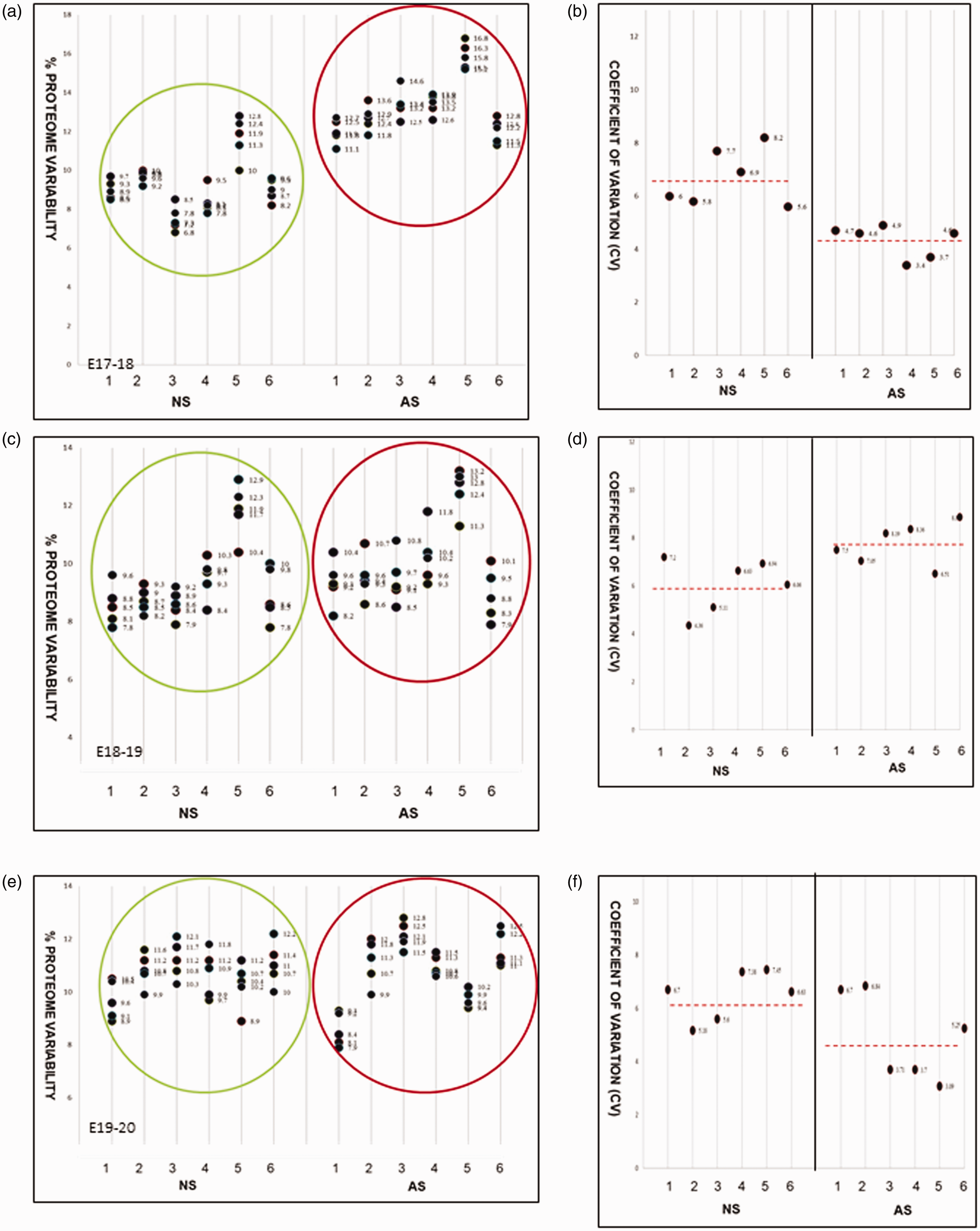
% Proteome variability between samples at E17-18 (a) E18-19 (c) and E19-20 (e). Normal (NS, in green circle) versus Abnormal (AS, in red circle); N = 6. The coefficient of variation (CV) between samples at each day is shown in b, d and f. The CV is less than 10 in each case indicating good reproducibility of the data.
Protein content percentage intervariability between controls and experimental samples as well as their means are shown in Table 1 and the means are shown graphically in Figure 2(a). CSF proteome composition analyses showed that, at E17-18, only 18.4% proteome (276 proteins) were common between normal and abnormal brain, whereas 36.7% (573 proteins) novel proteins were unique to abnormal CSF, and 38.4% (574 proteins) were unique to normal CSF. Common protein content became minimal (0.67%; 13 proteins) at E18-19 and rose again to 18.7% at E19-20. Abnormal CSF varied in composition from 40.2% (573 proteins) to 49.1% (948 proteins) between days 17 to 19. Similarly, normal unique CSF proteome rose from 38.4% (574 proteins) to 50.2% (969 proteins) between those days. However, between day 18 and 20, normal unique proteome decreased from 50.2% (969 proteins) to 23.74% (654 proteins) while abnormal unique proteome rose even further from 49.1% (948 proteins) to 57.4 (1583 proteins), which was the most significant change in CSF composition between day 19–20 and throughout this time- point study. Common protein content increased again at day 19–20 (18.7%; 517 proteins) and this last gestational age was used for the following IPA analysis.
Percentage of variability in protein content (normal unique, abnormal unique and common).
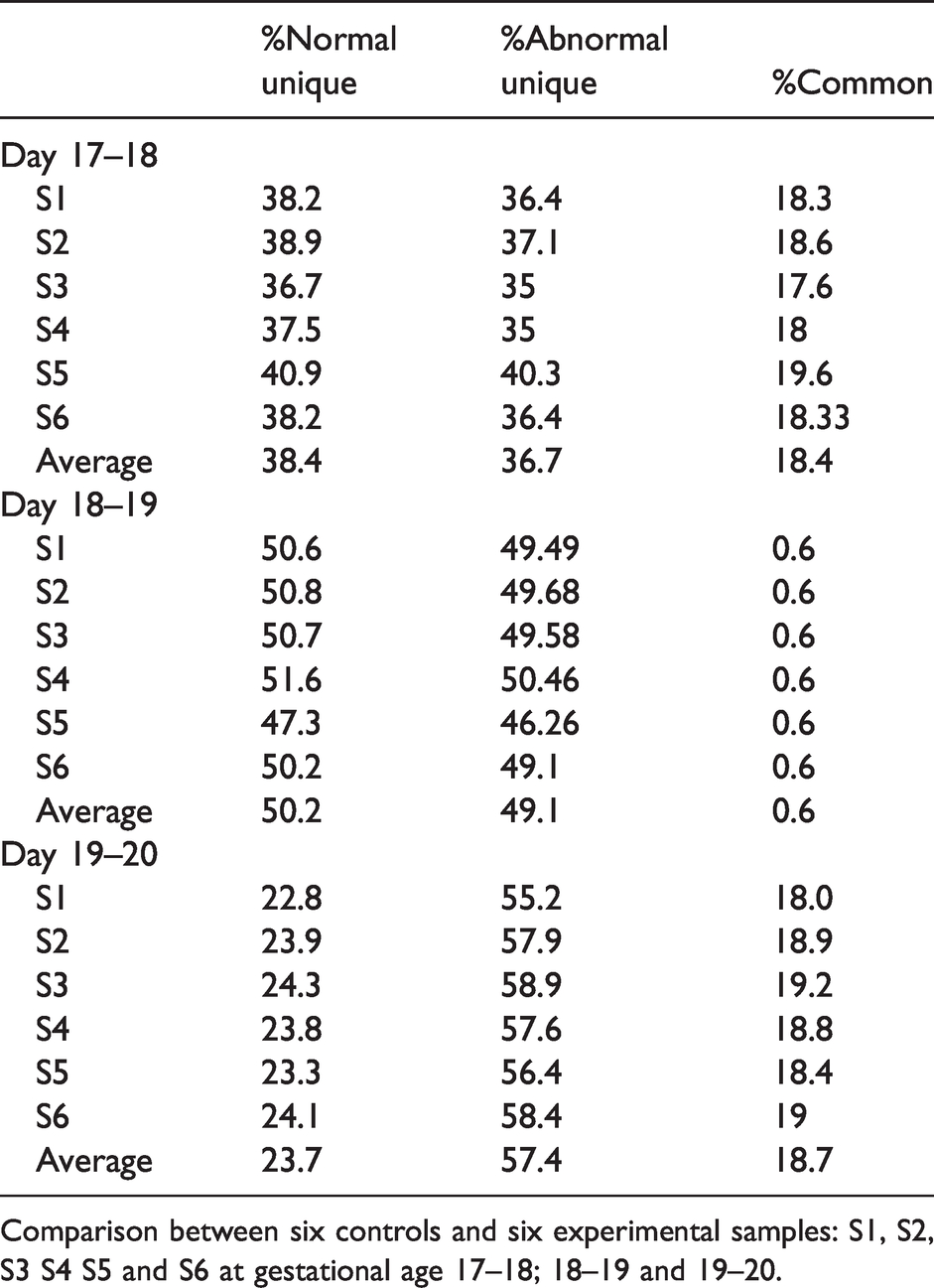
Comparison between six controls and six experimental samples: S1, S2, S3 S4 S5 and S6 at gestational age 17–18; 18–19 and 19–20.
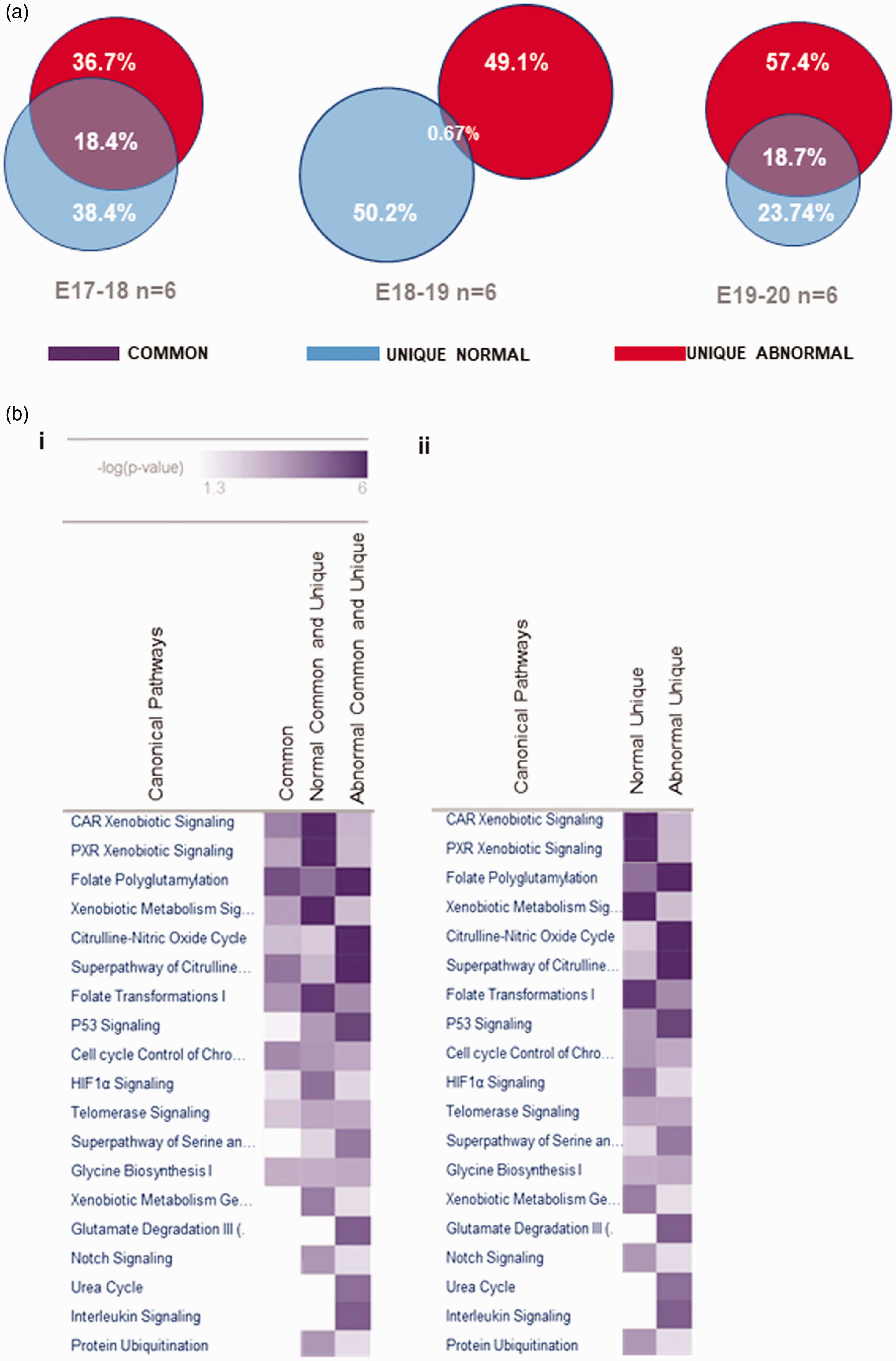
(a) CSF proteome data categorization and average protein content comparison analysis for gestational day E17 to E20. Average differential protein expression in normal and abnormal CSF are shown to highlight unique and common protein content. Content diverges throughout gestational ages leading to major changes between day 18 and 20 where the unique abnormal proteome diverges from the unique normal proteome and increases in proportion to the total. (b) Comparative analysis of selected canonical pathways identified in the proteomes. Common proteome and full (common and unique) CSF proteome were analysed simultaneously and compared as shown in 2bi. heat map, where normal versus abnormal CSF canonical pathways are displayed. Similarly, unique normal CSF proteome was compared to unique abnormal CSF as illustrated in 2bii. heat map. Intensity of colour purple is equivalent to the significance of the identified pathways in relation to IPA reference canonical pathways. Significance or -Log (p-value is measured with the Fisher Exact Test; p < 0.05.
IPA metabolic pathway analysis of the CSF proteome
IPA was used to perform metabolic pathway analysis of the categorized LCMS proteome, identifying distinctive pathway profiles for the common proteome (common proteins in normal and abnormal CSF) and whole CSF proteome (common and unique proteins in normal and abnormal CSF) as shown in Figure 2(a). The folate polyglutamylation pathway appears as the only significantly enriched pathway (intense purple) within the common proteome, whereas full normal and unique normal CSF proteome shared the same pathway patterns, as was the case for full abnormal and unique abnormal proteome (Figure 2(b)).
Normal metabolic phenotype
The normal CSF metabolic profile (Figure 3(a)) consisted of the xenobiotic-related pathways: CAR, PXR, general signalling and general xenobiotic metabolism with overlapping or score values (–Log (p-value)) in the range 6–4. Following in degree of significance were HIFα1 signalling (p3.9), folate transformations I (p3), folate polyglutamylation (p2), superpathway of serine and glycine (p1.9) and cell cycle control of chromosomal activation (p1.8). Less significant but still above the threshold was P53 signalling (p1.4). Notably, ALDH1L1 is a protein member shared by the top four xenobiotic-related canonical pathways (Figure 4), and importantly also shared with folate transformations I, which is part of the folate metabolic cycle. Significantly, ALDH1L1 is reduced in hydrocephalic CSF, and is the marker we first identified for congenital hydrocephalus in the HTx rat. 7 Heat Shock Protein 90 Alpha family subunit A member 1 (HSP90AA1) was also a molecular component of all the top Xenobiotic pathways. Interestingly, our analysis identified neural precursor cell-expressed developmentally downregulated gene 4, (NEDD4) also called E3 ubiquitin ligase Rsp5, which is part of the ALDH1L1 molecular network (Figure 4). NEDD4 was exclusively detected in the unique normal CSF proteome and within the protein ubiquitination pathway. However, this pathway, along with Notch, telomerase signalling as well as Glycine synthesis, was only at the threshold of significance (-log (p-value) =1.3; p < 0.05 and it remains inconclusive whether these pathways are significantly represented in normal CSF.
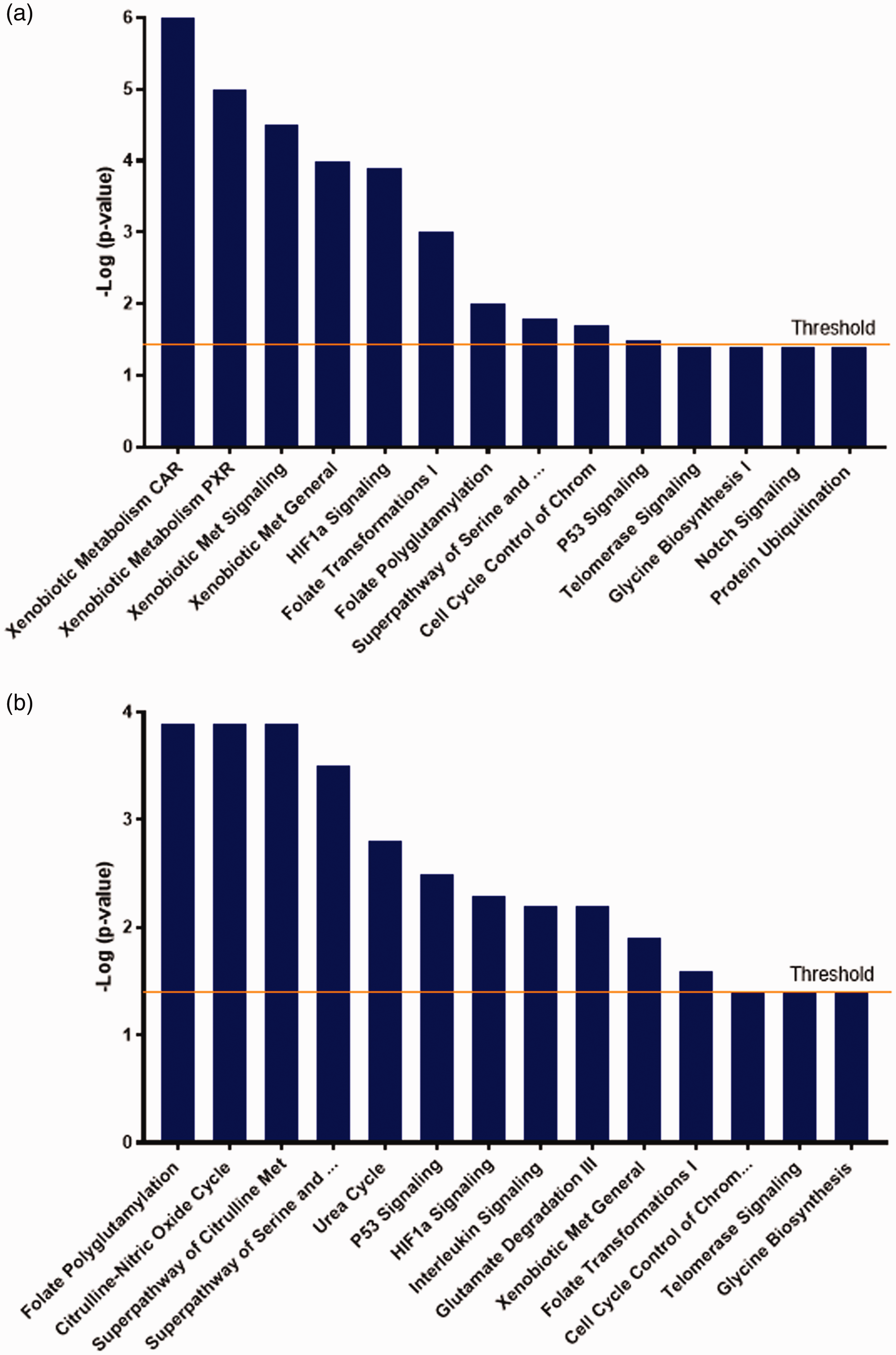
(a) Normal CSF unique metabolic phenotype. Canonical pathways generated by IPA on proteins solely detected in normal CSF are shown in this figure in order of significance from left to right. IPA predicted activation of top significant CAR Xenobiotic Signalling followed by PXR, General Xenobiotic Signalling, General Xenobiotic Metabolism, HIF1α Signalling and Folate related pathways (Folate Transformations I and Folate Polyglutamylation). Less significant and just above the threshold were Superpathway of Serine and Glycine, Cell Cycle Control of Chromosomal Activation, and P53 Signalling. Also shown in this figure were non-significant pathways: Telomerase Signalling, Glycine Biosynthesis I and Notch Signalling. Fisher Exact Test; Threshold 1.3; P < 0.05). (b) Abnormal CSF unique metabolic phenotype. Metabolic pathways predicted by IPA in abnormal CSF unique proteome are shown in this figure. In order of significance, Folate Polyglutamylation was the top pathway followed by Citrulline-Nitric Oxide Cycle, Superpathway of Citrulline Metabolism, Superpathway of Glycine and Serine Metabolism, Urea Cycle, P53 Signalling, HIF1 α Signalling, Interleukin Signalling, Glutamate Degradation III (via aminobutyrate), General Xenobiotic Metabolism and Folate Transformations I. Non-significant pathways also retrieved by IPA were Cell Cycle Control of Chromosomal activation, Telomerase Signalling and Glycine Biosynthesis. Fisher Exact Test; Threshold 1.3; P < 0.05).
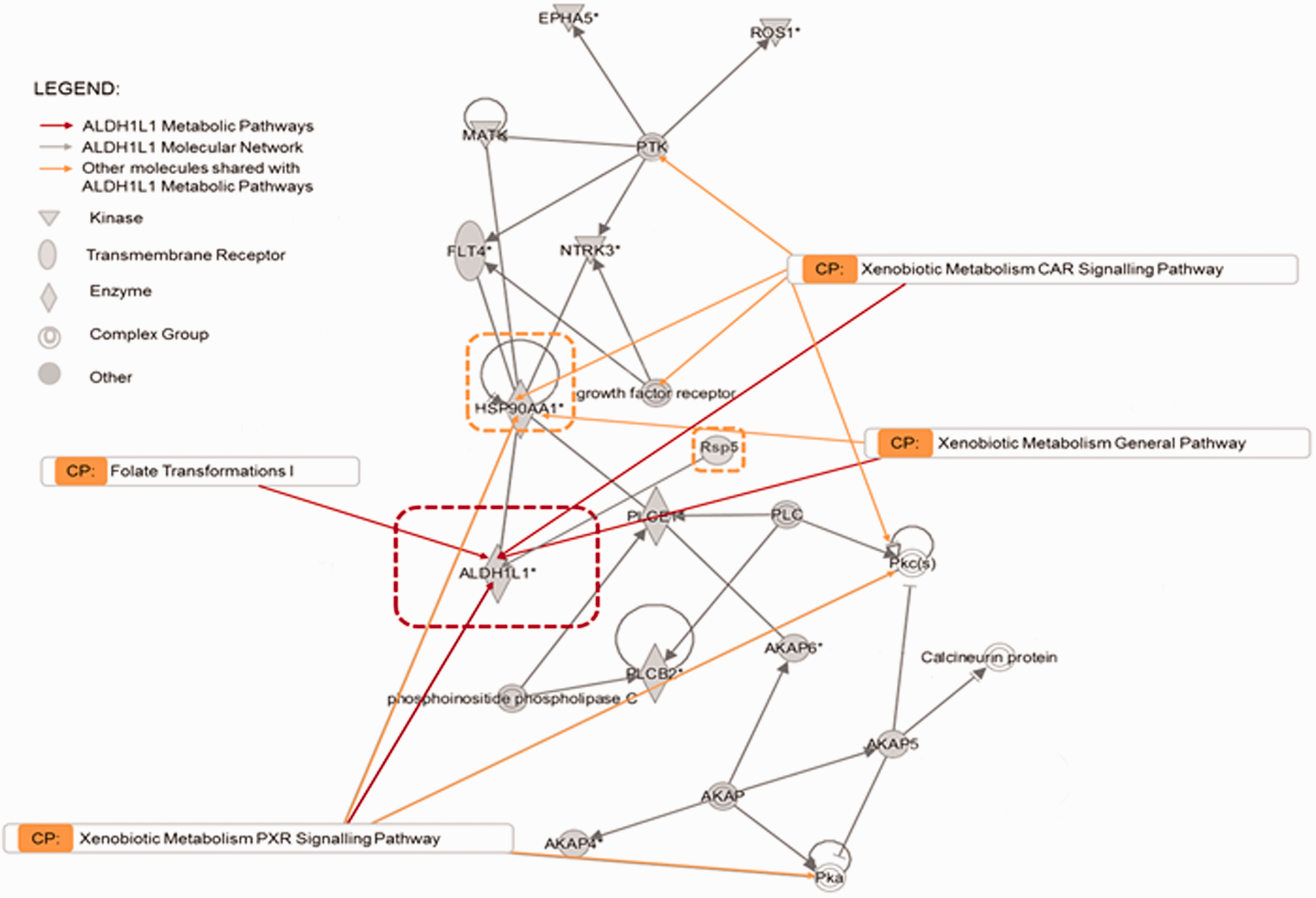
ALDH1L1 I in silico protein-protein IPA network analysis and its canonical pathways. ALDH1L1 molecular network and interrelationships with Xenobiotic pathways and Folate Transformation I. Orange arrows indicate the molecules that fall in the specific canonical pathway. Grey arrows show network intrarelationships. Ephrin type A receptor 5 precursor (EPHA5); Reactive Oxygen Species 1 (ROS1); Protein Tyrosine Kinase (PTK); Megakaryocyte-Associated Tyrosine Kinase (MATK); FMS-related tyrosine Kinase 4 (FLT4); Neurotrophic Receptor Tyrosine Kinase 3 (NTRK3); Heat Shock Protein 90 Alpha; family class A; member 1 (HSP90AA1); Aldehyde Dehydrogenase 1 L1 (ADH1L1); 1-Phospholipase C Epsilon 1 (PLCE1); E3 Ubiquitin-protein ligase type Rsp5 (Rsp5) Phospholipase (PLC); Protein Kinase c (PKc); Phospholipase C Beta 2 (PLCB2); A Kinase anchoring protein (AKAP);Protein Kinase a (PKa).
Abnormal metabolic phenotype
The abnormal hydrocephalic metabolic phenotype is shown in Figure 3(b). IPA identified three top canonical pathways in hydrocephalic CSF: folate polyglutamylation, citrulline-nitric oxide cycle and superpathway of citrulline, all with significance of overlap value 3.9. Superpathway of serine and glycine, urea cycle, P53 signalling, interleukin signalling, and glutamate degradation III (via aminobutyrate) followed in significance with scores 2.7, 2.5, 2.3, 2.3 and 2.2 respectively. Less enriched but still significant were xenobiotic general metabolism (p1.9) and folate transformations I (p1.6). Finally, IPA identified common metabolic pathways to normal and abnormal unique CSF proteome. However, these were enriched with different protein members of the pathway. Folate polyglutamylation, superpathway of serine and glycine, and P53 Signalling, were all shared pathways significantly more enriched in abnormal than normal unique proteomes. By contrast, common pathways HIF1α signalling and folate transformations I were relatively more represented in unique normal CSF.
IPA predicted tumor protein (TP53) and specificity 1 protein (SP1) as master regulators leading to the abnormal cHC metabolic phenotype
IPA predicted top rated regulators and their molecular interactions are shown in Figure 5(a) and (b). IPA assigned the highest score of significance (p15.4) to TP53; p < 0.05. The second top master regulator was SP1 (p11.3), followed by lower scored regulators with scores 8 to 1.3. These were: NFE2L2, HNF4A, PPARG, CREB1, AR, SP3 and AHR. Proposed molecular interactions and molecular network for TP53 and SP1 are shown in Figure 5(b). TP53 is suggested to directly exert its effect on SP1 inhibiting Histone Deacetylase 1 (HDAC1) and 3 (HDC3), which at the same time inhibit TP53.
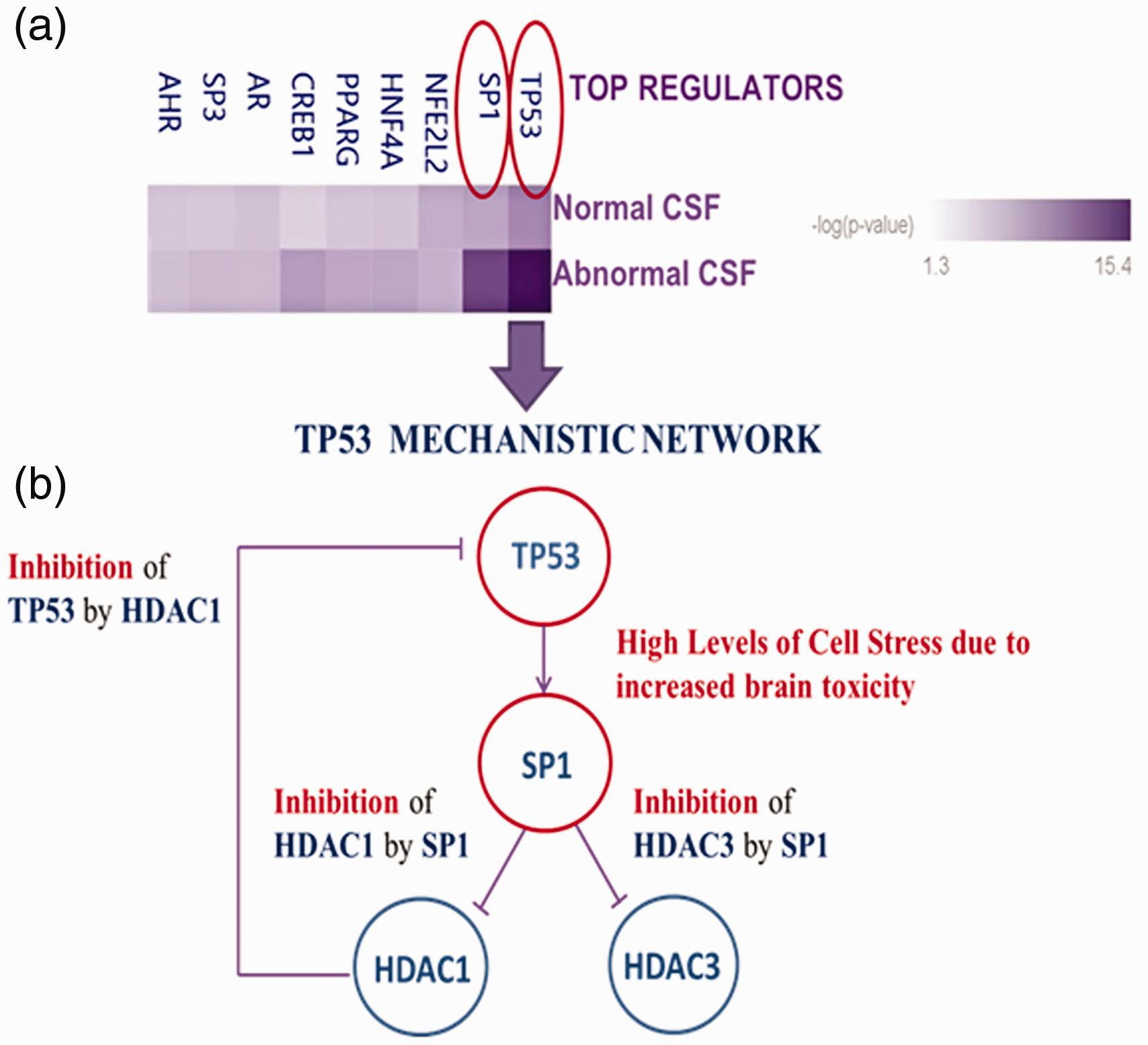
Master Regulators and proposed interlinked mechanistic networks predicted by IPA. Master regulators Heatmap suggested by IPA in order of significance (5a); Threshold: 1.3; p < 0.05. TP53 mechanistic network (5b). TP53 and SP1 were identified as top regulators with top score 15.4 for Tumor Protein 53 (TP53) and second top score 11.3 for Specificity Protein 1 (SP1). Lower scores ranging from 8 to 1.3 were assigned to Nuclear Factor Erythroid 2 like 2 (NFE2L2), Hepatic Nuclear Factor 4 (HNF4A), Peroxisome Proliferator Activated Receptor Gamma (PPARG), CAMP Responsive Element Binding Protein (CREB1), Androgen Receptor (AR), Specificity Protein 3 (SP3) and Aryl Carbon Receptor (AHR).
Folate hydrolase 1 (FOLH1) is unique to the abnormal hydrocephalic proteome and is associated with histone deacetylation and folate polyglutamylation
FOLH1, also called Glutamate Carboxypeptidase II, was a unique feature of the hydrocephalic proteome. IPA molecular network analysis of FOLH1 is described in Figure 6 where the interrelationship of FOLH1 with HDAC1 is highlighted. This hydrolase converts polyglutamated folate into monoglutamated folate and it is interrelated with the Folate Polyglutamylation pathway (www.targetexplorer.ingenuitypathanalysis.com). Additional details are illustrated in the pathway analyses shown in supplementary figures.
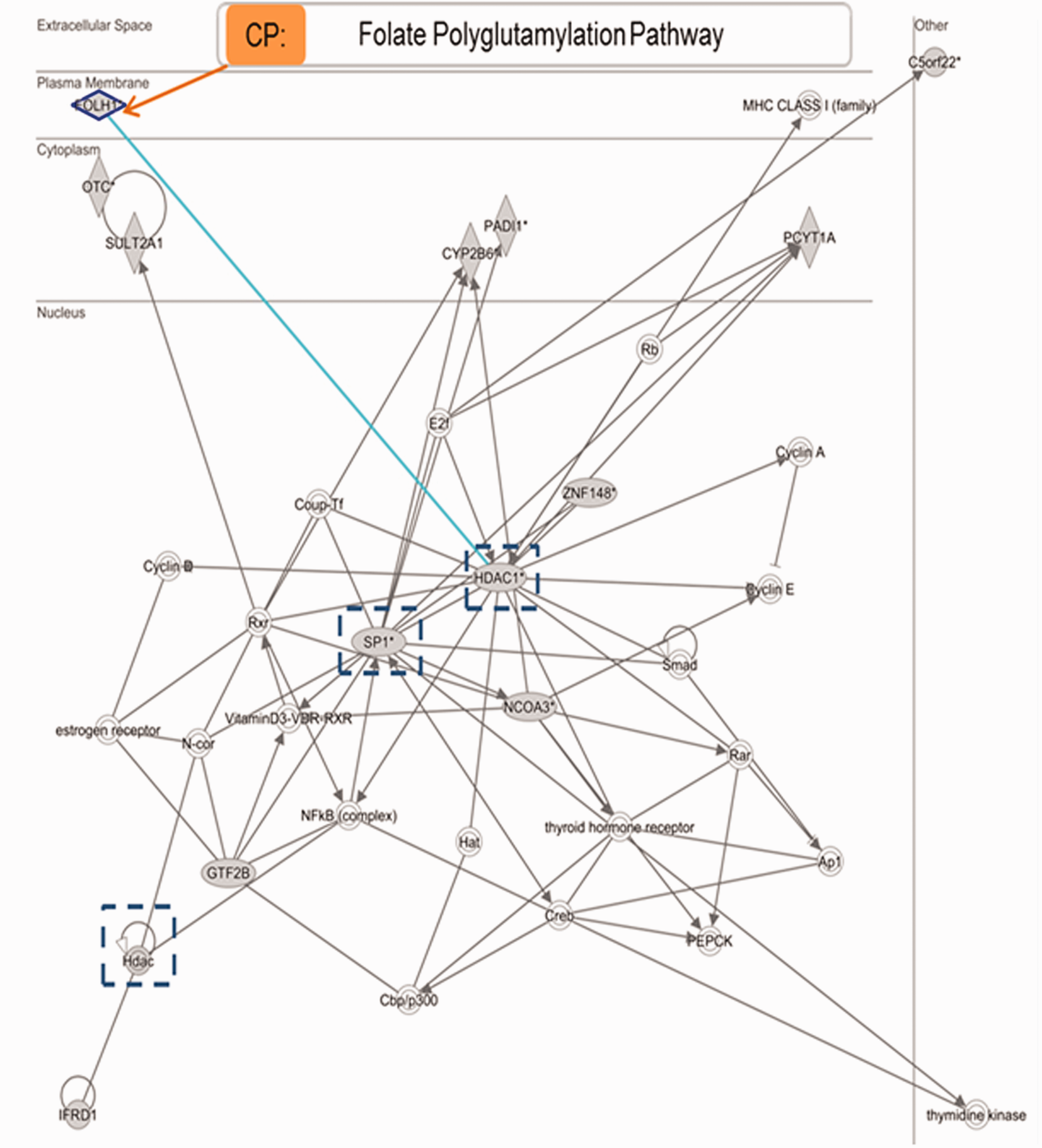
FOLH1 Molecular network and metabolic pathway overlay. FOLH1 direct interrelationship with HDAC1 (light blue) and indirect relationships with SP1 and general HDACs (dark blue dotted squares) are detailed in this diagram as well as relationship with related pathway (Folate Polyglutamylation).
Discussion
Proteomic profiling is suggested to supersede genomic and transcriptomic analysis to reflect the actual metabolic phenotype of diseases more accurately. This is due to the fact that gene expression levels do not always correlate with protein expression levels, and genetic analysis is not suitable for identification of posttranslational modifications like ubiquitination, acetylation/deacetylation and methylation. 25 The pathophysiology of cHC is still unclear and a high proportion of affected pregnancies result in terminations, stillbirths and live births with lifelong neurological deficits because of the lack of a predictive tool early in pregnancy 5 or a preventive therapy such as that for neural tube defects. Surgical treatment for hydrocephalus involves shunting CSF or endoscopic third ventriculostomy, the main treatments of choice to remove damaging excess CSF, but these are not free from life-threatening complications. At the same time, CSF is a promising source of information to identify metabolic patterns and biomarker discovery. 26 , 27 In this scenario, it was reasonable to perform CSF proteomic analysis followed by “in silico” computational analysis for identification of the cHC metabolic phenotype to further understand the complex pathophysiology of cHC and to discover novel candidates for drug therapies. In this study, no genetic testing was performed to identify proteome variability due to sex differences. Our LCMS analysis rather focused on exposed differentially expressed protein profiles between controls and experimental samples at different gestational ages. Previous studies have found no differences between sexes in the incidence or severity of hydrocephalus. 14 , 15 Thus, we postulate that these differences are the result of abnormal gene replication, transcription, translation and/or changes in metabolism triggered by a CSF drainage insufficiency. IPA complementary analysis further identified the metabolic signature of cHC as well as associated master regulators responsible for the metabolic changes observed.
The normal metabolic phenotype of fetal CSF consisted of, among others, xenobiotic- related pathways. Xenobiotics, such as aldehyde compounds, are natural and highly reactive molecules that are either intermediates or products of numerous metabolic reactions. 28 Free aldehydes induce genotoxic damage and cellular stress in neural stem cells 29 triggering xenobiotic metabolism, in which at least 17 types of Aldehyde Dehydrogenase are involved. Mutations in these genes are responsible for inborn errors of metabolism. 30 , 31 The Aldehyde dehydrogenase ALDH1L1 is a xenobiotic metabolizing enzyme that is part of the general Xenobiotic Metabolism Signalling Pathway and the CAR and PXR Metabolism Signalling Pathways. 32 , 33 These were all top-rated pathways by IPA analysis of normal CSF. CAR and PXR are protein receptors present in the cytoplasm and nuclei of brain cells. In cytoplasm, these receptors, in the absence of ligand, bind to the HSP90 chaperon protein forming a complex that is eventually translocated to the nucleus, where target genes related to the xenobiotic metabolizing enzymes Phase I (ALDH1L1) are activated for stimulation of xenobiotics metabolism. ALDH1L1 is activated by its own substrates through signalling cascades that are involved in its binding to CAR and PXR receptors in the nuclei. Aryl Hydrocarbon Receptor (AHR) ligand and Aryl hydrocarbon receptor nuclear translocator (ARNT) or HIF-B are receptors that regulate xenobiotic-metabolizing enzymes such as ALDH1L1. 34 , 35 Similar to CAR and PXR receptors, AHR and ARNT bind to HSP90 chaperon protein and translocate to nuclei where target genes like ALDH1L1 are transcribed in order to trigger the xenobiotics metabolism, promoting cell proliferation and differentiation, and enhancing cell survival in a toxic cell environment. Null or negligible levels of ALDH1L1 in hydrocephalic CSF are suggested to be involved in the pathogenesis of HC. 7 Importantly, in the present study, all ALDH1L1-related xenobiotic-pathways were identified in “control” or normal CSF, but not detected in abnormal CSF. Since, ALDH1L is a key enzyme for aldehyde detoxification, its absence in CSF suggests that all the canonical pathways involved in Xenobiotic metabolism might be compromised, resulting in brain toxicity and cellular stress. Yet, what is observed in the fetal hydrocephalic brain is an arrest of development thought to be a consequence of aspects of folate metabolism involving ALDH1L1 in synthesising DNA. 7 , 36 This arrest in development may be a mechanism to prevent the brain toxicity that might otherwise occur. Indeed, no cell death is observed in the cHC fetal HTx brain. 37
ALDH1L1 is a member of the folate transformations I pathway, which is part of the folate metabolic cycle. This cycle involves folate transformation reactions where folates act as carriers of one-carbon units in several oxidation states. 38 These reactions are important for the synthesis of glycine, methionine, formyl-methionine and purines and pyrimidine nucleotides for DNA synthesis. This pathway is shared with both normal and abnormal CSF, yet greater protein enrichment in normal CSF implies delayed activation of the Folate Metabolic Cycle. This finding confirms a fault in folate metabolism already identified in previous investigations 7 , 39 that demonstrated how reduced CSF levels of the folate enzyme ALDH1L1 are associated with impaired folate metabolism and poor brain folate provision.
Another commonly identified folate-related pathway was folate polyglutamylation, which, in contrast with folate transformations I, was significantly less represented in normal unique CSF, and the highest top-rated pathway in the abnormal unique proteome (Figure 3). Folates are either free in normal CSF, bound to just one molecule of glutamate (monoglutamated state), or stored intracellularly transformed into folyl-polyglutamated folates by folylpoly-gamma-glutamate synthase or converted into dihydrofolate and tetrahydrofolate in the folate metabolic cycle. 40 High representation of the folate polyglutamation pathway in the hydrocephalic CSF points to an elevated use of glutamate, probably provided by the tricarboxylic cycle in mitochondria. This cycle is linked with the superpathway of serine and glycine in the sense that 2-oxoglutarate is synthesized in the former pathway to feed the latter to produce the glutamate required for folate polyglutamylation. 41 These notions indicate that folate polyglutamylation and superpathway of serine and glycine are intertwined (Figure 3), which explains why they appear both highly represented in hydrocephalic CSF with similar significance of overlap. Importantly, abnormally increased folate polyglutamylation in CSF would effectively make folate unavailable for metabolism. In addition, FOLH1 or glutamate carboxypeptidase II (GCP II); a γ-glutamylhydrolase commonly found in glial cells and neurons, was uniquely detected in abnormal CSF. 42 This enzyme is interrelated with the Folate Polyglutamylation pathway (Figure 6) and involved in the degradation of folate polyglutamate chains in the peripheral nervous system. The interplay between FOLH1 and folylpoly-gamma-glutamate synthase is crucial to regulate dietary folate homeostasis. 43 , 44 Nevertheless, FOLH1 up-regulation is associated with brain disease, specifically with glutamate excitotoxicity, traumatic brain injury and neuropathic pain. 45 Consequently, FOLH1 presence in abnormal brain might well point to neurotoxicity associated with enhanced glutamate transmission from neuron to neuron and neuron to glia signalling. 46
Another common pathway was HIF1α signalling. This pathway is induced to maintain homeostasis in situations of hypoxia, stimulating transcriptional activations and translation into proteins involved in angiogenesis, cell proliferation and glucose metabolism.47–49 This pathway was second top-rated in normal CSF, but relatively less enriched in abnormal brain, suggesting delayed vascularization and other pathway-associated functions. HIFα is also one of the subunits of the Aryl Hydrocarbon Receptor Nuclear Translocator (ARNT), an essential component of the Xenobiotic Metabolism, which highlights the interrelationship between xenobiotic-related pathways and HIF1α signalling 50 ; both pathways highly represented in normal CSF (Figure 3).
The citrulline-nitric oxide cycle and superpathway of citrulline metabolism were significantly protein-enriched pathways solely identified within the unique abnormal CSF proteome (Figure 3(b)). As well as being key in vascular homeostasis, nitric oxide (NO) plays a crucial role in cell signalling in the nervous system and is involved in synaptic plasticity. NO release depends on arginine bioavailability provided by the general blood circulation from exogenous sources or by regeneration in the Citrulline-Nitric Oxide Cycle, where arginine is transformed into citrulline and NO. 51 Arginine is at the same time regenerated from citrulline by arginosuccinate synthase and lyase, which involves production of fumarate. 52 In addition, superpathway of citrulline metabolism is interlinked with the Citrulline-NO cycle as this pathway is involved in the generation of citrulline from glutamine, glutamate and arginine, among other precursors feeding the Citrulline-NO Cycle. Importantly, high levels of fumarate lead to neurotoxicity, apoptosis and altered global methylation profiles, and NO contributes to neuronal death. 53 , 54 In this context, brain toxicity due to accumulation of fumarate is proposed. Similarly, potential neurotoxicity is also suggested by exclusive detection of Glutamate Degradation Pathway (via aminobutyrate) in abnormal CSF. Glutamate is converted into 4-aminobutyrate and ammonia by glutamate decarboxylase. 4-aminobutyrate transaminases then converts 4-aminobutyrate into succinate semialdehyde and this is converted to succinate by succinate semialdehyde dehydrogenase. High levels of succinate like fumarate also lead to neurotoxicity and altered methylation status. 55 , 56 Lastly, in our metabolic pathways analysis, is the interleukin signalling pathway, which was also a unique metabolic feature of abnormal CSF. This outcome was unexpected as cHC is generally associated with neuroinflammation, and this pathway is linked to anti-inflammatory processes (Figure 3(b)).
In summary, canonical pathway analysis of the normal and abnormal unique proteomes gives compelling evidence of the striking CSF metabolic transformations occurring in the early stages of cHC. Our analysis identifies a hydrocephalic CSF metabolic signature characterized by absent or delayed detoxification (undetected xenobiotic-related pathways), genotoxicity and cytotoxicity (citrulline-related pathways), and differences in the time window for activation of the folate metabolic cycle, which might be potentially delayed and/or detrimental for later brain development and fits with the developmental deficit and cell cycle arrest we have observed in the developing hydrocephalic brain. 7 , 36 , 39
“In Silico” ALDH1L1 molecular network analysis (Figure 4) describes HPS90AA1-ALDH1L1 protein interactions and their relationships with Xenobiotic Metabolism. Cytoplasmic HPS90AA1 functions as a chaperon that assists to stabilize protein tridemensional structure unfolding and refolding proteins to regain the normal folding as well as preventing their aggregation due to the occurrence of misfolded proteins in heat-associated cell stress as well as cell stress due to the presence of toxic chemicals, oxidative stress and inflammation. 57 Importantly, HPS90AA1 also aids in protein quality control through the ubiquitination process: a posttranslational modification needed for removal of, among others proteins misfolded enzymes, which is required for protein homeostasis. Failed protein quality control is associated with neurodegenerative diseases and protein misfolding diseases. 58 Interestingly, normal proteomic profile analysis identified ubiquitinase NEDD4 or Rsp 5 as part of the ALDH1L1 molecular network (Figure 4). NEED4 is a type of ubiquitin E3 Ligase that was unique to normal CSF and not present in abnormal CSF. E3-Ligases are responsible for ubiquitination marking proteins like ALDH1L1 for proteosomal degradation. This important result highlights NEDD4 as a novel candidate biomarker of cHC that might explain why ALDH1L1 is at normal concentrations in brain tissue but absent in CSF in the hydrocephalic brain. Previously published IHC staining revealed ALDH1L1 at normal levels in brain tissue and reduced or null levels in CSF of hydrocephalic brains; however, IHC staining does not discern between intact and misfolded ALDH1L1. Therefore, depletion of ALDH1L1 in CSF might suggest permanent binding of misfolded ALDH1L1 to HPS90A11 caused by a lack of NEDD4 in brain tissue that otherwise in normal conditions would have recognized the ALDHL1-HPS90AA1 complex and destroyed it in the proteosome. Fang et al. 59 and Khan et al. 60 described a similar process in the literature. Moreover, ALDH1L1 is a multicomplex enzyme with an intermediate domain, rich in phospho-serine and NEDD4 is a special type of ligase that acts on enzymes rich in this amino acid. 61 , 62 These facts together with our findings suggest that misfolded ALDH1L1 followed by impaired ubiquitination might lead to cHC. However, whether the protein ubiquitination pathway is truly represented in normal CSF remains inconclusive as the significance for this pathway was at the threshold level (1.3; p < 0.05) (Figure 6). A plausible explanation is that day 19–20 of gestation is still early for complete expression of all the numerous proteins involved in ubiquitination and proteosomal degradation in cHC.
In this study, we were also interested in the molecular driving force or master regulators leading to cHC. IPA software identified two top main top regulators P53 and SP1 (Figure 5(a) and (b)). The transcriptional regulator TP53 responds to cellular stress to regulate expression of target genes resulting in potential cell cycle arrest, cell cycle progression, angiogenesis, DNA repair, apoptosis as well as changes in metabolism. Increased cell stress induces genotoxicity and cytotoxicity to trigger TP53 activation 63 promoting abnormal histone acetylation/deacetylation and methylation status. 64 , 65 Moreover, oxidative stress leads to hyperacetylation of TP 53, which activates SP1, the second top-rated master regulator in our study. SP1 activation is also the result of histone acetylation through Histone Acetylation Transferases (HAT) 66 as well as inhibition of HDCA1 and HDAC3 activity, which promotes transcription and erasure of methylation. 67 , 68 We have found a profound loss of methylation in the HTx rat supporting the activity of this pathway ((Naz et al., 2021, in preparation). These concepts agree with the IPA predicted mechanistic network for TP53 and SP1 master regulators as shown in Figures 5 and 6 where TP53, SP1, HDAC1 and HDCA3 molecule-molecule interactions are described. In this context, lack of DNA methylation and therefore gene silencing by promotion of acetylation is suggested as well as exacerbated transcription caused ultimately by TP53 activation of SP1and inhibition of deacetylation. Finally, evidence of increased transcription activity in cHC is demonstrated by the sheer amount of protein expression in the abnormal proteome.
TP53 however, develops a myriad of responses acting as “good cop” or “bad cop” depending on situations of low or high cell stress.63–65 This duality of function might explain why TP53 is the same top regulator predicted by IPA in the normal or “control” proteome. Since toxicity-associated pathways characterize the abnormal proteome, it is plausible that TP53 might act as “bad cop” compromising cell metabolism and inducing cell damage in cHC due to a shutdown of xenobiotic metabolic pathways. Conversely, in situations of low stress TP53 cell survival mechanisms such as detoxification activation are stimulated to promote cell survival.
Furthermore, HDAC1 is suggested to interact with FOLH1 to regulate stability by modulating its lysine acetylation status. 69 As previously mentioned, SP1-dependent-trancription activation by acetyl transferases is associated with inhibition of histone deacetylation (HDAC1). In our study, HDAC1 appears to be repressed, implying that FOLH1 is bound to be unstable with negative repercussions for folate polyglutamylation and, hence, folate homeostasis.
Interestingly, the work of Eide and Ringstad in 2018 and 2020 70 , 71 importantly revealed how the brain is prone to increased toxicity in the entorhinal cortex and lateral ventricles by a faulty glymphatic system as well as a dysfunctional choroid plexus promoting spontaneous normal pressure HC. Moreover, the apical surface of the choroid plexus (epiplexus) is acknowledged as a macrophage-like tissue. Macrophage activation in this area was associated with choroid plexus cell death by the release of macrophage toxic substances leading again to choroid plexus dysfunction, CSF blockade and hydrocephalus in spontaneous hypertensive rats. 72 These findings are concurrent with our study that suggests how brain toxicity is linked to CSF obstruction, which hampers the clearance of toxic waste leading to the unique metabolic changes represented in the CSF of hydrocephalic Texas (HTx) rats.
Conclusions
This non-target metabolic profile analysis provides a comprehensive protein screening for identification of cHC-related metabolic pathways represented in CSF. Overall, 1583 proteins differentially expressed in abnormal CSF were rigorously detected (CV < 10; N = 6 in each gestational age) by LCMS and investigated further by complementary biocomputing IPA analysis to elucidate the metabolic phenotype of this condition, which was mainly characterized by toxicity-related pathways and differences in folate metabolism when compared to control CSF. This body of work also clarifies the key pathways mediating the pathogenesis of cHC, provides a tool for disease prediction and prognosis early in pregnancy, and proposes novel focus proteins like NEDD4, FOLH1, TP53, SP1, HDAC1 and HDAC3 as potential therapeutic targets in the endeavour to discover a non-invasive treatment that might prevent all-together the need for life-threatening surgical procedures later at birth. The findings of this in-depth analysis confirm many of our previous findings of folate metabolic imbalance, which are, among others, the specific loss of ALDH1L1, also known as 10-formyl tetrahydrofolate dehydrogenase, in hydrocephalic CSF and its relationship to severity of hydrocephalus. Moreover, a study we are currently preparing for publication demonstrates significant loss of methylation in the HTx rat across systems including the brain, which taken together, support the findings of the current study, and the methodology to investigate in depth changes associated with this condition. A further, important aspect of this work is to demonstrate the physiological role of CSF in the developing brain and no doubt also in the adult brain considering its continuous production and association with other conditions later in life.
Supplemental Material
sj-pdf-1-jcb-10.1177_0271678X211039612 - Supplemental material for Profound changes in cerebrospinal fluid proteome and metabolic profile are associated with congenital hydrocephalus
Supplemental material, sj-pdf-1-jcb-10.1177_0271678X211039612 for Profound changes in cerebrospinal fluid proteome and metabolic profile are associated with congenital hydrocephalus by Alicia Requena-Jimenez, Mohammad Nabiuni and Jaleel A Miyan in Journal of Cerebral Blood Flow & Metabolism
Footnotes
Acknowledgements
We thank The Charles Wolfson Charitable Trust for generous support and Dr Leo Zeef, of the Bioinformatics Core Facility at The University of Manchester, for his expertise and training on the Qiagen IPA analysis platform.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
JAM, ARJ and MN planned and executed the project. JAM conceived the initial project with MN and then the IPA analysis with ARJ. MN performed sample preparation and liquid chromatography with tandem mass spectrometry (LCMS). He also analysed the data through MASCOT to produce the list of proteins in order of abundance. ARJ analysed the data to assess proteomic reliability, reproducibility, and repeatability of the LCMS study. She categorized the data with algorithms to make it compatible with IPA software for protein mapping and analysis, designing and customizing IPA analysis to query for methylation and related pathways. ARJ wrote the original draft of the manuscript, but all authors edited and then approved the final draft and submission.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.