
Abstract
Traumatic brain injury (TBI) is commonly followed by long-term cognitive deficits that severely impact the quality of life in survivors. Recent studies suggest that microglial/macrophage (Mi/MΦ) polarization could have multidimensional impacts on post-TBI neurological outcomes. Here, we report that repetitive intranasal delivery of interleukin-4 (IL-4) nanoparticles for 4 weeks after controlled cortical impact improved hippocampus-dependent spatial and non-spatial cognitive functions in adult C57BL6 mice, as assessed by a battery of neurobehavioral tests for up to 5 weeks after TBI. IL-4-elicited enhancement of cognitive functions was associated with improvements in the integrity of the hippocampus at the functional (e.g., long-term potentiation) and structural levels (CA3 neuronal loss, diffusion tensor imaging of white matter tracts, etc.). Mechanistically, IL-4 increased the expression of PPARγ and arginase-1 within Mi/MΦ, thereby driving microglia toward a global inflammation-resolving phenotype. Notably, IL-4 failed to shift microglial phenotype after TBI in Mi/MΦ-specific PPARγ knockout (mKO) mice, indicating an obligatory role for PPARγ in IL-4-induced Mi/MΦ polarization. Accordingly, post-TBI treatment with IL-4 failed to improve hippocampal integrity or cognitive functions in PPARγ mKO mice. These results demonstrate that administration of exogenous IL-4 nanoparticles stimulates PPARγ-dependent beneficial Mi/MΦ responses, and improves hippocampal function after TBI.
Introduction
Traumatic brain injury (TBI) is a major cause of fatality and disability in young adults in the United States and other developed countries. According to the Centers for Disease Control and Prevention, 2.87 million TBI cases were reported in 2014 and approximately 13.5 million TBI survivors live with a disability in the United States (https://www.cdc.gov/traumaticbraininjury/index.html). The neurological sequelae of moderate or severe TBI include sensorimotor and cognitive deficits. While sensorimotor functions after TBI can be improved at least in part through rehabilitation training, 1 cognitive impairments are long-lasting and adversely impact quality of life of TBI survivors. 2 Currently, there is no approved therapy to prevent post-TBI cognitive dysfunction.
Components of innate immunity, including brain resident microglia and monocyte-derived infiltrating macrophages (Mi/MΦ), are rapidly activated after TBI.3,4 Mi/MΦ are highly dynamic cells that can polarize to various functional phenotypes in response to specific microenvironmental signals, thereby influencing short and long-term TBI outcomes.5,6 Under certain conditions, Mi/MΦ may shift their functional state toward an inflammation-resolving phenotype associated with improved cognitive function in models of TBI or stroke.4,7
Interleukin-4 (IL-4) is an anti-inflammatory cytokine and a well-established inducer of Mi/MΦ polarization toward inflammation-resolving and pro-repair phenotypes. 8 IL-4 is released into the interstitial fluid from injured neurons following cerebral ischemia and is known to polarize microglia via receptor binding.9,10 IL-4 deficiency is associated with poor outcomes in models of brain injury,7,11,12 whereas central administration of IL-4 protein improves functional outcomes after CNS injuries by promoting Mi/MΦ phagocytosis, inflammation resolution, and tissue repair.9,13,14 In a recent study, we demonstrated that brain delivery of IL-4 promoted sensorimotor recovery in a murine model of TBI by activating oligodendrocyte-mediated white matter repair. 15 However, the impact of IL-4 on long-term cognitive functions after TBI is not known. To fill these gaps, we tested the hypothesis that nose-to-brain delivery of IL-4 recombinant protein improves long-term hippocampus-dependent cognitive functions in a murine model of TBI. Second, we tested the hypothesis that IL-4 restores cognition by enhancing microglial beneficial responses and maintaining the functional and structural integrity of the hippocampus.
Materials and methods
For further methodological details on the CCI model, IL-4 nanoparticle production, neurobehavioral tests, diffusion tensor imaging, and immunofluorescence and image analysis, please consult the SI Appendix. Key resources that are essential to reproduce the results are including in the S Table 1 and all statistical analyses are summarized in S Table 2.
Animals
All animal experiments were approved by the University of Pittsburgh Institutional Animal Care and Use Committee and the VA Institutional Animal Care and Use Committee, in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Animal data are reported in accordance with ARRIVE guidelines. 16 All animals were housed in a temperature and humidity-controlled 12-h light/dark cycle with ad libitum food and water. Every attempt was made to minimize animal suffering and reduce the numbers of biological replicates. Male wildtype (WT) mice and Mi/MΦ-specific PPARγ conditional knockout (mKO) mice were used at 10–12 weeks of age (see SI Appendix).
Murine model of traumatic brain injury
TBI was performed by unilateral controlled cortical impact (CCI) as previously described. 15 Details and procedures are described in the SI Appendix. Surgeries and all outcome assessments were performed by investigators blinded to mouse genotype and experimental group assignments.
LTP measurements
Measurements of long-term potentiation (LTP) were performed to assess synaptic function and plasticity. At 35 days after TBI, mice were euthanized, and brains were rapidly extracted. Transverse hippocampal slices (350 µm) were cut on a Vibratome (Leica) in pre-gassed (95% O2/5% CO2) NMDG-HEPES solution (92 mM N-Methyl-D-Glucamine, 2.5 mM KCl, 1.2 mM NaH2PO4, 20 mM HEPES, 30 mM NaHCO3, 25 mM glucose, 5 mM sodium ascorbate, 3 mM sodium pyruvate, 2 mM thiourea, 10 mM MgSO4, and 0.5 mM CaCl2; pH7.3-7.4) at 4 °C and then recovered in artificial cerebrospinal fluid (aCSF; 119 mM NaCl, 26.2 mM NaHCO3, 2.5 mM KCl, 2.5 mM CaCl2, 1.3 mM MgSO4, 1.0 mM NaH2PO4, and 11.0 mM glucose; pH 7.3–7.4) at 32 °C for 0.5 h. Brain slices were further recovered by incubation at room temperature for 1 h, and then transferred to a recording chamber where they were submerged and constantly perfused with aCSF (3–4 mL/min) at 26–28°C.
Field excitatory postsynaptic potentials (fEPSPs) were evoked with concentric bipolar electrodes placed in the Schaffer collaterals and recorded in the CA1 stratum radiatum using an aCSF-filled microelectrode with 3–7 MΩ resistance. Both the stimulation electrodes and the recording electrode were placed at a depth of 100 µm within the brain slices, and a series of ascending stimuli (0–60 μA) were delivered for input/output recordings. fEPSPs were evoked by a 0.2-ms single pulse delivered every 30 s and baseline was recorded with stimuli intensity eliciting 40–60% of maximal response. After at least 20 min of stable baseline recordings, LTP was induced by a train of high-frequency stimulation (100 pulses at 100 Hz, delivered at 0.5 Hz, repeated 3 times) using the same stimulation intensity as above, and fEPSPs were recorded for 60 min.
Statistical analysis
All statistics were performed using GraphPad Prism 8 or SPSS software. The Kolmogorov-Smirnov normality test was initially performed on all datasets. Datasets adhering to a Gaussian distribution are presented as mean ± SD. Non-normally distributed data are presented as box-and-whisker plots showing the 25th, 50th, and 75th percentiles and the maximum and minimum values. For normally distributed data, comparisons between two groups were conducted by the two-tailed Student’s t-test, and comparisons between multiple groups were conducted by one or two-way ANOVA followed by the Bonferroni post hoc test. For continuous variables with non-normal distributions, the two-tailed Mann-Whitney U rank sum test was used. Correlation analyses between continuous data with normal distributions were performed using Pearson analyses. A p-value of <0.05 was deemed significant. Detailed information on all data analyses is listed in S Table 2.
Results
A total of 232 C57BL6 male mice (96 WT mice, 27 PPARγ OPC-cKO mice, and 109 Mi/MΦ-specific PPARγ mKO mice) were used in this study, including 171 mice subjected to CCI, and 61 mice subjected to sham operation. Seven mice were excluded by observers blinded to mouse genotypes and/or experimental grouping due to sampling errors (subpar brain slicing, insufficient brain perfusion, or other random procedural errors) or death, including via TBI-induced epilepsy. In addition, 12 mice were excluded due to unsuccessful pre-TBI training or failed performance in neurobehavioral tests (unwilling to swim or failed to discover the hidden platform in the MWM; never touched either the old or new object in the NOR). Finally, 10 mice were excluded due to failure in the electrophysiology experiments (failed to induce LTP, static interference, etc.), or other experimental problems (power outage, etc.).
Intranasal administration of IL-4 nanoparticles improves cognitive function after TBI
Long-term cognitive impairments, such as memory and executive dysfunction, are among the most disabling functional outcomes in TBI. To assess the therapeutic effects of IL-4 on TBI-induced cognitive deficits, we employed the Morris water maze (MWM), passive avoidance test, and novel object recognition (NOR) test to examine spatial and non-spatial cognitive functions before and up to 35 days after TBI (Figure 1(a)). Mice that received sham surgery showed no differences in cognitive function across vehicle and IL-4-treated groups (Supplemental Figure 1). Therefore, the sham control groups were pooled for statistical analysis for all in vivo experiments throughout the study. The MWM was conducted 22–27 days after TBI to assess spatial learning and memory. Representative MWM track plots (Figure 1(b)) and quantitative data on learning (Figure 1(c)) and memory are illustrated (Figure 1(d)). Consistent with our previous results in this model, 17 CCI led to severe impairments in both learning and memory in the vehicle-treated mice, as reflected by longer times until escape in the cued learning phase (vehicle-treated TBI group vs. sham group, p = 0.0012) and less time spent in the target quadrant relative to all quadrants (vehicle-treated TBI group vs. sham group, p = 0.0008) as well as fewer platform crossings (vehicle-treated TBI group vs. sham group, p = 0.0086) in the spatial memory test phase. In contrast, IL-4-treated TBI mice displayed superior performance in cued learning at day-25 (IL4- vs. vehicle-treated TBI group, p = 0.0083) and memory at day-27 (target quadrant time, IL-4- vs. vehicle-treated TBI group, p = 0.0024). Unlike vehicle-treated mice, IL-4-treated mice did not display a statistically significant drop in platform crossings after TBI compared to sham-injured controls (sham group vs. IL-4-treated TBI group, p = 0.4402). All three groups had similar swim speeds (Figure 1(e)), indicating that the cognitive test results were not influenced by intergroup differences in gross locomotor function.
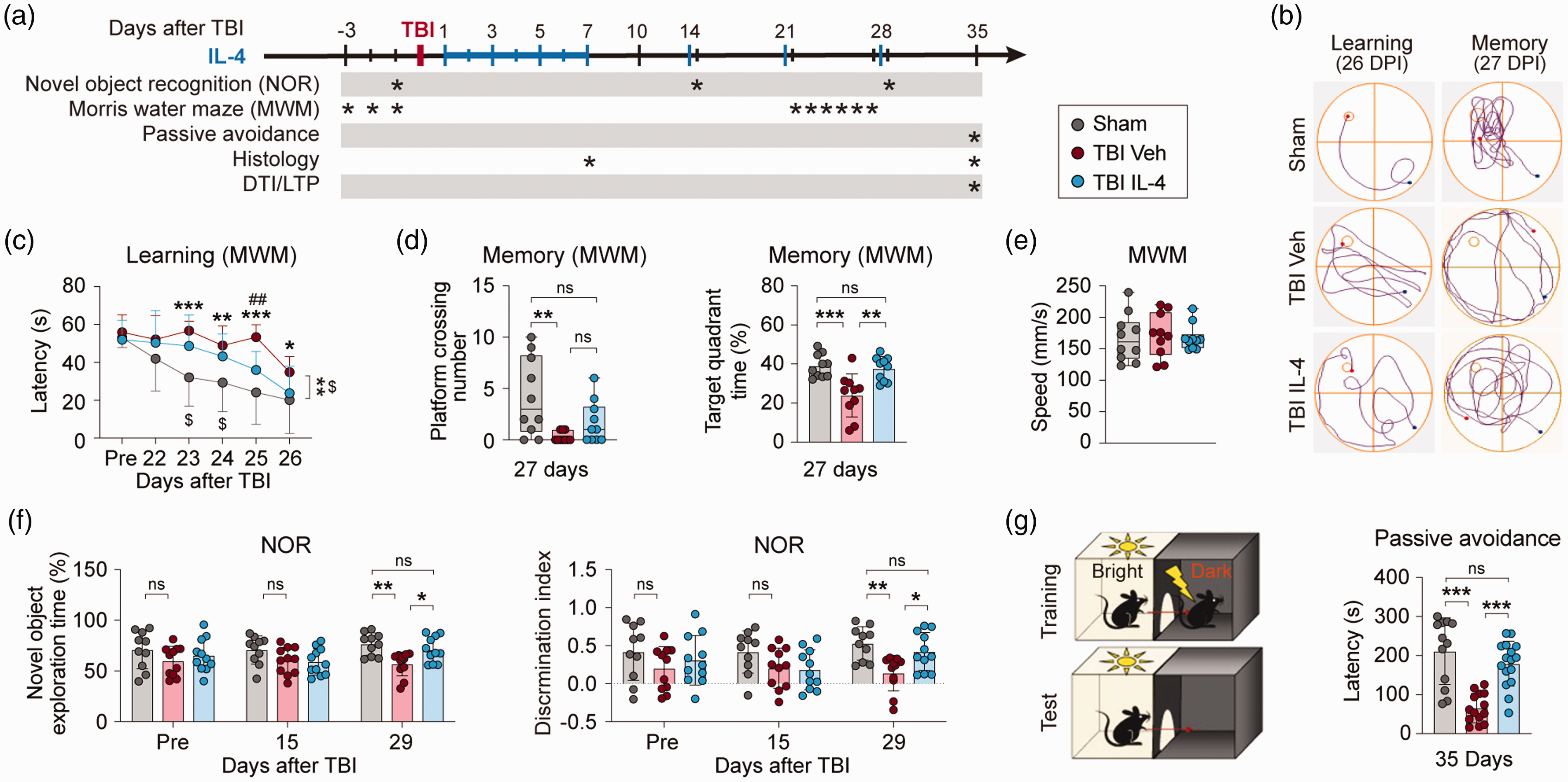
IL-4 treatment improves long-term cognitive functions after TBI. (a) Illustration of experimental timeline. Mice received intranasal administrations of IL-4 (50 µg/kg) or vehicle starting at 6 h after TBI and repeated daily at 1-7 days and then weekly at 2, 3, and 4 weeks after TBI. Sham, sham surgery group; TBI Veh, vehicle-treated TBI group; TBI IL-4, IL-4-treated TBI group. (b–e) Morris water maze (MWM) test. Representative track plot of learning and memory phases of test (b), the escape latency during cued learning phase pre-TBI and 22-26 days after TBI (c), the number of platform crossings and time spent in target quadrant in the memory test on day 27 (d), and swim speed (e). n = 10 per group. (f) Novel object recognition (NOR) test. Exploration times and discrimination indices were calculated. n = 10, sham group; n = 11 per TBI group. (g) Passive avoidance test. Illustration of test design (left panel). Step-through latency (right panel) was recorded on the test day. n = 12, sham group; n = 14, TBI Veh group; n = 16, TBI IL-4 group. Statistical analyses: Two-way repeated measures ANOVA with Bonferroni post hoc test for c (*p < 0.05; **p < 0.01; ***p < 0.001, TBI Veh vs. Sham; $p < 0.05 TBI IL-4 vs. Sham; ##p < 0.01 TBI Veh vs. TBI IL-4) and f. One-way ANOVA followed by Bonferroni post hoc test for d (target quadrant time) and g. Kruskal-Wallis test with Dunn post hoc for d (platform crossings) and e. Shown are mean ± SD or box plots. *p < 0.05, **p < 0.01, ***p < 0.001 as indicated, ns: no significance.
The NOR test was performed 15 and 29 days after TBI to assess hippocampus-dependent non-spatial cognitive function.18,19 All three groups showed similar abilities to recognize a novel object at 15 days after TBI (Figure 1(f)), as measured by time spent exploring the novel object. However, by 29 days after TBI, vehicle-treated TBI mice exhibited significant declines in NOR performance (vehicle-treated TBI group vs. sham group, p = 0.0025) and lower novel object discrimination indices (vehicle-treated TBI group vs. sham group, p = 0.0025) compared to the sham group. Compared to vehicle-treated TBI mice, IL-4-treated TBI mice spent significantly more time exploring the novel object 29 days after injury (IL4- vs. vehicle-treated TBI group, p = 0.0374).
The passive avoidance test was conducted at 35 days after TBI to assess fear-motivated memory as another hippocampus-dependent non-spatial cognitive function. 20 Vehicle-treated TBI mice showed significantly decreased latencies to enter the shock compartment compared to the sham group ((Figure 1(g), vehicle-treated TBI group vs. sham group, p = 0.0002), indicating impaired memory of fear-associated events after TBI. In comparison to vehicle-treated TBI mice, IL-4-treated TBI showed increased fear memory (Figure 1(g), IL-4- vs. vehicle-treated TBI group, p < 0.0001).
Together, these results demonstrate that nose-to-brain IL-4 delivery significantly improves long-term spatial and non-spatial cognitive functions after TBI.
Conditional deletion of PPARγ within oligodendrocyte precursor cells does not prevent IL-4-mediated improvements in cognitive function after TBI
White matter lesions can be a major contributing factor in the pathogenesis of cognitive impairments. 21 Our recent study showed that oligodendrocyte precursor cell-specific PPARγ conditional knockout (PPARγ OPC-cKO) significantly hinders white matter repair and weakens IL-4-afforded improvement in sensorimotor functions after TBI. 15 To determine whether IL-4-afforded cognitive recovery is also impaired in PPARγ OPC-cKO mice, the Morris water maze and passive avoidance tests were performed on IL-4- and vehicle-treated PPARγ OPC-cKO mice after TBI. There was no difference in swim speed among the three groups (Supplemental Figure 2(a)). TBI induced cognitive deficits in vehicle-treated PPARγ OPC-cKO mice, as demonstrated by significant increases in escape latency during the cued learning phase (Supplemental Figure 2(b), vehicle-treated OPC-cKO TBI group vs. OPC-cKO sham group, p < 0.0001), less time spent in the target quadrant (vehicle-treated OPC-cKO TBI group vs. OPC-cKO sham group, p = 0.0025) and fewer platform crossings (vehicle-treated OPC-cKO TBI group vs. OPC-cKO sham group, p = 0.0001) (Supplemental Figure 2(c) and (d)) in the spatial memory phase of the Morris water maze test, and significantly decreased fear latency in the passive avoidance test (vehicle-treated OPC-cKO TBI group vs. OPC-cKO sham group, p < 0.0001) (Supplemental Figure 2(e)). However, IL-4-treated TBI PPARγ OPC-cKO mice showed significantly improved cognitive performance (Supplemental Figure 2(b) to (d), Escape latency, p < 0.0001; Quadrant time, p = 0.0321) or a strong trend toward improvement in spatial memory (Supplemental Figure 2(d), Platform crossings, p = 0.0590) compared to vehicle-treated TBI mice. Moreover, IL-4-treated TBI PPARγ OPC-cKO mice also showed significantly lower passive avoidance deficits at 35 days after TBI compared to vehicle-treated mice (Supplemental Figure 2(e), p < 0.0001). These data demonstrate that oligodendroglial PPARγ deficiency does not hinder IL-4-mediated cognitive improvements after TBI.
IL-4 Treatment preserves the functional integrity of the hippocampus after TBI
The hippocampus is an essential centerpiece of the telencephalic neural network subserving higher-order cognitive functions.22,23 As IL-4 treatment markedly attenuates both spatial and non-spatial cognitive deficits after TBI, we tested the hypothesis that IL-4 treatment improves cognitive functions by promoting the functional and structural integrity of the hippocampus. Hippocampal long-term potentiation (LTP) is considered to be one of the major physiological substrates underlying learning and memory acquisition. 24 Hence, we assessed LTP in the CA1 stratum radiatum evoked by high-frequency stimulation (HFS) of the Schaffer collateral pathway at 35 days after TBI or sham surgery. The data reveal a lower input/output relationship (magnitude of fEPSP vs. stimuli) in the two TBI groups compared to the sham control group (Figure 2(a), TBI group vs. sham group, p < 0.0001), and no differences in this measure were observed between the IL-4 and vehicle treated groups after TBI. These data suggested that IL-4 treatment did not reverse TBI-induced deficits in basal synaptic transmission at the Schaffer collateral-CA1 synapse. Next, LTP was examined following a train of high-frequency stimulation of the Schaffer collateral-CA1 projections. HFS produced a robust and immediate potentiation in sham hippocampal slices, and maintained normal short- and long-term potentiation, which decayed over a 10 min period, stabilizing at ∼170% of the pre-HFS baseline (Figure 2(b)). Compared to vehicle, IL-4 treatment partially prevented the LTP impairments observed after TBI (Figure 2(b), IL-4- vs. vehicle-treated TBI group, p = 0.0125), suggesting that IL-4 treatment improves synaptic plasticity in the injured hippocampus. A Pearson correlation coefficient analysis to test for associations between hippocampal LTP and cognitive behavior data revealed that the slope of fEPSP% (in the last 2 min of recording) was indeed negatively correlated with the latency in the MWM learning phase (Figure 2(c), r = −0.596, p = 0.015), and positively correlated with the target quadrant time in the MWM memory test (Figure 2(c), r = 0.602, p = 0.014) and latency in the passive avoidance test (Figure 2(d), r = 0.5, p = 0.049), but not significantly correlated with exploration time in the NOR test (Figure 2(e)).
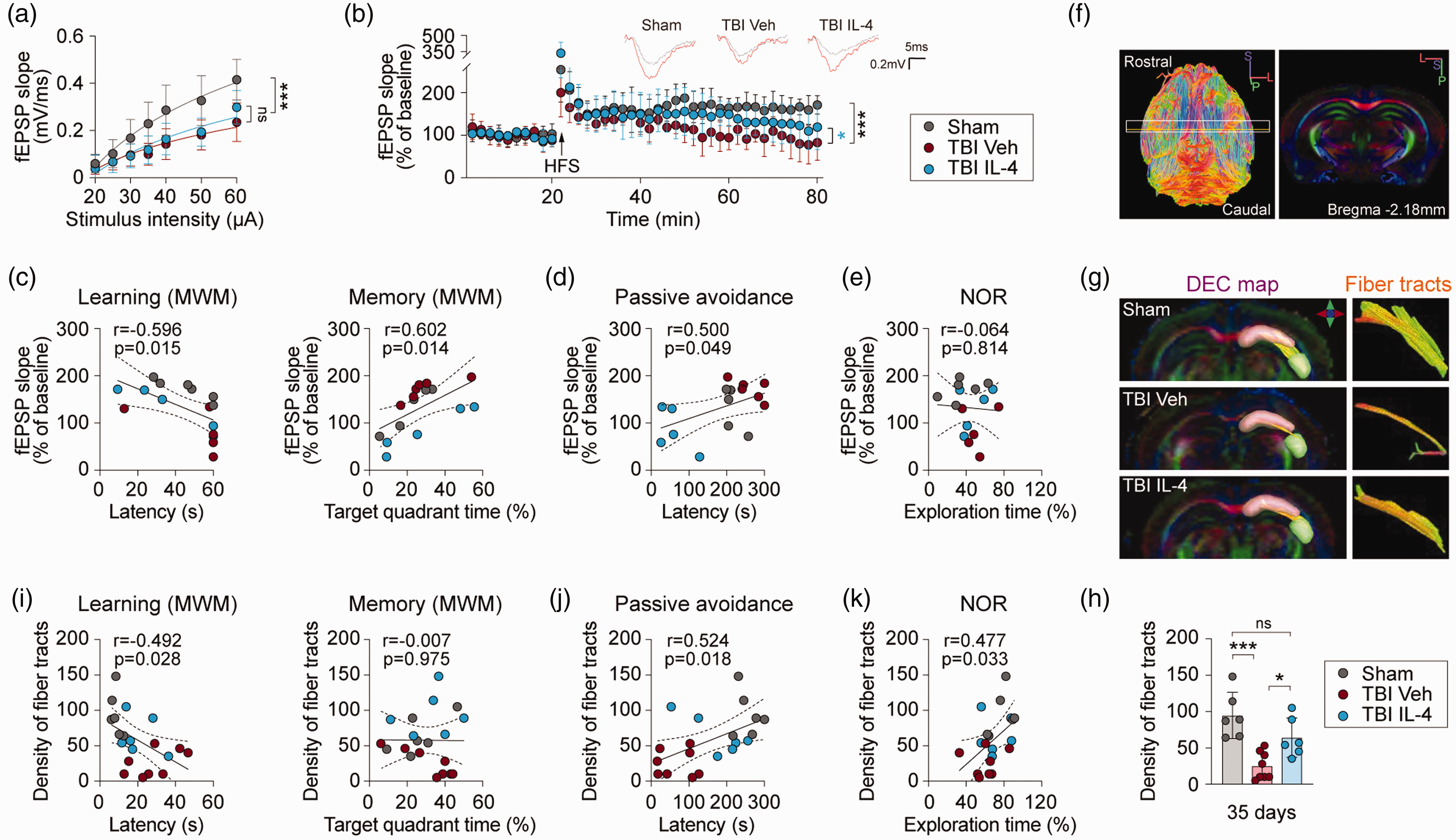
IL-4 treatment promotes the functional and structural integrity of the hippocampus after TBI. (a and b) Electrophysiological analysis of hippocampal LTP at 35 days after TBI or sham surgery. (a) The relationship between field excitatory postsynaptic potential (fEPSP) magnitude and stimulus intensity (in microamperes). (b) Representative traces and cumulative data plots of fEPSP slope (normalized from baseline) prior and up to 60 min after high-frequency stimulation (HFS). n = 6, sham group; n = 5 per TBI group. (c–e) Pearson correlation between fEPSP slope (the final 2-min recording) and learning or memory phase in the Morris water maze (MWM) test (c), latency in the passive avoidance test (d), or exploration time in the novel object recognition (NOR) test (e). n = 4-6 per group. (f–h) Protective effect of IL-4 treatment on hippocampal fiber tracts after TBI, assessed using ex vivo DTI. (f) Left panel: a 3 D reconstruction based on one axial DTI scanning plane that encompasses the hippocampus. The white box depicts the approximate coronal levels for DTI scanning of Schaffer collaterals, and the yellow line indicates the coronal level of AP −2.18 mm. Right panel: A representative 2 D directional color-encoded image showing the dorsal hippocampal formation at the level of AP −2.18 mm. Color hues indicate directions of neural fibers. (g) 3 D reconstruction of DTI images showing the fiber tracts of Schaffer collaterals between CA1 (pink) and CA3 (green) of the hippocampus. (h) Quantification of fiber tract density of two serial sections (AP: −1.58 mm and −2.18 mm). n = 6, sham group; n = 8, TBI Veh group; n = 6, TBI IL-4 group. (i–k) Pearson correlation between density of fiber tracts and learning or memory phase in the MWM test (i), latency in the passive avoidance test (j), or novel object exploration time (% of total exploration time) in the NOR test (k). n = 6–8 per group. Statistical analyses: Two-way repeated measures ANOVA with Bonferroni post hoc test for a and b. One-way ANOVA followed by Bonferroni post hoc test for h. Pearson linear regression analysis for c-e and i-k. Shown are mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001 as indicated, ns: no significance.
Ex vivo DTI was performed to assess the integrity of hippocampal white matter at 35 days after TBI or sham surgery. The results showed that TBI did not cause gross changes in hippocampal sizes, in terms of absolute hippocampal volume (Supplemental Figure 3(a) and (b)) or relative hippocampal volume (fold vs. contralateral non-injured hippocampus) (Supplemental Figure 3(a) and (c)) across the three groups (sham group, vehicle-treated TBI group, IL-4-treated TBI group). TBI (vehicle-treated) also did not lead to significant alterations in several other DTI parameters, including fractional anisotropy (FA), axial diffusivity (AD), and radial diffusivity (RD) (Supplemental Figure 3(d)). However, TBI (vehicle-treated) mice showed significant increases in mean diffusivity (MD) (p = 0.0330), indicating the uncontrolled diffuse of water molecules in the voxel; in contrast, MD was not increased in IL-4-treated TBI mice (p = 0.0843) (Supplemental Figure 3(d)). Notably, IL-4-treated TBI mice showed significantly increased AD (p = 0.0234) compared to the sham control group (Supplemental Figure 3(d)), suggesting less axonal injury. 25
Next, we assessed the fiber tracts of the Schaffer collaterals – the axons that transmit signals from CA3 to CA1 pyramidal neurons. As shown (Figure 2(f) to (h)), there was substantial (∼70%) loss of these fiber tracts after TBI with vehicle treatment (p = 0.0003) compared to the sham control group, whereas IL-4-treated TBI mice exhibited significantly denser fiber tracts (p = 0.0369) compared to vehicle-treated TBI mice. Pearson correlation coefficient analyses showed a significant negative correlation between the density of fiber tracts and the latency to learn new spatial information in the MWM (Figure 2(i), r = 0.492, p = 0.028), and a significant positive correlation between the density of fiber tracts and latency in the passive avoidance test (Figure 2(j), r = 0.524, p = 0.018) or new object exploration time in the NOR test (Figure 2(k), r = 0.477, p = 0.033).
Overall, these results suggest that IL-4 treatment partially preserves the structure and function of the Schaffer collaterals after TBI, which is likely to contribute to superior learning and memory in IL-4-treated TBI mice.
IL-4 treatment protects hippocampal CA3 neuronal somata and their schaffer collaterals
Immunohistochemical staining was performed at 35 days after TBI to determine whether IL-4 preserves hippocampal synaptic plasticity (see above) by preventing CA3 neuronal loss, a pathological hallmark of the CCI model.26,27 NeuN staining showed that TBI resulted, on average, in 30% neuronal loss in CA3 (Figure 3(a) and (b), p = 0.0093 vs. sham group), but no cell loss in CA1 or the DG. Notably, hippocampal CA3 neurons remained at sham control levels in IL-4-treated TBI mice (Figure 3(a) and (b)). Pearson correlation coefficient analyses showed that there was a significant positive correlation between the number of CA3 neurons and fear memory in the passive avoidance test (Figure 3(d), r = 0.518, p = 0.005) or the novel object exploration time in the NOR test (Figure 3(e), r = 0.706, p < 0.001). However, the number of CA3 neurons was not significantly correlated with learning or memory performance in the MWM (Figure 3(c)).
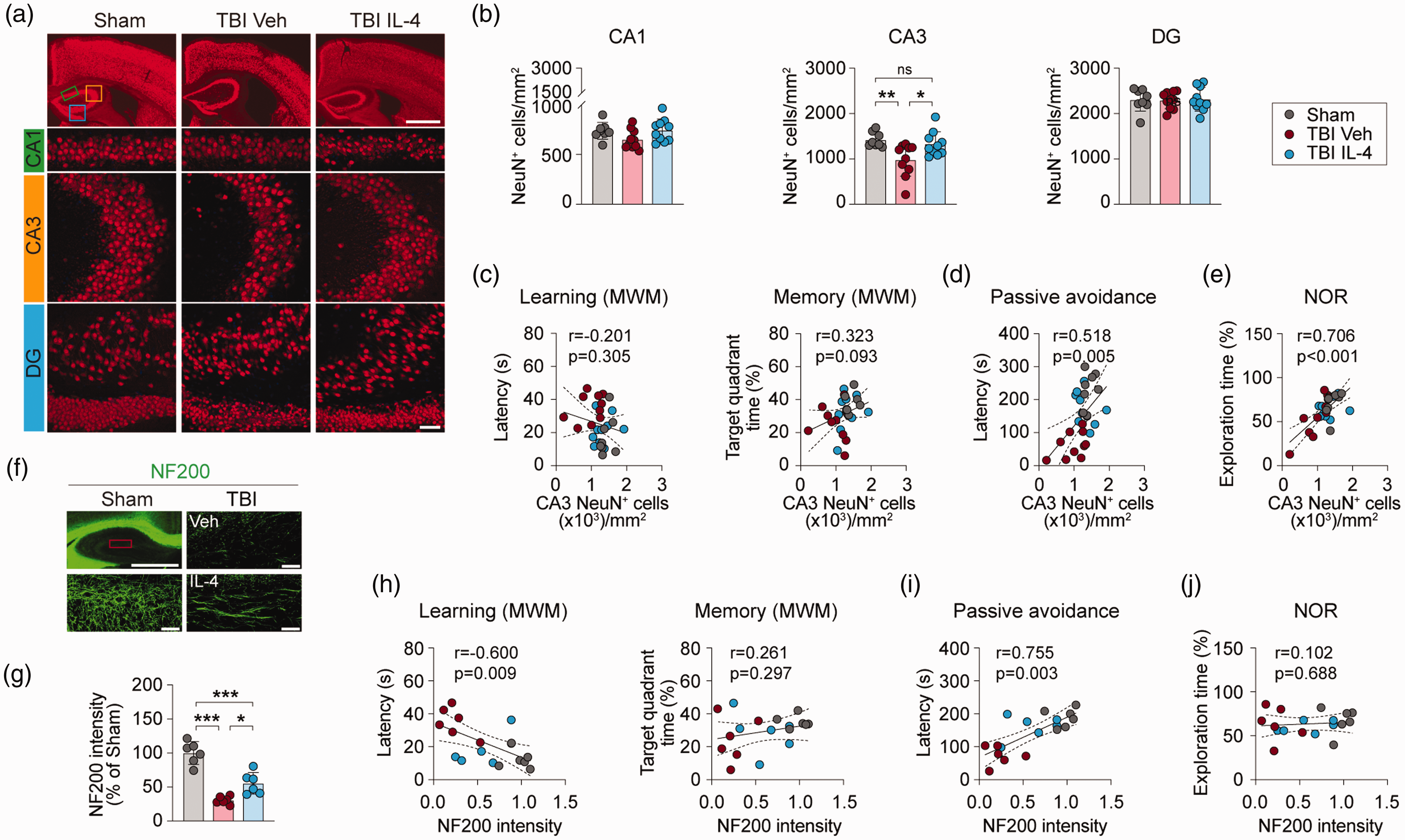
IL-4 treatment protects against neuronal loss in hippocampal CA3 after TBI. (a) Representative images of NeuN immunofluorescence (red) showing the regions of interest in CA1 (green rectangle), CA3 (yellow square), and DG (blue square). Scale bar = 1 mm (upper panel) and 50 µm (lower panel). All images are captured from mice sacrificed at 35 days after TBI or sham surgery. (b) Quantification of NeuN+ cells in CA1, CA3, and DG regions at 35 days after TBI or sham surgery. n = 8, sham group; n = 10 per TBI group. (c–e) Pearson correlation between NeuN+ cell counts in CA3 and learning or memory phase in MWM test (c), latency in the passive avoidance test (d), or novel object exploration time in the NOR test (e). n = 8, sham group; n = 10 per TBI group. (f) Representative images of NF200 immunofluorescence (green) in the dorsal hippocampus at 35 days after TBI. The upper left image is a low-power view of the dorsal hippocampus (scale bar = 1 mm), in which the red rectangle depicts where high-power images were captured. The other three images are high-power views of region of interest from different experimental groups as indicated (scale bar = 50 µm). (g) Quantification of relative NF200+ neural fiber intensity in CA1. n = 6 per group. (h–j) Pearson correlation between NF200+ neural fiber intensity and learning or memory phase results in MWM test (h), latency in the passive avoidance test (i), or novel object exploration time in the NOR test (j). n = 6 per group. Statistical analyses: One-way ANOVA followed by Bonferroni post hoc test for b and g. Pearson correlation coefficient analyses for c-e and h-j. Shown are the mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001 as indicated, ns: no significance.
Semi-quantitative analyses of immunofluorescent staining of neurofilament protein H (NF200) in the hippocampal CA1 region showed that NF200 immunofluorescence intensity was markedly decreased in vehicle-treated (∼75%, p < 0.001 vs. sham control mice) and IL-4-treated (∼50%, p < 0.001 vs. sham control mice) TBI mice (Figure 3(f) and (g)). IL-4 treatment significantly increased the intensity of NF200-positive nerve fibers in the CA1 region after TBI compared to vehicle treatment (p = 0.0205). The loss of CA1 NF200-positive nerve fibers was negatively correlated with behavioral performance in the learning phase of the MWM test (Figure 3(h), r = −0.600, p = 0.009) and positively correlated with fear memory in the passive avoidance test (Figure 3(i), r = 0.755, p = 0.003), but not novel object exploration time in the NOR test (Figure 3(j), r = 0.102, p = 0.688). These correlation analyses suggest that the enhancement in neuronal survival in CA3 and attenuation of the loss of Schaffer collaterals is likely to contribute to IL-4-mediated improvement in some but not all cognitive functions after TBI.
Intranasal delivery of IL-4 polarizes brain microglia and inhibits brain inflammation after TBI
The IL-4 cytokine is well characterized for its ability to polarize Mi and MΦ to an inflammation-resolving and pro-repair phenotype. 28 Therefore, we hypothesized that IL-4 treatment might also polarize Mi/MΦ after TBI and thereby exert potent anti-inflammatory actions. First, we performed triple-label immunofluorescent staining for Iba1 (a pan-cell marker for Mi/MΦ), CD16/32 (a pro-inflammatory marker), and Arginase-1 (Arg1, an inflammation-resolving and pro-repair marker) at 7 days after TBI (Figure 4(a)). In sham control mice, there were few CD16/32+/Iba1+ or Arg1+/Iba1+ cells in all hippocampal sub-regions examined (Figure 4(a) to (c)). In vehicle-treated TBI mice, however, there were robust increases in the numbers of CD16/32+/Iba1+ cells in the CA1, CA3, and DG areas; as hypothesized, these numbers were significantly attenuated in these three brain regions in IL-4-treated TBI mice (Figure 4(a) and (b), p < 0.001). In contrast, the numbers of Arg1+/Iba1+ cells were significantly increased in IL-4-treated TBI mice in CA1 (p = 0.0003), CA3 (p = 0.0109), and the DG (p = 0.0052) compared to vehicle-treated TBI mice (Figure 4(a) and (c)).
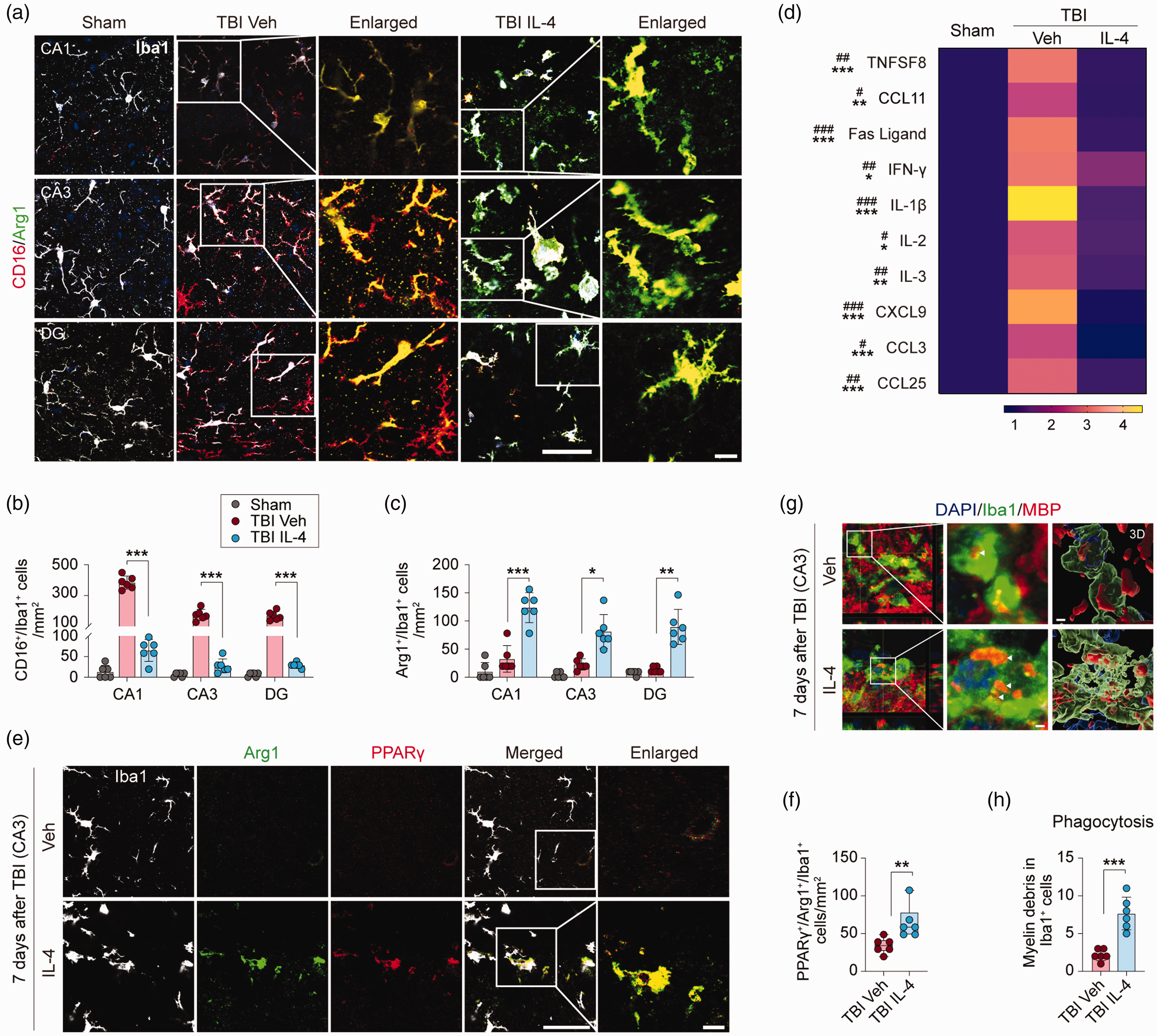
IL-4 polarizes microglia and enhances phagocytosis after TBI. (a) Representative images of triple-label immunofluorescence for Iba1 (gray), CD16 (red), and Arg1 (green) in CA1, CA3, and DG at 7 days after TBI or sham surgery. The region of interest (ROI) depicted by white squares in the second and fourth columns are enlarged and presented in the third and fifth columns, respectively. Scale bar = 50 µm (fourth column) and 10 µm (fifth column). (b and c) Quantification of CD16+/Iba1+ (b) and Arg1+/Iba1+(c) cells in CA1, CA3, and DG after TBI or sham surgery. n = 6 per group. (d) A panel of 40 inflammatory makers were measured in hippocampal extracts at 5 days after TBI or sham surgery. Heatmap showing mean expression levels of 10 makers that were significantly upregulated in TBI Veh brains compared to sham control brains, and that were significantly reduced in TBI IL-4 brains. (e) Representative images of triple-label immunofluorescence for Iba1 (gray), Arg1 (green), and PPARγ (red) in CA3 at 7 days after TBI. The ROI depicted by white squares in the fourth column is enlarged and presented in the fifth column and the Arg1(green) and PPARγ (red) merged images are shown. Scale bar = 50 µm (fourth column) and 10 µm (fifth column). (f) Quantification of PPARγ+/Arg1+/Iba1+ cells in CA3 region. n = 6 per group. (g) Representative images of immunofluorescence for MBP (red) and Iba1 (green) in CA3 at 7 days after TBI. Nuclei were stained with DAPI (blue). The ROIs depicted by white squares in the first column are enlarged (middle column) or 3 D-rendered (right column). Arrowheads point to myelin debris inside the microglia. Scale bar = 2 µm. (h) Quantification of MBP+/Iba1+ cell numbers. n = 6 per group. Statistical analyses: One-way ANOVA followed by Bonferroni post hoc test for b (CA1). Welch ANOVA followed by Dunnett T3 post hoc test for b (CA3 and DG). Brown-Forsythe ANOVA followed Dunnett T3 post hoc for c. Two-way ANOVA with Bonferroni post hoc test for d. Mann-Whitney test for f. Welch’s t-test for h. Shown are the mean ± SD or box plots. #p < 0.05, ##p < 0.01, ###p < 0.001 TBI Veh vs. Sham. *p < 0.05, **p < 0.01, ***p < 0.001, ns: no significance vs. TBI IL-4.
To further determine if Mi or MΦ cells display alterations in CD16/32 and Arg1 expression after TBI, we performed double-label immunofluorescent staining for Iba1 and TMEM119, a microglia-specific marker. The results showed that more than 95% of Iba1+ cells in various hippocampal subregions were also TMEM119+ (Supplemental Figure 4). In contrast, TMEM119+ immunofluorescence was observed in ∼60% Iba1+ cells in the peri-lesion cortical areas (Supplemental Figure 4). These results suggest that TBI-induced inflammation in the cortex encompasses both microglia and infiltrating macrophages, but microglia are the predominant source for inflammation in the hippocampus proper.
We assessed brain inflammation 5 days after TBI using a Mouse Inflammation Antibody Array. Among the 40 inflammation markers tested, 10 were significantly elevated in vehicle-treated TBI mice compared to sham control mice, including TNFSF8, Fas ligand, INF-γ, IL-1b, IL-2, IL-3, IL-12 p70, CXCL9, CCL3, and CCL25 (Figure 4(d)). IL-4 treatment significantly reduced these inflammation markers (Figure 4(d)).
IL-4 upregulates microglial PPARγ and promotes myelin debris clearance after TBI
Our recent studies identified PPARγ as a master regulator of phagocytosis and inflammation-resolving actions by Mi/MΦ.29,30 Therefore, we examined the temporal profile and cellular distribution of PPARγ expression in the hippocampus at 1, 3, and 7 days after TBI using immunofluorescent staining (Supplemental Figure 6). Compared to sham control mice, the number of PPARγ+ mature oligodendrocytes (APC+) were significantly increased one day after TBI, and the numbers of PPARγ+ microglia (Iba1+), neurons (NeuN+), astrocytes (GFAP+), and oligodendrocytes (APC+) were significantly increased 3 days after TBI. At 7 days after TBI, the number of PPARγ+ microglia were lower, whereas the expression of PPARγ+ remained elevated in all other cell types (Supplemental Figure 6). As IL-4 is known to activate the STAT6/Arg1/PPARγ pathway after ischemic brain injury, 12 we investigated the effect of IL-4 treatment on PPARγ protein expression in the hippocampal CA3 region at 7 days after TBI. IL-4 treatment significantly increased the number of microglia expressing both PPARγ and Arg1 in the CA3 region (Figure 4(e) and (f), p = 0.0065).
Myelin debris clearance by microglia is a prerequisite for remyelination in white matter repair. 31 The cellular levels of PPARγ and Arg1 are positively correlated with the phagocytotic activity of Mi/MΦ.29,32 Thus, we investigated the effect of IL-4 treatment on myelin debris clearance by microglia in hippocampal CA3 after TBI. To this end, double-label immunofluorescent staining for Iba1 and MBP was followed by Z-stack collection and volumetric analyses. Notably, the results showed that microglia of IL-4-treated TBI mice phagocytosed approximately 3 times more myelin debris than microglia of vehicle-treated TBI mice (Figure 4(g) and (h), p = 0.0009).
PPARγ is essential for IL-4-induced microglia polarization after TBI
To determine the role of PPARγ in microglial polarization after TBI, we constructed Mi/MΦ-specific PPARγ conditional knockout (mKO) by crossing PPARγflox/flox mice and CX3CR1CreER mice (Figure 5(a)) as described previously. 19 Tamoxifen was administrated for 4 consecutive days to induce PPARγ mKO, and TBI was performed in mKO mice and CX3CR1CreER (wild type control) mice 10 days following the final tamoxifen injection. Double-label immunofluorescent staining for PPARγ and Iba1 at 5 days after TBI confirmed 95% reduction (median of the box plots) of microglial PPARγ protein expression in PPARγ mKO mice compared to CX3CR1CreER mice (Figure 5(b) and (c), p = 0.0004).
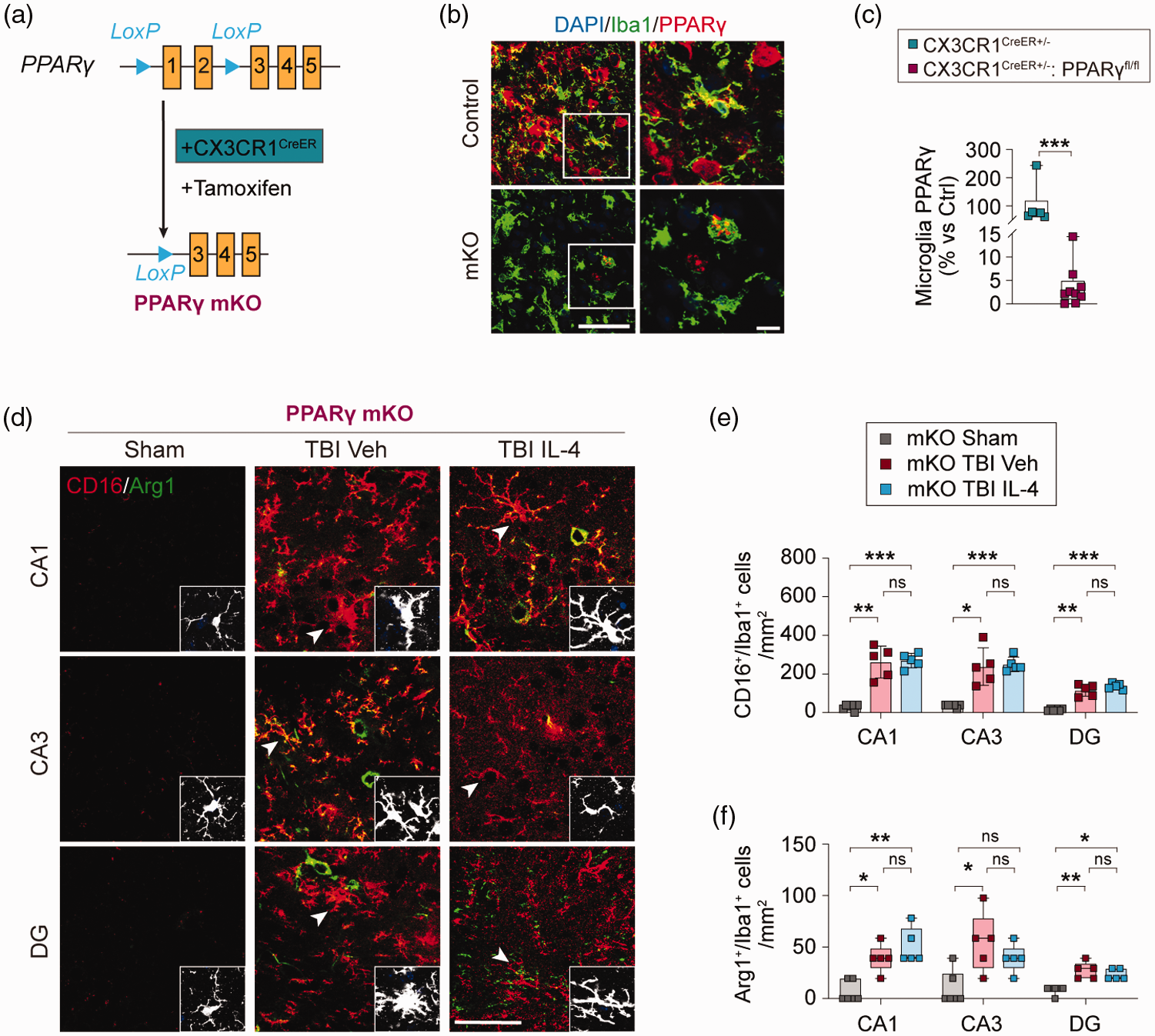
PPARγ is obligatory for IL-4-induced microglial polarization after TBI. (a) Generation of tamoxifen inducible Mi/MΦ-specific PPARγ knockout (PPARγ mKO) mice. (b) Representative images of immunofluorescence for Iba1 (green) and PPARγ (red) in CA3 at 7 days after TBI. Nuclei were stained with DAPI (blue). The ROIs depicted by white squares (left column) are enlarged (right column). Scale bar = 50 µm (left column) and 10 µm (right column), respectively. (c) Quantification of PPARγ relative immunofluorescence intensity in CA3 microglia. n = 6, control group; n = 9, mKO group. (d) Representative images of triple-label immunofluorescence for Iba1 (gray), CD16 (red), and Arg1 (green) in CA1, CA3, and DG at 7 days after TBI or sham surgery in PPARγ mKO mice. Iba1 immunofluorescence (gray) from the selected cells (pointed by arrow heads) in the second and third columns is shown in the inserts. Scale bar = 50 µm. For more details, see Supplemental Figure 7(e) and (f). Quantification of CD16+/Iba1+ (e) and Arg1+/Iba1+ (f) cells in CA1, CA3, and DG at 7 days after TBI or sham surgery. n = 6, sham group; n = 5 per mKO TBI group. Statistical analyses: Mann-Whitney test for (c). Brown-Forsythe ANOVA followed by Dunnett T3 post hoc for (e). Kruskal-Wallis with Dunn post hoc for (f). Shown are the mean ± SD or box plots. *p < 0.05, **p < 0.01, ***p < 0.001 as indicated, ns: no significance.
Triple-label immunofluorescent staining for CD16/Arg1/Iba1 was performed to determine the effect of IL-4 treatment on microglial polarization in PPARγ mKO mice after TBI. Compared to sham control mice, TBI induced robust increases in the number of CD16/32+/Iba1+ cells (CA1, p = 0.0088; CA3, p = 0.0244; DG, p = 0.0048) and modest increases in the number of Arg1+/Iba1+ cells (CA1, p = 0.0457; CA3, p = 0.0186; DG, p = 0.0052) in vehicle-treated PPARγ mKO mice (Figure 5(d) to (f), Supplemental Figure S7). IL-4 treatment in post-TBI PPARγ mKO mice failed to significantly alter the numbers of CD16+/Iba1+ or Arg1+/Iba1+ cells in the hippocampus, as compared to vehicle-treated TBI mice (Figure 5(d) to (f)). These results thus confirmed an essential role for PPARγ in mediating IL-4-induced microglial polarization in the injured hippocampus.
Mi/MΦ-specific PPARγ knockout abolishes IL-4-afforded hippocampal protection
Because IL4 treatment failed to polarize microglia in post-TBI PPARγ mKO mice, we hypothesized that IL-4 afforded hippocampal protection would also be impaired in PPARγ mKO mice. To test this hypothesis, ex vivo DTI analysis of the Schaffer collaterals was performed at 35 days after TBI. As hypothesized, TBI-induced loss of fiber tracts in PPARγ mKO mice was not ameliorated by IL-4 treatment (Figure 6(a) and (b), p = 0.9012). Moreover, NF200 immunofluorescent staining of the hippocampus showed ∼75% loss of nerve fibers in the CA1 region in post-TBI PPARγ mKO mice (Figure 6(c) and (d), TBI vehicle vs. sham group, p = 0.0227; TBI IL-4 vs. sham group, p = 0.0120), an effect that was not attenuated by IL-4 treatment (Figure 6(c) and (d), TBI vehicle vs. TBI IL-4, p > 0.9999). Finally, immunofluorescent staining for NeuN was performed at 35 days after TBI to test the effect of IL-4 treatment on neuronal loss in PPARγ mKO mice. TBI resulted in ∼50% and ∼20% neuronal loss in CA3 (p < 0.0001) and DG (p = 0.0096), respectively, but did not lead to significant neuronal loss in CA1 of the hippocampus (p = 0.0759). As expected, IL-4 treatment failed to prevent neuronal loss in CA3 or DG in post-TBI PPARγ mKO mice (Figure 6(e) and (f)).
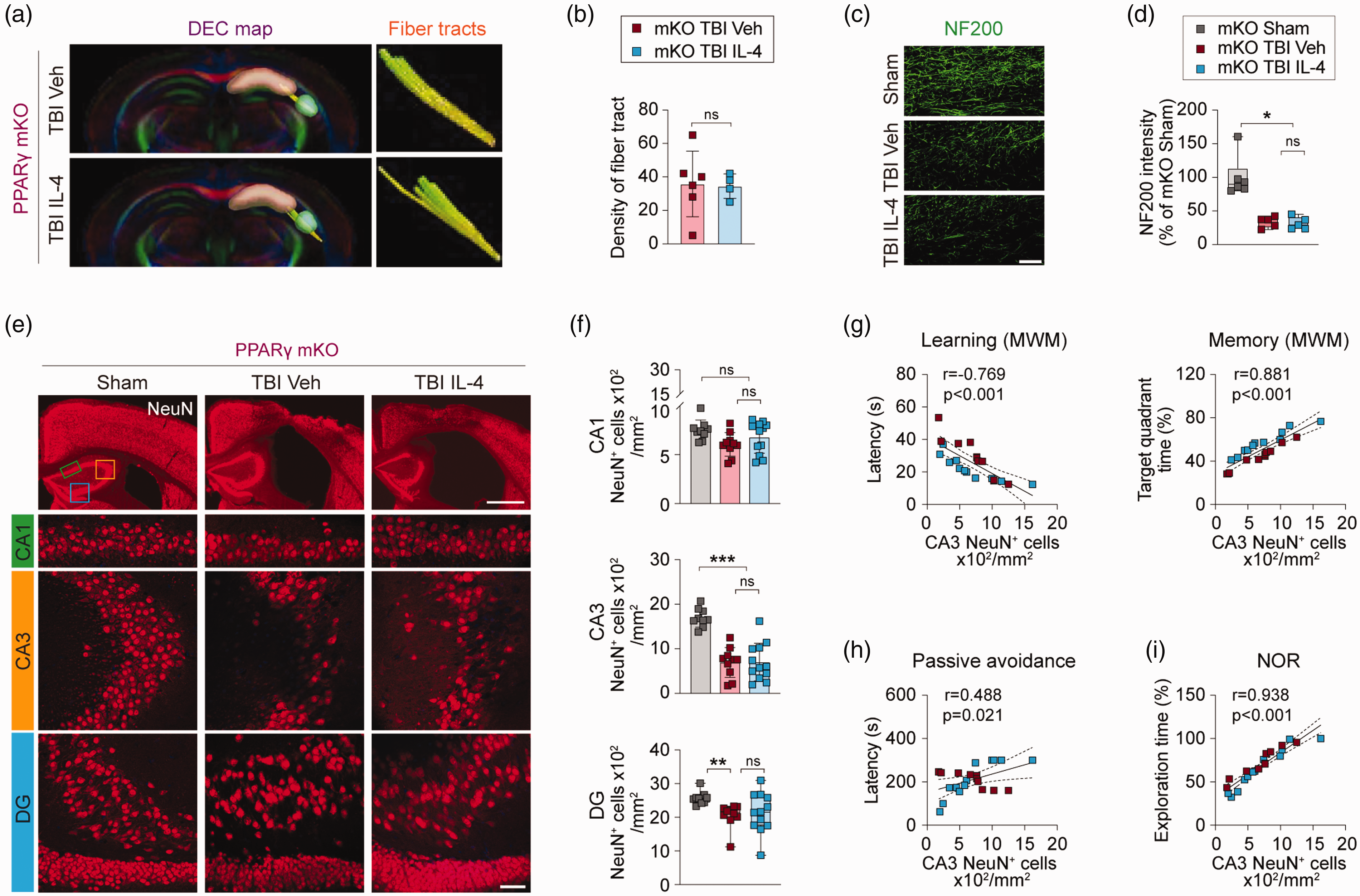
PPARγ mKO abolishes IL-4-afforded hippocampal protection. (a) 3D reconstruction of DTI images showing fiber tracts of Schaffer collaterals between CA1 (pink) and CA3 (green) in the hippocampi of PPARγ mKO mice at 35 days after TBI. (b) Quantification of fiber tract density. n = 6, TBI Veh group; n = 4, TBI IL-4 group. (c) Representative images of NF200 immunofluorescence (green) in hippocampal CA1 of PPARγ mKO mice at 35 days after TBI. Scale bar = 50 µm. (d) Quantification of relative NF200+ immunofluorescent signal intensity in CA1. n = 6, mKO sham group; n = 5 per mKO TBI group. (e) Representative images of NeuN immunofluorescence (red) showing the ROIs in CA1 (green rectangle), CA3 (yellow square), and DG (blue square). Scale bar = 1 mm (upper panel) and 50 µm (lower panel). All images are from PPARγ mKO mice at 35 days after TBI or sham surgery. (f) Quantification of NeuN+ cells in CA1, CA3, and DG of PPARγ mKO mice at 35 days after TBI or sham surgery. n = 9, sham group; n = 10, TBI Veh group; n = 12, TBI IL-4 group. (g–i) Pearson correlation between NeuN+ cell counts in CA3 and learning or memory phase in MWM test (g), latency in passive avoidance test (h), or exploration time in NOR test (i). n = 10, TBI Veh group; n = 12, TBI IL-4 group. Statistical analyses: Student’s t-test for (b). Kruskal-Wallis test with Dunn post hoc test for (d) and (f) (DG). One-way ANOVA followed by Bonferroni post hoc test for (f) (CA1, CA3). Pearson correlation coefficient analyses in (g) to (i). Shown are the mean ± SD or box plots. *p < 0.05, **p < 0.01, ***p < 0.001 as indicated, ns: no significance.
Pearson correlation analyses revealed robust linear correlations between the number of CA3 neurons and behavioral performance in the learning (r = −0.769, p < 0.0001) and memory (r = 0.881, p < 0.001) phases of the MWM test (Figure 6(g)), in the fear memory test (Figure 6(h), r = 0.488, p = 0.021), or in the NOR test (Figure 6(i), r = 0.938, p < 0.001).
These collective data reveal that IL-4 treatment cannot promote the structural integrity of the hippocampus after TBI when PPARγ is conditionally deleted from microglia and that microglia are no longer able to exert a protective effect on neuron viability. Thus, microglia PPARγ is essential for IL-4-stimulated protection against hippocampal degeneration after TBI.
Mi/MΦ-specific PPARγ knockout abolishes IL-4-afforded cognitive enhancement after TBI
Finally, we determined if microglial PPARγ is essential for IL-4-afforded improvement of cognitive functions after TBI. PPARγ mKO mice were subjected to TBI or sham surgery and then received vehicle or IL-4 treatment as before (see Figure 1). Cognitive functions were assessed using MWM, passive avoidance, and NOR tests before and up to 35 days after TBI. Supplemental Figure 8 shows no significant differences in any of the neurobehavioral parameters between vehicle- and IL-4-treatment sham control mice. Therefore, the two groups of sham control mice were pooled for statistical analyses. Following TBI, PPARγ mKO mice exhibited significant impairments in learning (Figure 7(a), p = 0.0077) and memory (Figure 7(b), Platform crossing, p = 0.0001; Target quadrant time, p = 0.0099) phases of the MWM test, deficits in fear memory (Figure 7(d), p = 0.0081), but no decrease in novel object exploration times (Figure 7(e)). IL-4 treatment did not alter cognitive functions in PPARγ mKO mice after TBI, as compared to vehicle-treated PPARγ mKO mice (Figure 7(a) to (e)). In sum, these collective results confirm an essential role of microglial PPARγ in IL-4-stimulated enhancement of spatial and non-spatial cognitive functions after TBI.
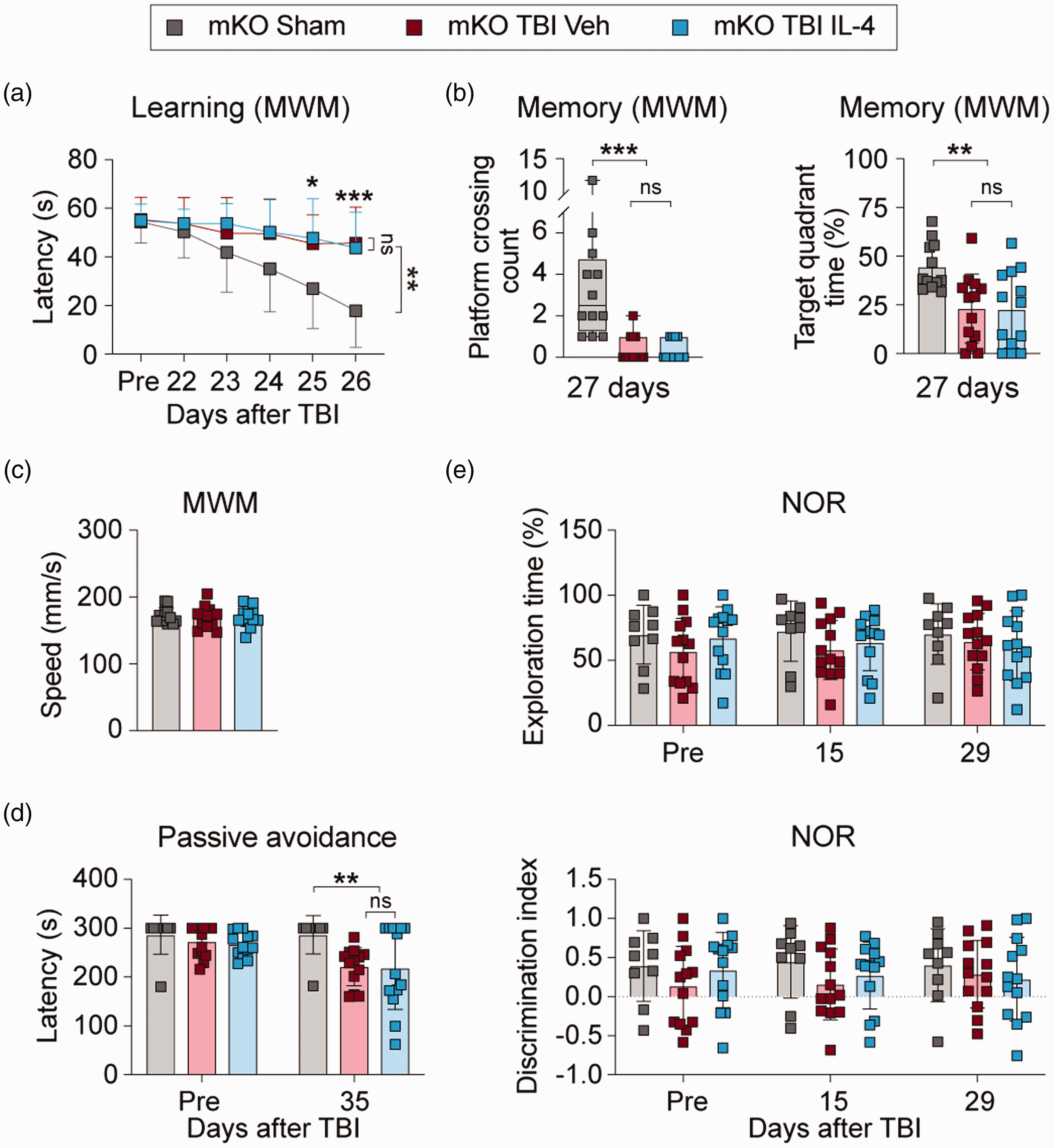
PPARγ is essential for IL-4-afforded improvement of long-term cognitive functions after TBI. (a–c) Morris water maze test. The escape latency during the cued learning test, pre-TBI and 22-26 days after TBI (a), the number of platform crossings and time spent in target quadrant in the memory test on day 27 (b), and swim speed (c) are presented. n = 12, mKO sham group; n = 13 per mKO TBI group. (d) Passive avoidance test. Step-through latency was recorded on the test day. n = 9, mKO sham group; n = 13, mKO TBI Veh group; n = 13, mKO TBI IL-4 group. (e) Novel object recognition test. Novel object exploration time (upper panel) and discrimination index (lower panel) were calculated. n = 12, mKO sham group; n = 13, mKO TBI Veh group; n = 13, mKO TBI IL-4 group. Statistical analyses: Two-way repeated measures ANOVA with Bonferroni post hoc test for (a), (d), and (e). One-way ANOVA followed by Bonferroni post hoc test for (b) (Target quadrant time) and (c). Kruskal-Wallis test with Dunn post hoc test for (b) (platform crossing count). Shown are the mean ± SD or box plots. *p < 0.05, **p < 0.01, ***p < 0.001 as indicated, ns: no significance.
Discussion
The present study contributes three new observations to the TBI and neuroimmunology fields: First, intranasal delivery of IL-4 nanoparticles, initiated as late as 6 hours after TBI, markedly improved hippocampus-dependent cognitive functions, as assessed using the Morris water maze, novel object recognition, and passive avoidance tests. Second, post-TBI IL-4 treatment promoted the functional and structural integrity of the hippocampus, as demonstrated by electrophysiological, DTI, and histological parameters. Third, IL-4 activated microglial PPARγ and Arginase1, thereby apparently shifting global microglial responses to inflammation-resolving and pro-neurorestorative phenotypes, which elicited protection against hippocampal damage.
Cognitive disorders, including impairments in attention, memory, information-processing and executive functioning, are common and long-lasting clinical sequelae of TBI.33–37 In this study, three well characterized neurobehavioral tests were employed to assess hippocampus-dependent cognitive functions in TBI mice and sham surgery control mice. In the Morris water maze test, which examines spatial cognitive functions, TBI mice exhibit severe deficits in both learning and memory test phases, whereas IL-4 treatment significantly attenuated these deficits (Figure 1(b) and (c)). In the novel objection recognition and passive avoidance tests, which evaluate recognition memory38,39 and fear-motivated memory, 40 respectively, IL-4 treatment nearly completely restored neurobehavioral performances in TBI mice (Figure 1(f) and (g)). Our previous studies have similarly found that IL-4 treatment improves spatial cognitive functions of post-stroke mice in the Morris water maze.7,12,41 The results presented here expand those prior findings on IL-4-mediated improvement of spatial cognition to non-spatial cognitive tasks. In sum, the evidence strongly supports the conclusion that IL-4 treatment prevents both spatial and non-spatial functional decline after TBI.
Consistent with the protective effects of IL-4 treatment on hippocampus-dependent cognitive functions after TBI, our data demonstrate that IL-4 treatment promotes the functional and structural integrity of the hippocampus proper in the injured brain (Figures 2 and 3). First, IL-4 treatment partially preserves hippocampal LTP 35d after TBI. LTP experiments were carried out by stimulating presynaptic fibers of Schaffer collaterals at the stratum radiatum and recording responses from a collection of postsynaptic cells of the CA1 pyramidal neuron layer, thereby reflecting the excitatory synaptic strength of the Schaffer collaterals and responsiveness of their neuronal targets. Hippocampal LTP is required for both spatial memory42,43 and fear processing and memory.44,45 Accordingly, the Pearson correlation coefficient analyses demonstrated significant correlations between the fEPSP slope of hippocampal LTP and spatial and fear memory function, as assessed using the Morris water maze and passive avoidance test, respectively. Second, the results of DTI and immunostaining for NeuN and NF200 indicate that, in the CCI model of moderate to severe TBI, the decline in hippocampal LTP is due to loss of CA3 pyramidal neurons and their axons projecting to CA1 (Schaffer collaterals), rather than loss of CA1 or DG neurons per se. Indeed, selective degeneration of hippocampal CA3 neurons is a well-known pathological feature of the CCI model.26,27 As CA3 neurons are crucial for information processing and memory formation,46–48 it is probable that IL-4 treatment prevents TBI-induced behavioral dysfunction at least partly by attenuating CA3 neuronal degeneration and preserving LTP in the Schaffer collaterals.
How does IL-4 prevent CA3 neurodegeneration after TBI? It is unlikely that IL-4 plays a direct role in protecting against neuronal death in CA3, as we failed to find a direct protective effect of IL-4 in primary cultures of cortical and hippocampal neurons against NMDA toxicity or oxygen-glucose deprivation-induced neuronal death (data not shown). Alternatively, IL-4 may achieve neuroprotection and neurorestoration after TBI by modulating the functional states of Mi/MΦ, thereby creating a beneficial microenvironment in the central nervous system that is permissible for neuronal survival, tissue repair, and functional recovery.13,28 This potential mechanism is supported by prior reports that IL-4 is released from injured neurons and polarizes microglia in peri-infarct areas after ischemic stroke. 10 T cell-derived IL-4 enhances cognitive functions by polarizing meningeal myeloid cells and releasing brain-derived neurotrophic factor. 49 In models of hemorrhagic stroke, IL-4 polarizes Mi/MΦ to enhance erythrocyte clearance, inflammation resolution, and tissue protection. 13 In the current study, we found that IL-4 treatment reduces the number of CD16+ pro-inflammatory microglia but increases the number of Arg1+ or Arg1+/PPARγ+ inflammation-resolving and phagocytosis-active (MBP-containing) microglia in the hippocampus 7 days after TBI (Figure 4). Moreover, the protein array data show that IL4 suppressed expression of pro-inflammatory cytokines after TBI (Figure 4). This collective body of work supports the view that IL-4 polarizes microglia toward a phenotype favoring inflammation resolution and tissue repair, thereby contributing to functional restoration after brain injury.
IL-4 binds to its receptor IL-4Rα or IL-4Rα/IL-13Rα and activates signal transducer and activator of transcription 6 (STAT6). 13 Activated STAT6 then drives the additional production of IL-4 and IL-13 in a positive feedback loop. 50 On the other hand, the IL-4/STAT6 axis promotes alternative activation of tissue macrophages into M2 cells, resulting in increased secretion of IL-10 and TGFβ to inhibit inflammation as well as secretion of arginase, proline, and polyaminases, which contribute to tissue repair. 51 Many of the inflammation-resolving and pro-repair actions by IL-4/STAT6 are achieved through interactions with signaling molecules such as CREB-binding protein, 52 NFKB1, 53 and PPARγ. 15 Our study using the Mi/MΦ-specific PPARγ mKO mice confirms the obligatory role for Mi/MΦ PPARγ in mediating the beneficial effects of IL4 on hippocampus-dependent cognitive functions after TBI. The data presented here show that IL-4 can neither polarize Mi/MΦ toward the beneficial phenotype nor promote the functional and structural integrity of post-TBI hippocampi in PPARγ mKO mice. As a master gatekeeper of the response to brain injury, 54 PPARγ activation leads to inhibition of neuroinflammation and oxidative stress,55,56 enhancement of Mi/MΦ phagocytotic activity, 57 and production and release of neurotrophic factors. 58 These collective factors likely contribute to its neurorestorative effects against TBI.
In the current study, intracerebral delivery of IL-4 was achieved through the intranasal route, a drug administration method increasingly used for central nervous system studies. 59 We have shown that a single intranasal delivery of IL-4 nanoparticles elevates IL-4 levels in the cortical parenchyma and subcortical brain regions for 12-24 hours. 12 Intranasal delivery is non-invasive and painless; it allows the drug to avoid first-pass liver metabolism,60–62 and is particularly relevant to prolonged drug administration regimens. For example, in the present study, IL-4 is administrated repeatedly over a 4-week period. In addition, the use of nanoparticles for drug delivery encourages slow drug release, thereby elevating drug concentration in target tissues for a prolonged period of time. 59
There are limitations of this study that warrant future consideration. First, we did not address biological sex as an experimental variable. Sex dimorphisms in microglial reactions, brain metabolism and cerebrovascular responses after brain injuries have been reported.63,64 Additional studies are now warranted to determine if IL-4 is equally effective against cognitive dysfunction after TBI in female animals and if the same signaling pathway underlies IL-4-afforded protection in both sexes. Second, the approach taken in the current study to classify the pro-inflammatory or inflammation-resolving Mi/MΦ after TBI is oversimplified. Based on our recent bulk RNA-seq analysis in microglia and macrophages sorted from injured brains,65,66 we have identified several clusters of genes to define various microglial phenotypes, including proinflammatory and anti-inflammatory cytokines (e.g., Il6, Tnf, Il4, Il10), phagocytosis-related genes (e.g., Gas6, Cd14, Axl, Lamp2), and trophic factors (e.g., Bdnf, Tgfb1, Cntf). Focused expression analyses of the above genes and subsequent validation using flow cytometry combined with immunohistochemistry will help us define the in situ phenotypic changes in Mi/MΦ in IL-4-treated mice vs. vehicle-treated mice after TBI. Third, IL-4 delivery was initiated 6 hours after TBI. Although this time frame may be clinically manageable, future studies should test whether further delay in IL-4 administration can still be effective against TBI-induced cognitive dysfunctions. Given that Mi/MΦ reactions may peak a few days after brain injury, 3 this also raises our hope that approaches targeting Mi/MΦ with IL-4 may offer a considerably wider time window of opportunity for intervention.
In summary, the present study demonstrates that post-injury nose-to-brain delivery of IL-4 nanoparticles improves hippocampus-dependent long-term cognitive functions in a murine model of TBI. Mechanistically, IL-4 suppresses pro-inflammatory responses and enhances debris clearance by microglia by activating Mi/MΦ PPARγ, thereby promoting the structural and functional integrity of the mammalian hippocampus. These results support the continued study of IL-4 as a potential therapeutic for TBI and, perhaps, other brain disorders involving neuroinflammation.
Supplemental Material
sj-pdf-1-jcb-10.1177_0271678X211028680 - Supplemental material for Intranasal delivery of interleukin-4 attenuates chronic cognitive deficits via beneficial microglial responses in experimental traumatic brain injury
Supplemental material, sj-pdf-1-jcb-10.1177_0271678X211028680 for Intranasal delivery of interleukin-4 attenuates chronic cognitive deficits via beneficial microglial responses in experimental traumatic brain injury by Hongjian Pu, Cheng Ma, Yongfang Zhao, Yangfan Wang, Wenting Zhang, Wanying Miao, Fang Yu, Xiaoming Hu, Yejie Shi, Rehana K Leak, T Kevin Hitchens, C Edward Dixon, Michael VL Bennett and Jun Chen in Journal of Cerebral Blood Flow & Metabolism
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Merit Review grant (I01 BX003377) from the US Department of Veterans Affairs. JC is the Richard King Mellon Professor of Neurology at the University of Pittsburgh and also supported by the Senior Research Career Scientist Award from the Department of Veterans Affairs.
Acknowledgements
We thank Lesley M Foley for technical assistance with MRI experiments and Patricia Strickler for administrative support.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest concerning the research, authorship, and/or publication of this article.
Authors’ contributions
HP, CM, and YZ contributed equally to this project. JC conceptualized the study. HP, CM, YZ, and YW performed experiments. JC, XH, and YS designed the study and supervised the project. HP, WZ, WM, and FY analyzed data. HP, CM, YZ, and JC wrote the manuscript. JC, RKL, XH, YS, TKH, CED, and MVLB interpreted the data and critically revised the manuscript.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.