
Abstract
Intracranial hemorrhage (ICH) is a devastating disease which induces high mortality and poor outcomes including severe neurological dysfunctions. ICH pathology is divided into two types: primary brain injury (PBI) and secondary brain injury (SBI). Although there are numerous preclinical studies documenting neuroprotective agents in experimental ICH models, no effective drugs have been developed for clinical use due to complicated ICH pathology. Oxidative and inflammatory stresses play central roles in the onset and progression of brain injury after ICH, especially SBI. Nrf2 is a crucial transcription factor in the anti-oxidative stress defense system. Under normal conditions, Nrf2 is tightly regulated by the Keap1. Under ICH pathological conditions, such as overproduction of reactive oxygen species (ROS), Nrf2 is translocated into the nucleus where it up-regulates the expression of several anti-oxidative phase II enzymes such as heme oxygenase-1 (HO-1). Recently, many reports have suggested the therapeutic potential of Nrf2 activators (including natural or synthesized compounds) for treating neurodegenerative diseases. Moreover, several Nrf2 activators attenuate ischemic stroke-induced brain injury in several animal models. This review summarizes the efficacy of several Nrf2 activators in ICH animal models. In the future, Nrf2 activators might be approved for the treatment of ICH patients.
Introduction
Intracranial hemorrhage (ICH) is a devastating disease accounting for 10 to 20% of all stroke cases, which is mostly induced by hypertension. 1 ICH is associated with high mortality and neurological dysfunctions; 50% of patients will be dead in approximately one month, while the others will exhibit severe neurological disabilities including paralysis, aphasia, motor dysfunction and/or cognitive dysfunction.2–4 Although numerous preclinical studies were conducted to investigate the efficacy of several neuroprotective agents in experimental ICH animal models, no therapeutic agents are currently approved for the treatment of ICH. 5 , 6 Therefore, effective pharmaceutical therapy for patients with ICH has long been desired for over two decades.
In ICH pathology, both oxidative and inflammatory stress damage brain tissue including neurons and vascular components, which contributes to brain injury and poor outcomes. 4 ,7–9 Humans and other mammals possess the anti-oxidative stress defense system that protects against oxidative stress. In particular, the transcription factor nuclear factor-erythroid 2 related factor 2 (Nrf2) is an important regulator of that defense system. 10 , 11 The Nrf2 signaling pathway plays crucial roles in attenuating brain injury in experimental ICH models via its strong anti-oxidative and anti-inflammatory effects. 12 Therefore, it is anticipated that activation of Nrf2 may be a novel approach for treatment of patients with ICH. There are many reports which demonstrate the efficacy of Nrf2 and its downstream factors for treating other types of strokes such as ischemic stroke and subarachnoid hemorrhage (SAH). However, only a few review reports have focused on the efficacy of Nrf2 and its downstream factors for treating ICH. Thus, the aims of this review are (1) to summarize the mechanisms of ICH pathology specifically focusing on oxidative and inflammatory stresses and (2) review the efficacy of several Nrf2 activators for treating ICH in experimental ICH models.
Pathology of ICH
ICH is induced by cerebral vascular rupture and patients usually have comorbid physiological problems such as chronic hypertension, ageing and cerebral amyloid angiopathy. 1 , 4 Brain microvascular injury induces extravasation of blood components with subsequent hematoma formation. 13 After ICH onset, hematoma-induced brain damage occurs, and it is mainly divided into two types: (1) primary brain injury (PBI) and (2) secondary brain injury (SBI) (Figure 1).
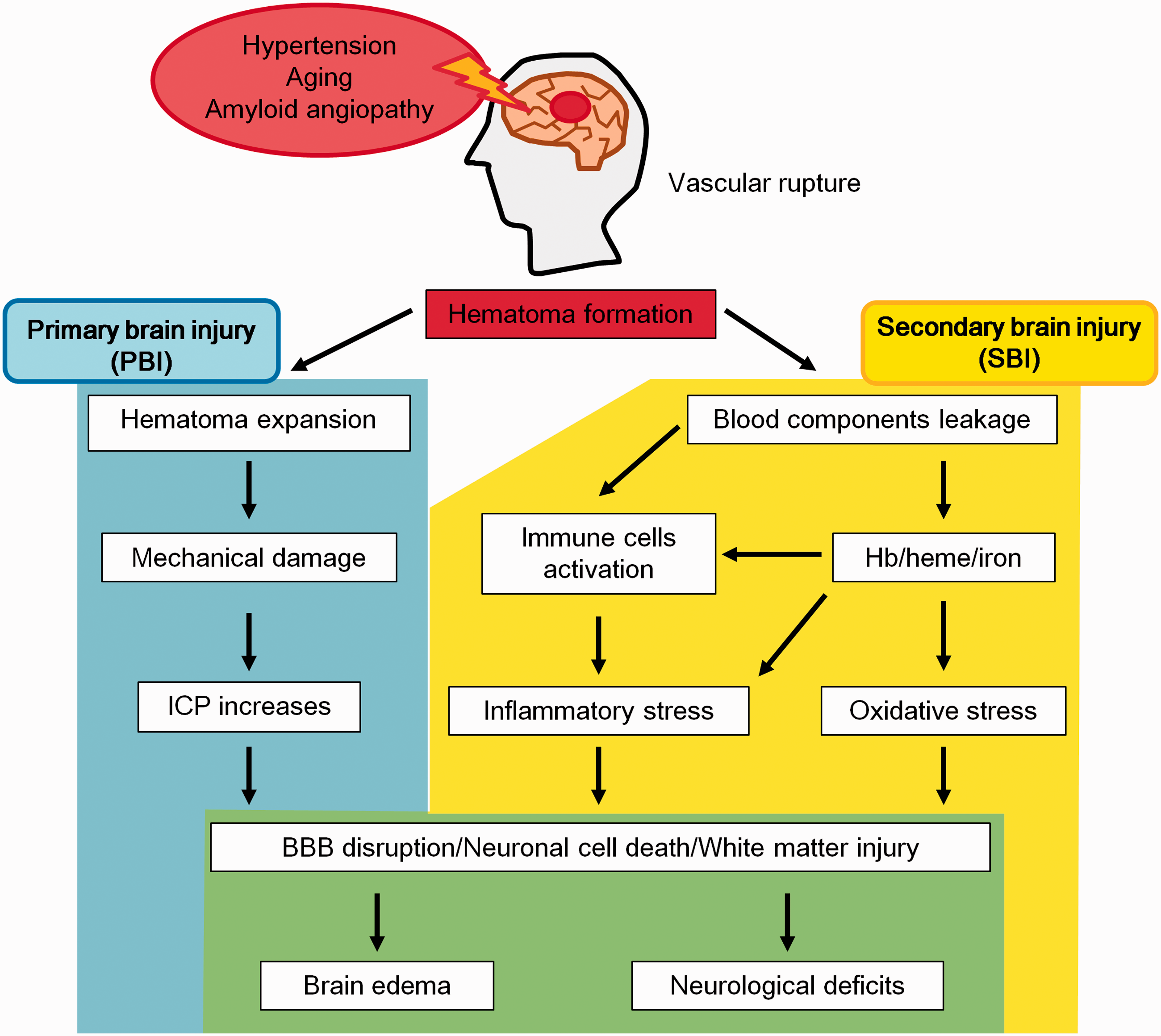
The pathology of ICH. Several diseases such as chronic hypertension and amyloid angiopathy induce vascular fragility that leads to the rupture of blood vessels. After ICH, a hematoma is formed in brain parenchymal tissue, which induces brain injury such as primary brain injury (PBI; blue area) and secondary brain injury (SBI; yellow area). These injuries ultimately lead to devastating functional brain impairments (green area).
PBI
In the early phase after ICH onset, the hematoma gradually enlarges in about 20% patients and intracranial pressure (ICP) rapidly increases due to the hematoma mass effect. 2 , 4 , 14 Increasing ICP by the hematoma induces mechanical damage to the normal brain parenchyma tissues and is referred to as PBI,. 14 In general, high ICP more than 20 mmHg induces high mortality and severe disability in ICH patients. 15 PBI mainly occurs within a few to 24 hours after ICH and progression of PBI induces brain herniation and oedema, which leads to neuronal cell death and neuronal network disruption. 2 In addition, using a gel transplantation animal model, gel-induced mass effect directly induced neuronal apoptosis without blood components. 16 , 17 Moreover, the intracranial hematoma volume strongly contributes to patients mortality and neurological disabilities. 18 Therefore, the progression of mechanical stress induces tissue dissociation and subsequently neuronal cell death, which triggers severe neurological deficits. In clinical, brain oedema could be prevented by management of blood pressure, use of osmotic diuretics and surgical intervention. 5 Currently, the use of novel surgical techniques and anti-hypertensive drugs has drastically improved PBI treatment.
SBI
Compared with PBI, a few days to weeks after ICH, various detrimental reactions contribute to SBI.
Toxicity of Hb and its metabolites
Along with hematoma formation, red blood cells (RBCs) are released from the hematoma into surrounding normal brain tissues, and they are subsequently disassembled to hemoglobin (Hb). 19 Hb is a tetrameric protein composed of alpha and beta subunit chains each containing a heme group bound to an iron atom. Hb supplies oxygen to the whole body by transporting oxygen from the lungs to the rest of the body via the circulation. 20 When extracellular Hb releases heme, which is captured by phagocyte cells such as macrophages and microglia, the heme is subsequently metabolized to iron, biliverdin and carbon monoxide by intracellular heme oxygenase (HO). 19 , 21 Hb metabolites such as heme and iron have direct cytotoxic effects on neurovascular units (NVU) including the blood-brain barrier (BBB) and neurons.22–24 In addition, treatment with Hb or its metabolites induced apoptosis via increasing reactive oxygen species (ROS) production. 23 , 25
Using ICH animal models, autologous blood injection into brain parenchyma tissue induced drastic brain damage. 19 This brain damage occurred via detrimental inflammatory reactions, BBB disruption, neuronal cell death and brain oedema, which ultimately led to neurological dysfunctions. 9 , 19 Moreover, hemin, the heme-related molecule containing the trivalent iron (Fe3+), also induced oxidative and inflammatory stresses via activating the toll like receptor (TLR) 2 or TLR4 as well as stimulating intracellular iron accumulation. 23 , 26 , 27
In ICH animal models, the intracellular iron regulation system might be disrupted. In support of this hypothesis, after ICH, iron accumulation was observed surrounding the hematoma tissue and phagocyte cells uptakes these irons. Moreover, iron-regulated proteins such as transferrin (an iron transfer protein) and ferritin (an iron storage protein) were significantly increased at 3 days after ICH. 21 , 23 , 28 , 29 Iron accumulation in the brain was maintained until 28 days after ICH. 28 In addition, an increase in ferritin in the sera of ICH patients was positively correlated to outcome severity. 30 The detrimental effects of iron mainly contribute to oxidative stress via the Fenton reaction in which a bivalent iron (Fe2+) promotes ROS production from hydrogen peroxide (H2O2) (Fe2+ + H2O2 → Fe3+-OH + •OH). 21 , 28 , 31
Recent studies have shown that iron derived from Hb/heme induced ferroptosis in experimental ICH models. 24 , 32 , 33 Ferroptosis is iron-dependent cell death and has different features compared to typical cell death such as apoptosis. 33 Ferroptosis significantly increases ROS production, lipid peroxidation and activates both the mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) pathway and the receptor-interacting serine/threonine-protein kinase (RIPK) pathway. 24 , 33 , 34 These pathways are not affected by anti-apoptosis reagents (e.g., caspase inhibitors) but are suppressed by iron chelators (e.g., deferoxamine; DFO), a necrosis inhibitor (e.g., necrostatin) and anti-oxidants (e.g., N-acetyl-L-cysteine). 24 , 33 , 34 Moreover, ferroptosis induced mitochondrial morphological changes in neuronal cells after ICH. 35 In the acute and sub-acute phases after ICH, the neuronal mitochondria were enlarged and swollen, which were maintained until 28 days after ICH. 35 Inhibition of ferroptosis by ferrostatin-1 administration attenuated neuronal cell loss, iron accumulation, lipid ROS accumulation and neurological deficits after ICH in both in vivo and in vitro models. 34 Therefore, the suppression of oxidative stress via inhibiting both iron accumulation and the subsequent increase in ROS production may be an effective therapy for ICH.
Based on these findings, a clinical trials were conducted in order to investigate whether DFO, an Fe3+ chelator, improved the outcomes in ICH patients. 36 DFO has neuroprotective effects via suppression of iron accumulation and ROS over-production. 37 Selim et al. assessed the safety and efficacy of DFO in the phase-II trial. In this trial, the efficacy of DFO on outcomes was evaluated by modified Rankin Scale at 90 and 180 days after ICH. As results, DFO (32 mg/kg/day) infusion administration for 3 consecutive days was safe and slightly good outcomes at 180 days after ICH. Unfortunately, there was no significant difference of patient neurological outcome between DFO and placebo group. 36 There are still challenges such as sample size, criteria and timing of endpoints, this result indicated that the neuroprotective effects via suppression of iron accumulation may be weakly to improve the neurological outcomes in ICH patients.
Inflammation
In addition to oxidative stress, hematoma formation also induces an inflammatory reaction via activation of macrophages/microglia, which subsequently leads to the induction of the immune cascade and secretion of pro-inflammatory cytokines. 9 , 19 , 21 , 38 These inflammatory reactions are initiated within 1 day after ICH and gradually progress. 39 The ionized calcium-binding adapter molecule 1 (Iba1; a macrophage/microglial cell marker) and glial fibrillary acidic protein (GFAP; an astrocyte marker) were accumulated around the hematoma in ICH patients and in experimental ICH animal models. 40 , 41 Observation of the glial cell dynamics in an animal ICH model revealed that macrophages/microglia and astrocytes were accumulated in the hematoma territory at 1 to 14 days after ICH. 40
Polarization of glial cells to the pro-inflammatory phase such as A1 astrocytes and M1 microglia contributes to neuroinflammation and ICH-related brain injury. 38 , 39 In cellular experiments, Hb or hemin treatment directly activated these glial cells and induced morphological changes (consistent with the inflammatory M1 subtypes) via activation of the TLR family, especially TLR4. 26 , 27 , 42 The activation of TLRs induced the expression of nuclear factor kappa B (NF-κB), a representative transcription factor that transcriptionally transactivates several pro-inflammatory molecules. 43 Liu et al. utilized a rat model system that induced ICH via autologous blood injection. Compared to the sham-treated group, the ICH group induced NF-κB expression in brain tissue at 3 hours after ICH. The NF-κB levels then peaked at 3 days and NF-κB was still present at 5 days. 44 The induction of NF-κB subsequently up-regulated the expression of several genes, including those from the matrix metalloproteinase (MMP) family, NOD-like receptor family pyrin domain containing 3 (NLRP3) inflammasome and several pro-inflammatory cytokines such as interleukin 1 beta (IL-1β) and tumor necrosis factor-alpha (TNF-⍺). Expression of these genes ultimately leads to neuroinflammation and BBB hyper-permeability. 39 , 45 In the chronic phase of ICH, iron accumulation and immune-derived inflammatory stress also injured white matter tracts and induced severe neurological dysfunctions; specifically motor and memory functions. 46
In parallel to the inflammatory response, macrophages/microglia and astrocytes also contribute to the tissue repair system. 39 The activation of macrophages/microglia promote phagocytosis and clean up both the hematoma and cellular debris, which decreases the hematoma size. 39 , 40 , 47 In addition, the M2 polarized microglia accelerates tissue repair via secreting several cytokines such as IL-4, IL-10 and transforming growth factor-β (TGF-β). 39 The expression of these cytokines was rapidly increased as early as 1 to 1.5 hours after ICH and temporarily declined at 6 hours, then increased again at 3 or 7 days. 40 M2 polarization up-regulates regulatory helper T cells and then ameliorates ICH-induced brain damage through an anti-inflammatory reaction. 39 , 40 Thus, the inflammatory reaction in ICH pathology is a highly complex mechanism because of its dual roles.
In summary, hematoma-induced SBI progresses with gradual aggravation of various insults including brain oedema and neuronal cell death. 9 Since ICH pathology involves numerous diverse pathways and cell types, no effective drugs have been developed to treat ICH. Therefore, agents which strongly suppress both oxidative and inflammatory stresses may be effective for ICH treatment.
The role of the Nrf2 anti-oxidant defense system
Regulation of Nrf2-Keap1 signaling
Nrf2 has a basic leucine zipper region which belongs to the Cap’n’Collar transcription factor group, and is activated by ROS and/or electrophilic substances. 48 , 49 Nrf2 is the central transcription factor in the biological anti-oxidative defense system. Under unstressed conditions, Nrf2 binds to kelch-like ECH-associated protein 1 (Keap1), and forms a complex in the cytoplasm. 48 , 50 Keap1 functions as the redox sensor and regulator of Nrf2 signaling. The Nrf2-Keap1 complex is rapidly ubiquitinated and degraded by the proteasome. 50 Therefore, Nrf2 is inactivated by Keap1. Under unstressed conditions, Nrf2 expression is tightly regulated and has a very low expression level.
On the other hand, during oxidative stress or inflammatory conditions, the Keap1 cysteine residues become oxidized in response to ROS or electrophilic substances, and Keap1 undergoes a conformational change. 50 , 51 This conformational change disrupts the Nrf2-Keap1complex and Nrf2 is translocated into the nucleus. 48 , 51 Nrf2 is then free to bind to the anti-oxidant response element (ARE) located in the DNA of several anti-oxidant enzymes. 52 The interaction of Nrf2 with the ARE promotes transcription of several anti-oxidant enzymes including heme oxygenase-1 (HO-1), superoxide dismutase (SOD), nicotinamide adenine dinucleotide phosphate [NAD(P)H], quinone oxidoreductase-1 (NQO1), catalase, glutathione S-transferase (GST) and thioredoxin reductase 52 (Figure 2). Therefore, agents that disrupt the formation of the Nrf2-Keap1 complex contribute to anti-oxidative effects. The Nrf2 signaling pathway also attenuates inflammatory stress via suppressing NF-κB transcription. 10 , 11
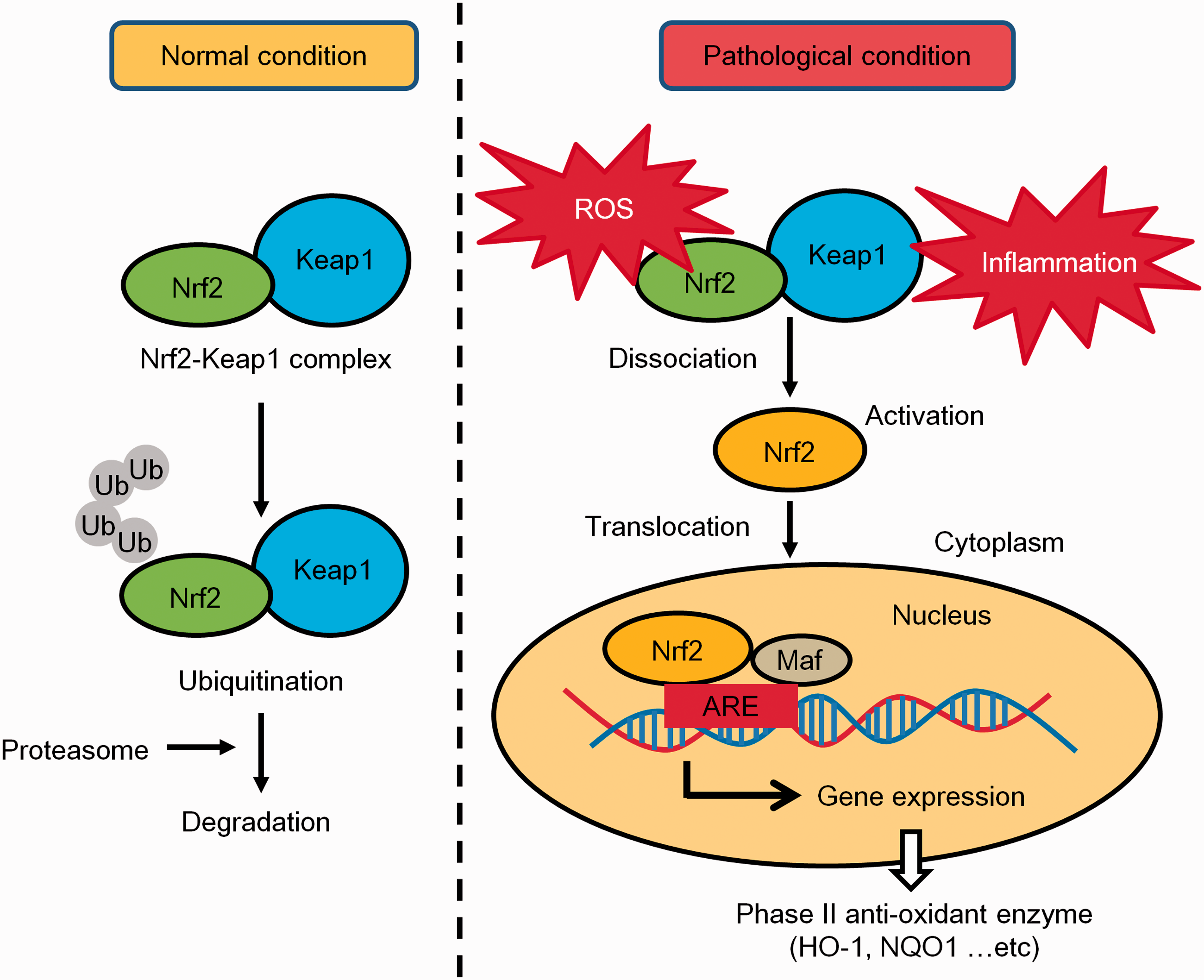
The regulation of the Nrf2 anti-oxidative stress defense system. Under normal conditions, Nrf2 binds to Keap1 and forms a complex, which is rapidly ubiquitinated and degraded by the proteasome. Under pathological conditions (e.g., exposure to oxidative stress and/or inflammation), Nrf2 is dissociated from Keap1 and is translocated into the nucleus. Subsequently, Nrf2 binds to anti-oxidant response elements (AREs) in phase II enzyme genes and activates their transcription. These phase II enzymes such as HO-1 and NQO1 mitigate the cellular stress-induced damage.
In the brain, Nrf2 is ubiquitously expressed in several types of cells in order to counteract both oxidative and inflammatory stresses. 10 , 53 In a mouse model of ischemic stroke, glial cells (astrocytes and microglia) and neurons robustly expressed Nrf2 at the peri-infarct area within 24 hours after transient middle cerebral occlusion (MCAO) surgery. 54 Moreover, activation of the Nrf2 signaling pathway had beneficial roles in an experimental SAH model. 55 Therefore, there is overwhelming evidence that Nrf2 plays crucial roles in protecting the brain against various types of strokes.
The contributions of Nrf2 and its downstream enzymes on ICH pathology
The crucial roles of Nrf2 in ICH pathology
Similar to the protective effects of Nrf2 against ischemic stroke, numerous preclinical studies suggested the therapeutic potential of Nrf2 signaling in experimental ICH animal models (Figure 3). In 2007, Wang et al. firstly reported that at 24 hours after collagenase-induced ICH, Nrf2 knockout (Nrf2-/-) mice had significantly larger hematomas and more severe neurological deficits compared to wild type (WT) mice 12 (Figure 3(a-c)). In addition, Nrf2-/- mice had more severe cellular damage including increased neuronal cell death, ROS production, DNA damage and cytochrome c release. 12 Zhao et al. also investigated the effects of Nrf2 on ICH pathology using Nrf2-/- mice. 56 At 7 days after ICH, Nrf2-/- mice exhibited severe neurological deficits compared to WT mice. 56 Furthermore, the Nrf2 activator sulforaphane (SFN) suppressed these ICH-induced neurological deficits in WT mice but its protective effects were not observed in Nrf2-/- mice. 56 These reports demonstrated that Nrf2 deficiency blocked the anti-oxidant defense system and increased the vulnerability to oxidative stress induced by ICH.
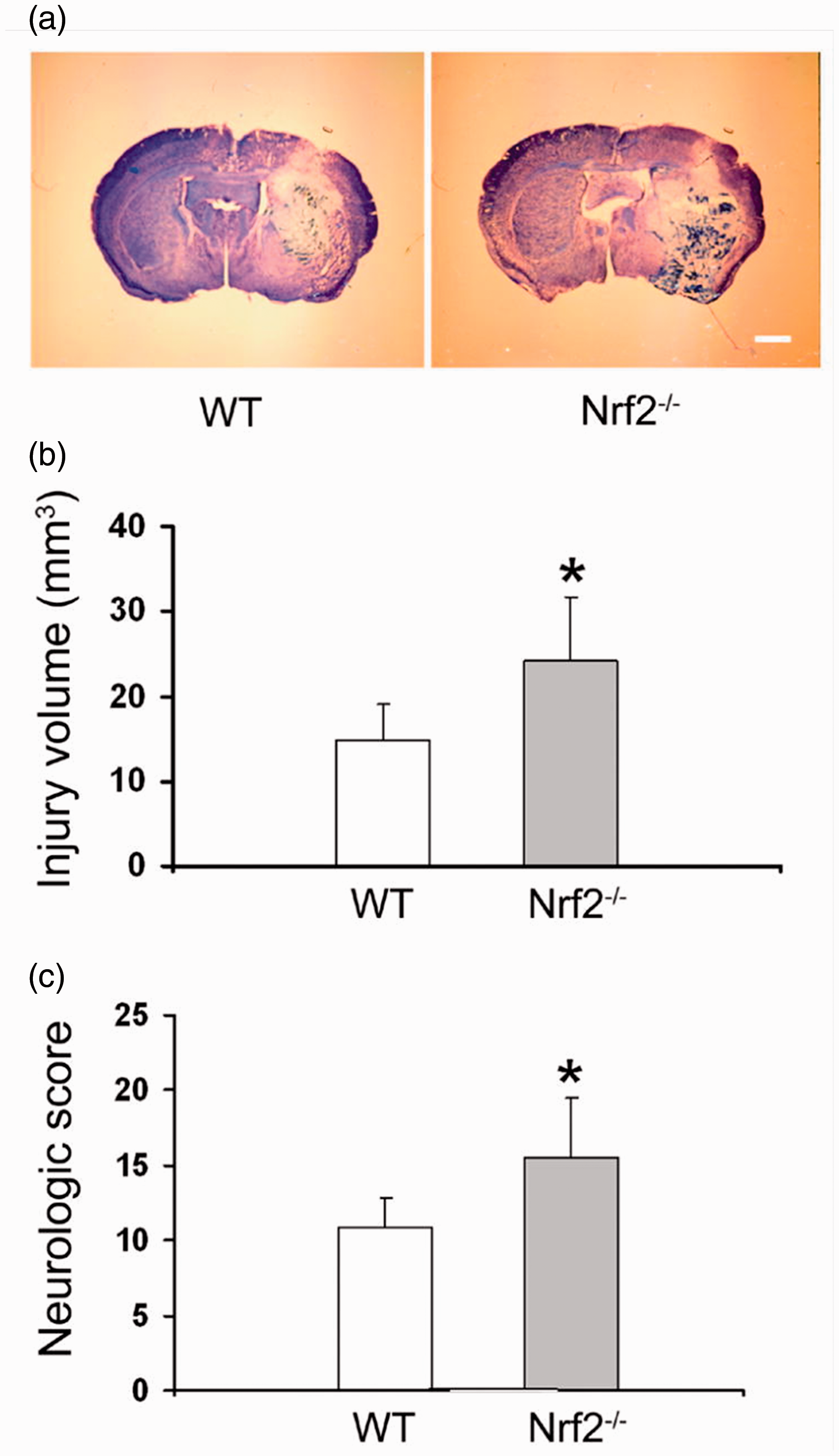
The essential role of Nrf2 in ICH pathology. (a) Images of representative ICH brain slices stained with luxol fast blue/cresyl violet. (b) After ICH, Nrf2-deficient (Nrf2-/-) mice exhibited a large injury volume compared to wild type (WT) mice. (c) Nrf2-/- mice exhibited severe neurological deficits compared to WT mice. The data is cited from a previous report (Jian Wang, Free Radic Biol Med. 2007;43(3):408-14), this citation is permitted by the journal.
In ICH, extravasated blood has detrimental effects through its metabolic process. 19 , 21 RBC lysates, which contain Hb/heme and iron, induce deadly oxidative stress in normal tissue near the hematoma. 7 , 21 Nrf2 is rapidly activated in response to these detrimental substances.
Shang et al. investigated the dynamics of Nrf2 signaling after ICH. 57 The mRNA expression level of Nrf2 and its downstream anti-oxidant enzymes such as HO-1, glutathione and thioredoxin (TRX) were increased at 2 hours after ICH and remained high up until 10 days. 57 Nrf2 protein expression was significantly increased at an early time point (2 hours) after ICH while the expression level of Keap1 gradually decreased beginning at 8 hours after ICH. 57 These results suggested that Nrf2 rapidly response to ICH-induced damage in order to protect normal tissues.
The contributions of HO on ICH pathology
The Nrf2 detoxification mechanism is mediated by an increase in the expression of phase II anti-oxidant enzymes. HO is a representative phase II anti-oxidant enzyme in the Nrf2 signaling pathway, which plays pivotal roles in heme degradation and in the anti-oxidant defense system. 21 , 22 This enzyme has two isoforms: HO-1 is rapidly induced by cellular stress and functions in the cellular stress response; HO-2 is constitutively expressed and maintains homeostasis. 21 , 22 In blood injection ICH rodent models, the HO-1 expression level was drastically increased at 3 to 5 days, and then declined. 57 , 58 The time-course HO-1 expression profile was similar to the progression of brain oedema. 57 In experimental ICH models, brain oedema peaked at 3 days after surgery-induced ICH because heme and its metabolites released from the hematoma damaged normal tissue near the hematoma. 19 After exposure to Hb or heme, HO-1 but not HO-2 was rapidly increased in various type-cells including neurons, endothelial cells and glial cells. 21 In both collagenase and blood-induced ICH models, an increase in HO-1 expression in astrocytes reduced hematoma size and attenuated brain damage. 59 , 60 In addition, systematic treatment with hemin, which induced HO-1, also attenuated BBB disruption in both ICH models. 61 Therefore, HO-1 is induced in response to heme and strongly contributes to brain protection after ICH.
The up-regulation of HO-1 increases the phagocytic capacity, and results in the production of anti-oxidants such as carbon monoxide (CO) and biliverdin through heme degradation. 21 , 22 A high concentration of CO is toxic, but inhalation of CO at low concentrations promoted hematoma clearance by the activation of microglia and suppressed vasospasm by affecting brain vascular smooth muscle cells, which attenuated neuronal death and brain damage after SAH. 62 , 63 In addition, CO-releasing molecules have directly protective effects on neurons and endothelial cells against excitotoxicity and oxidative stress via promoting anti-inflammatory and anti-apoptosis pathways. 64 , 65 However, Chen and Regan demonstrated that there was a discrepancy between the increases of HO-1 expression and CO production rate (HO activity) in ICH model. 58 Therefore, the contribution of induced HO-1 to CO synthesis via heme breakdown may be relatively minor in ICH condition. Biliverdin is quickly converted to bilirubin by biliverdin reductase and its reductase activity contributes to its anti-oxidant capability, which has protective effects on cerebral vasospasm after SAH. 66 Thus, HO-1 and its enzyme byproducts have protective effects against hemorrhagic stroke.
Even though there are many reports showing that HO-1 has protective effects against ICH-induced brain injury, Wang and Doré reported paradoxical results that HO-1 exacerbated early brain injury after ICH. 67 In HO-1 knockout (HO-1-/-) mice, the injury volume was smaller and both leukocyte infiltration and microglia activation were remarkably reduced compared to WT mice. 67 In addition, Wang et al. reported that in an iron injection model of ICH, HO-1-/- mice attenuated striatum damage compared to WT mice. 68 HOs catalyzes heme degradation and produces heme-derived iron, which is highly cytotoxic because it can induce oxidative stress. 21 , 22 , 28 Actually, Chang et al. found few cells with accumulated iron in HO-1 deficient mice after ICH. 32 Similar to HO-1 conflicting effects, HO-2 also has such effects. Knockout of HO-2 (HO-2-/-) aggravated brain injury following Hb-induced ICH, 69 which indicates that endogenous HO-2 may be important in brain protection after ICH. Conversely, HO-2 depletion attenuated brain damage via suppression of oxidative stress in Hb or blood injection ICH rodent models. 70 , 71 These results suggested that HOs activity may not only play protective roles via producing CO and biliverdin but also partially detrimental roles in ICH pathology via reinforcing iron toxicity.
Studies with HO inhibitors revealed the complex positive and negative effects of HO enzymes on ICH pathology. For example, zinc protoporphyrin (ZnPP), an HO inhibitor, attenuated white matter injury and improved neurological function after ICH. 72 , 73 Interestingly, contrary to these results, Zhao et al. demonstrated that in a autologous blood injection-induced ICH model, the inhibition of HO-1 by ZnPP treatment aggravated the neurological deficits and increased the number of apoptotic cells. 74 In this study, HO-1 protected the CNS by regulating phosphoinositide 3-kinase (PI3K)/Akt signaling pathway. 74 A recent study revealed an even greater complexity in the effects of HO-1 on ICH pathology. Zhang et al. found that after ICH, HO-1 activation induced brain damage in the early phase, but it promoted neurological function recovery in the later stage. 75 Specifically, in the early stage after ICH beginning at 1 day and lasting until 3 days, HO-1 induction by cobalt protoporphyrin IX (CoPP) induced severe brain damage including a large injury volume, brain oedema. In contrast, ZnPP suppressed these detrimental events. 75 However, CoPP treatment promoted hematoma clearance and angiogenesis in the later stage at 7 to 28 days after ICH. 75 Taken together, HO-1 has both detrimental and protective effects on ICH pathology. Regarding these HOs difference effects, the intracellular iron tolerated capacity may also be contribution. Glial cells can store more amount of iron than neurons. 76 Neurons may be more vulnerable to iron exposure compared to glial cells which play a role in clean-up the accumulated iron after ICH. Therefore, dramatically increases of HOs in neurons induce the intracellular iron overload and cause the detrimental effects.
Thus, even though there are many reports that HO-1 induction may be an effective approach for ICH therapy, the protective effects of HOs on ICH remains controversial.
Other Nrf2 downstream enzymes (catalase, SOD etc.) contribute to ICH pathology
After ICH, additional Nrf2 downstream enzymes also exerted protective effects on the brain. After ICH, the expression level of catalase was rapidly induced at 1 hour and was maintained for 24 hours, which protected neurons against oxidative stress. 77 In addition, after ICH, the up-regulation of catalase expression in macrophages promoted phagocytic capacity that facilitated hematoma clearance and improved neurological deficits. 78 The expression levels of SOD and NQO1 were significantly higher at 24 to 72 hours after ICH. 79 , 80 These increases were concomitant with nuclear Nrf2 expression and were regulated by miR-27b. 80 In addition, the ICH severity in SOD1 knockout (SOD1-/-) mice was aggravated via increased MMP-9 activity. 81 In ICH patient serum, an increase in TRX correlated with hemorrhagic severity and long-term mortality. 82 Therefore, after ICH, Nrf2 and its downstream enzymes are up-regulated to counteract brain injury.
Therapeutic potential of Nrf2 activators for the treatment of ICH
Many Nrf2 activators bind to the cysteine residues in Keap1 and induce a conformational change in the Nrf2-Keap1 complex. This conformational change induces the dissociation of the Nrf2-Keap1 complex (similar to what occurs under oxidative or electrophilic stress) and Nrf2 is subsequently translocated from the cytoplasm into the nucleus. Here, we summarize reports from the last 20 years describing the effects of several Nrf2 activators on in vivo experimental ICH models (Table 1).
The efficacy of Nrf2 activators in experimental ICH models.
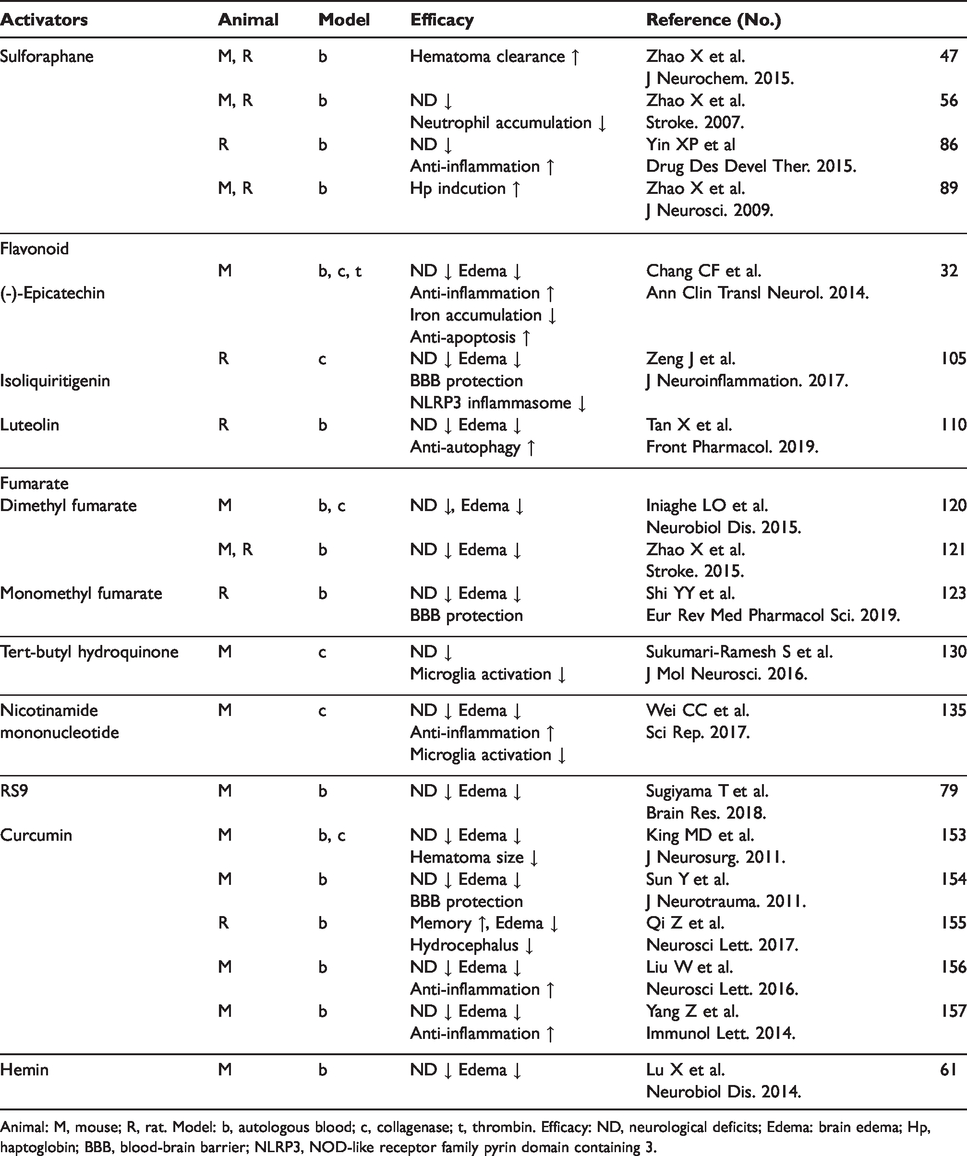
Animal: M, mouse; R, rat. Model: b, autologous blood; c, collagenase; t, thrombin. Efficacy: ND, neurological deficits; Edema: brain edema; Hp, haptoglobin; BBB, blood-brain barrier; NLRP3, NOD-like receptor family pyrin domain containing 3.
Sulforaphane (SFN)
SFN is an isothiocyanate that contains sulphur and is widely used as an Nrf2 activator. 49 , 83 Due to the Nrf2 activation effects, SFN alleviated infarct formation and cognitive impairment in ischemic stroke animal models. 84 , 85 In SAH animal models, SFN administration suppressed brain oedema and apoptosis via the up-regulation of Nrf2 downstream enzymes such as HO-1 and NQO1. 55 Zhao et al. showed that in a rat ICH model, intraperitoneal (i.p.) administration of SFN (5 mg/kg) after ICH induction suppressed neurological deficits at 10 days after ICH. 56 SFN activated Nrf2 and its downstream enzymes (e.g., catalase, SOD, GST and NQO1), which reduced neutrophil accumulation and oxidative damage near the hematoma region. 56 Yin et al. also demonstrated that SFN administration (1 mg/kg, twice daily, i.p.) after ICH induction promoted the gradual recovery of neurological deficits and suppressed pro-inflammatory factors after ICH. 86 In addition, administration of SFN not only increased the expression of both Nrf2 and HO-1, but also decreased the expression of both NF-κB and TNF-⍺ at 1 to 7 days after ICH. 86 Furthermore, SFN treatment promoted the phagocytosis of RBCs by cultured microglia/macrophages via activation of the Nrf2 pathway, which contributes to eliminate hematoma after ICH. 47 Taken together, these data indicated that Nrf2 activation by SFN not only improved neuronal function, but also promoted hematoma clearance through the up-regulation of anti-oxidant and anti-inflammatory pathways after ICH.
SFN treatment may also contribute to hematoma clearance via the up-regulation of haptoglobin (Hp). Hp is a plasma protein which is produced in the liver and subsequently secreted into circulating blood, where it functions as a Hb detoxification factor. 87 In the acute phase after ICH, Hp is recruited from the peripheral circulation and binds to Hb in order to eliminate the brain hematoma. 19 , 88 In an ICH rodent model, the lack of Hp aggravated neurological deficits while overexpression of Hp alleviated brain damage. 89 Injection of rats with SFN (5 mg/kg, i.p.) after ICH induction increased Hp expression at 3 hours to 7 days after ICH in the striatum. Moreover, SFN treatment also increased Hp expression in cultured oligodendrocytes in a dose-dependent manner. 89 Another report also demonstrated that SFN treatment increased Hp gene expression via activating Nrf2. 90 Taken together, these results indicated that SFN induces the expression of Hp, which plays a significant role in protecting the brain from ICH-induced injury.
Flavonoids: (-)-epicatechin (EPC), isoliquiritigenin (ILG) and luteolin
Flavonoids belong to a family of polyphenols which are present in fruits and vegetables, and their characteristic structure is two aromatic rings linked via a 3-carbon chain. 91 , 92 Polyphenols including flavonoids can induce Nrf2 activation via binding to the cysteine residues of Keap1. 93 After then, Keap1 is alkylated and Nrf2 is translocated into the nucleus. 94 Considering to the robust anti-oxidative effects of flavonoids, the long-term dietary flavonoids and flavonoid-rich foods intake may be associated with better cognitive function in dementia. 93 , 95
EPC, a member of the flavan-3-ols (catechins) family, is a brain-permeable flavanol that is abundant in cocoa and green tea. 96 , 97 EPC administration has protective effect on an ischemic stroke model via activation of the Nrf2 signaling pathway. 98 Two previous reports clarified that EPC treatment after ICH induction attenuated Hb-induced cellular toxicity and brain damage. 32 , 99 Chang et al. showed that EPC administration [15 mg/kg, intragastric (p.o.)] suppressed injury volume and neurological deficits at 72 hours after ICH in a collagenase-induced ICH model. 32 The efficacy of EPC administration was also observed in both autologous blood- and thrombin-induced ICH models. 32 The protective effects of EPC are mediated by various mechanisms including anti-oxidative, anti-apoptosis and anti-inflammatory pathways as well as mechanisms that suppress high-mobility group protein B1 (HMGB1) and MMPs. 32 Interestingly, the efficacy of EPC on ICH was not completely abolished in Nrf2-deficient mice. 32 Therefore, EPC might also induce Nrf2-independent effects on ICH. For example, EPC suppressed lipid peroxidation and iron accumulation via increasing lipocalin-2 which plays a role in brain iron clearance. 32 These results indicated that EPC might have anti-ferroptosis activity. EPC also suppressed the induction of activator protein-1 (AP-1) at 48 and 72 hours after ICH. 32 AP-1 is a stress-activated transcription factor that regulates inflammation, apoptosis and oxidative stress via the c-Jun and c-Fos pathway. 100 AP-1 activation leads to the up-regulation of MMPs and several pro-inflammatory cytokines. 100 Therefore, AP-1 contributes to SBI after ICH. Lan et al. also reported that pre-treatment with EPC protected cultured brain astrocytes against Hb-induced cytotoxicity via the induction of Nrf2 and AP-1 signaling pathways. 99 In summary, these data suggested that EPC has multiple protective effects on ICH due to its anti-oxidative, anti-inflammatory, anti-apoptosis and anti-ferroptosis activities.
ILG, which is present in the liquorice root Glycyrrhiza uralensis, is a flavonoid with a chalcone structure. 101 Previous studies reported that ILG treatment attenuated brain damage in ischemia stroke, traumatic brain injury, inflammation-induced cognitive impairment models. 102 , 103 The protective effects of ILG are conferred by its anti-oxidative activity, which is mediated by Nrf2 activation and its anti-inflammatory activity, which is mediated by NF-κB suppression. 104 In a transient (2 hours) MCAO-induced ischemic stroke rat model, ILG pre-treatment suppressed infarct formation, brain oedema and neurological deficits via preserving both anti-oxidant capacity and brain ATP content. 102 In addition, pre-treatment with ILG before ischemic stroke maintained the activity of several Nrf2 downstream anti-oxidative enzymes including SOD, catalase and glutathione peroxidase. 102 , 104 Zeng et al. firstly demonstrated that in a collagenase-induced ICH rat model, ILG administration (20 and 40 mg/kg, i.p.) markedly ameliorated neurological deficits, histological damage, BBB disruption, brain oedema, and neuronal degeneration. 105 This study also demonstrated that ILG administration after ICH induction suppressed the expression of oxidative stress markers (3-nitrotyrosine and 8-hydroxyguanosine) and induced both HO-1 and NQO1 expression. 105 This report also clarified that ILG suppressed the NLRP3 inflammasome pathway, which is increased after ICH in rats lacking ILG treatment. 105 In the early phase after ICH, cleaved caspase-1 and several pro-inflammatory cytokines including IL-1β are increased. These events activate both NF-κB-mediated transcription and formation of the NLRP3 inflammasome that accelerates brain injury. 45 , 106 Nrf2 activation suppresses these inflammatory cascades via regulating NF-κB. 11 Since ILG has potent anti-oxidative and anti-inflammatory activities, the dietary uptake of ILG from liquorice and supplements may be an effective approach for treating ICH.
Luteolin is another member of the flavonoid family, which is present in various vegetables. Luteolin has anti-oxidative, anti-inflammatory, anti-autophagic, anti-apoptosis, and anti-tumor activities. 107 In an experimental ischemic stroke model, luteolin has neuroprotective effects via modulating the MAPK and PI3K/Akt signaling pathways, as well as via suppression the expression of various MMPs and TLRs. 108 , 109 Tan et al. reported that luteolin administration (10 and 20 mg/kg, i.p.) attenuated brain oedema and neurological deficits including memory impairment in a rat autologous blood injection-induced ICH model. 110 Luteolin induced Nrf2 activation and up-regulated Nrf2 downstream anti-oxidative factors. 110 Moreover, luteolin enhanced autophagy by inducing the expression of autophagy-related proteins such as microtubule-associated protein 1 A/1B-light chain 3 (LC3) and the autophagy-adaptor protein, p62. 110 p62 plays a major role in Nrf2 activation by binding to Keap1 and sequestering it into autophagosomes, where it is subsequently degraded. 111 Furthermore, the promoting autophagy via up-regulation of both Nrf2 and LC3 proteins reduced oxidative stress and improved neurological deficits in a SAH rat model. 112 Therefore, luteolin might protect the brain against ICH via promoting autophagy, which leads to Keap1 degradation and Nrf2 activation. In neuronal cells, luteolin treatment suppressed oxyHb-induced injury via preserving both mitochondrial function and the p62-Keap1-Nrf2 signaling pathway. 110
Taken together, these flavonoids have multiple pharmacological effects that are mediated by activation of the Nrf2 signaling pathway. Therefore, these flavonoids may exhibit protective effects against ICH-induced brain injury. Daily flavonoid intake from foods and supplements may be led to prevention of ICH.
Fumarates: dimethyl fumarate (DMF) and monomethyl fumarate (MMF)
DMF is a fumaric acid ester that has been approved for multiple sclerosis therapy.113–115 DMF and its primary metabolite MMF have protective effects on central nervous system (CNS) cells via promoting Nrf2 activation. 115 , 116 In an MCAO-induced ischemic stroke model, treatment with fumarates at post ischemia attenuated neurological deficits and infarction via their anti-inflammatory and anti-oxidative activities mediated by activation of the Nrf2-NF-κB pathway.117–119 The mechanism of Nrf2 activation occurs via stabilization and phosphorylation of Nrf2, which is mediated by fumarate binding to cysteine residues in the Keap1 protein. This event inhibits the formation of the Nrf2-Keap1 complex. 116 Two studies reported the efficacy of DMF in experimental ICH models. 120 , 121 Zhao et al. firstly demonstrated that DMF administration (15 mg/kg, i.p.) suppressed neurological deficits and brain oedema at 3 days after ICH, but these protective effects were diminished in Nrf2-deficient mice. 121 These authors also reported that DMF induced several Nrf2 downstream enzymes such as HO-1, catalase and NQO1. 121 Moreover, DMF enhanced the expression of various detoxification and hematoma clearance proteins such as CD36, CD163 and Hp, which contribute to Hb phagocytosis by microglia. 121 In addition, DMF suppressed the expression of various pro-inflammatory mediators such as IL-1β and inducible nitric oxide synthase (iNOS). 121 A subsequent report by Iniaghe et al. investigated the effects of DMF in two types of rodent ICH models. 120 In a collagenase-induced ICH model, DMF administration (10 or 100 mg/kg, i.p.) after stroke induction reduced intercellular adhesion molecule-1 (ICAM-1) expression levels, but enhanced casein kinase 2 expression levels, which led to the suppression of brain oedema and neurological deficits. 120 ICAM-1 expression is up-regulated y NF-κB signaling and ICAM-1 aggravates the inflammatory response. 11 In addition, only the high dose of DMF (100 mg/kg) attenuated brain injury and microglia activation in an autologous blood injection-induced ICH model. 120 Moreover, DMF treatment (100 mg/kg/day, p.o.) for 2 weeks decreased the incidence of aneurysm formation and rupture via Nrf2 activation in an elastase aneurysm mice model. 122 Therefore, DMF may suppress the vascular rupture and subsequent brain injury.
Recently, Shi et al. reported that MMF administration (15 mg/kg, p.o.) after stroke induction alleviated cerebral oedema and neurological deficits in an autologous blood injection-induced ICH rat model. 123 The efficacy of MMF in this model was dependent on microRNA-139-mediated regulation of the Nrf2-NF-κB pathway. 123 MicroRNA-139 is involved in the repair of tissue damage. 124 MMF treatment significantly increased the levels of microRNA-139, and overexpression of microRNA-139 increased both the nuclear levels of Nrf2 and the viability of human neuroblastoma SH-SY5Y cells. 123
DMF is an oral treatment approved for use in patients with relapsing forms of multiple sclerosis. 113 , 114 Therefore, DMF and MMF may also be effective for other CNS diseases. Based on the evidence described above, treatment with fumarates may protect brain tissue and improve neurological function in patients with ICH.
Tert-butyl hydroquinone (TBHQ)
TBHQ is a phenolic anti-oxidant, which is used in food as an anti-septic. 125 TBHQ possesses a 1,4 diphenolic-type electrophilic structure. 126 Due to this structural feature, TBHQ binds to cysteine residues in Keap1 and promotes Nrf2 translocation into the nucleus. 126 , 127 TBHQ has neuroprotective effects on both experimental ischemic stroke and SAH models via activating the Nrf2 signaling pathway. 128 , 129 In the ischemic stroke model, TBHQ continuously administration [1 mM, intracerebroventricular (i.c.v.)] for 72 hours before stroke induction attenuated both the infarct area and neurological deficits at 24 hours after surgery via the induction of phase II enzymes such as NQO1 in astrocytes. 128 The neuroprotective effect of TBHQ was maintained for 30 days after surgery. 128 Moreover, the glutathione content in the cortex area was increased by TBHQ administration at 24 hours after stroke induction. 128 However, the protective effects of TBHQ in the ischemia stroke model were diminished in Nrf2-deficient mice. 128 Therefore, the efficacy of TBHQ depends on the Nrf2 signaling pathway. Similar to the ischemic stroke model, in a SAH model, oral administration of TBHQ (12.5 mg/kg) after stroke induction ameliorated brain oedema, BBB disruption, necrotic and apoptotic cell death as well as motor and memory impairment. 129 TBHQ significantly increased the expression levels of NQO1 and GST. 129 In a collagenase-induced ICH model, post-injury TBHQ administration (50 mg/kg, i.p.) attenuated acute neurological deficits and oxidative brain damage. 130 Moreover, this same study showed that TBHQ treatment mitigated microglia activation and neurodegeneration at 24 hours after ICH. 130 Taken together, TBHQ has protective effects on all types of stroke models via up-regulation of the Nrf2 signaling pathway.
Nicotinamide mononucleotide (NMN)
NMN is an intermediate compound in nicotinamide adenine dinucleotide (NAD+) biosynthesis. 131 NMN is produced from nicotinamide in a reaction catalyzed by phosphoribosyl transferase. 131 NAD+ is an essential coenzyme for maintaining mitochondrial electron transfer activity and contributes to the regulation of cell proliferation, death and metabolism. 131 Depletion of intracellular NAD+ is observed as a common pathological condition in aging and ischemic stroke. 132 , 133 A previous study found that NAD+ replenishment by NMN treatment after tissue plasminogen activator-induced hemorrhagic transformation in a cerebral ischemia model suppressed BBB disruption and hemorrhagic transformation. 134 Therefore, NMN may have potential protective activity against ICH-induced brain damage. In fact, Wei et al. showed that in a collagenase-induced ICH model, NMN administration [300 mg/kg, i.p. or intravenous (i.v.)] after stroke induction attenuated brain oedema, neurological deficits, cell death and neuroinflammation without affecting the hematoma. 135 NMN treatment rapidly supplied NAD+ to the brain and increased HO-1 and nuclear Nrf2 expression, which led to the suppression of oxidative and inflammatory stresses. 135 Although these reports found that supplementation of NAD+ by NMN leads to Nrf2 activation, the mechanism of Nrf2 activation by NMN treatment is still unclear. Harlan et al. hypothesized that NAD+ supplementation might activate Nrf2 via the up-regulation of endogenous sirtuins (SIRTs) including SIRT6. 136 SIRT6 was shown to be directly involved in both Nrf2 activation and in the increase of HO-1 expression in cultured astrocytes. 136 SIRT6 plays important roles in both DNA repair and in the anti-oxidative stress system, and up-regulation of SIRT6 attenuated brain injury in cerebral ischemia. 137 Thus, NMN treatment may have protective effects against ICH-induced brain injury via the replenishment of NAD+ and activation of the Nrf2 signaling pathway.
Synthesized triterpenoids: bardoxolone methyl (BARD) and RS9
Triterpenoids are natural compounds synthesized by plants, which have been used in medicine for decades because of their specific anti-bacterial, anti-oxidative, anti-diabetic and cardio-protective activities. 138 , 139 Chemically synthesized triterpenoids derived from oleanolic acid and ursolic acid react with the cysteine residues of Keap1, which disrupt the Nrf2-Keap1 complex and promote Nrf2 activation. 140 , 141 BARD is a well-known chemically synthesized triterpenoid which has anti-oxidative and anti-inflammatory activities via activating the Nrf2 signaling pathway. 141 BARD has protective effects in several animal models of disease including kidney, retinal, cerebral and cardiovascular diseases as well as cancer.142–144 For example, type 2 diabetes patients treated with BARD exhibited significantly improved renal function. 145 , 146 Therefore, BARD may soon be approved as a therapeutic agent to treat kidney disease with diabetes. Additionally, in a MCAO-induced ischemia stroke model, BARD administration before reperfusion induction attenuated brain damage that occurred after ischemia reperfusion injury. Takagi et al. reported that in a tMCAO-induced focal brain ischemia model, BARD administration (2 mg/kg, i.v.) just before reperfusion decreased the infarct volume and improved neurological deficits. 54 Nrf2 activation by BARD treatment was observed in both neurons and astrocytes. 54 Furthermore, Imai et al. also showed that BARD suppressed hemorrhagic transformation after MCAO in anti-coagulant-treated mice. 147 In this study, BARD attenuated neurological deficits and protected BBB integrity via activating Nrf2 in BBB component cells such as vascular endothelial cells and pericytes. 147 Based on these results, synthesized triterpenoids might be useful reagents for treating stroke patients, but the effects of these compounds on ICH are unclear.
Recently, a more effective Nrf2 activator, referred to as RS9, was developed by a microbial transformation method. RS9 induced Nrf2 activation and promoted the expression of Nrf2 downstream anti-oxidant factors at a lower concentration compared to BARD. 148 In addition, RS9 treatment had protective effects in several eye disease models both in vivo and in vitro. 148 , 149 Moreover, the protective effects of RS9 may be mediated by the regulation of autophagy via the Nrf2-p62 signaling pathway. 149 BARD or RS9 administration (0.2 mg/kg, i.p.) immediately after reperfusion, attenuated infarct formation and neurological deficits as well as prolonged the survival rate after transient MCAO via activating the Nrf2-NF-κB signaling pathway. 150 The Nrf2 expression levels in the RS9 treatment group were significantly increased at 2 hours after MCAO. 150 On the other hand, in an autologous blood injection-induced hemorrhagic stroke model, Sugiyama et al. reported that RS9 administration (2 mg/kg, i.p.) after stroke induction suppressed neurological deficits and brain oedema. 79 RS9 promoted the phosphorylation of Akt in the early phase and subsequently increased the expression of HO-1 and SOD1 at 3 days after ICH. 79 In addition, RS9 has a direct neuroprotective effect in a hemin-induced neuronal cell damage model, which may depend on HO-1 activity because co-treatment with ZnPP diminished the protective effects of RS9. 79 In summary, synthesized triterpenoids (especially RS9) might be useful therapeutic reagents for treating SBI after ICH due to their potent anti-oxidative and anti-inflammatory activities.
Other protective agents related Nrf2-HO-1 pathways: curcumin and hemin
Curcumin is an electrophilic polyphenol in Indian spice Turmeric (Curcuma longa Linn) and has multiple effects such as anti-oxidant and anti-inflammatory properties. 151 The anti-oxidative effect of curcumin is derived in Nrf2 activation. 151 In ischemic stroke model, curcumin attenuated brain edema and neurological dysfunction via elevation of Nrf2 and down-regulation of NF-κB. 152 Moreover, five reports show that curcumin administration also attenuated brain injury in animal ICH models.153–157 However, there was no evidence in these reports that curcumin activated Nrf2-HO-1 pathway in ICH condition.
Hemin is a famous HO-1 inducer and used in clinical to treat acute porphyria. 158 In ICH condition, hemin released from hematoma is metabolized to iron by HO-1 and promotes ROS production. 21 , 22 Therefore, hemin treatment damaged brain tissues including neurons and endothelial cells.22–24, 26 However, systemic hemin administration (4 or 26 mg/kg, i.p.) after ICH attenuated BBB disruption in both collagenase and blood injection models. 61 Moreover, a low-dose hemin therapy also improved brain oedema and neurological outcome at chronic phase in a collagenase-induced ICH model. 61 Previous reports demonstrated that hemin treatment (50-100 µM) for 6 h promoted the translocation of Nrf2 and increased downstream anti-oxidant enzymes such as HO-1 and TXR in cellular experiments. 159 , 160 Therefore, the positive effects of hemin in ICH models might contribute to Nrf2 activation. In brief, hemin therapy may be effective for patients with ICH via activating Nrf2 pathways.
Conclusion
This review describes the efficacy of Nrf2 activators on ICH (Figure 4).
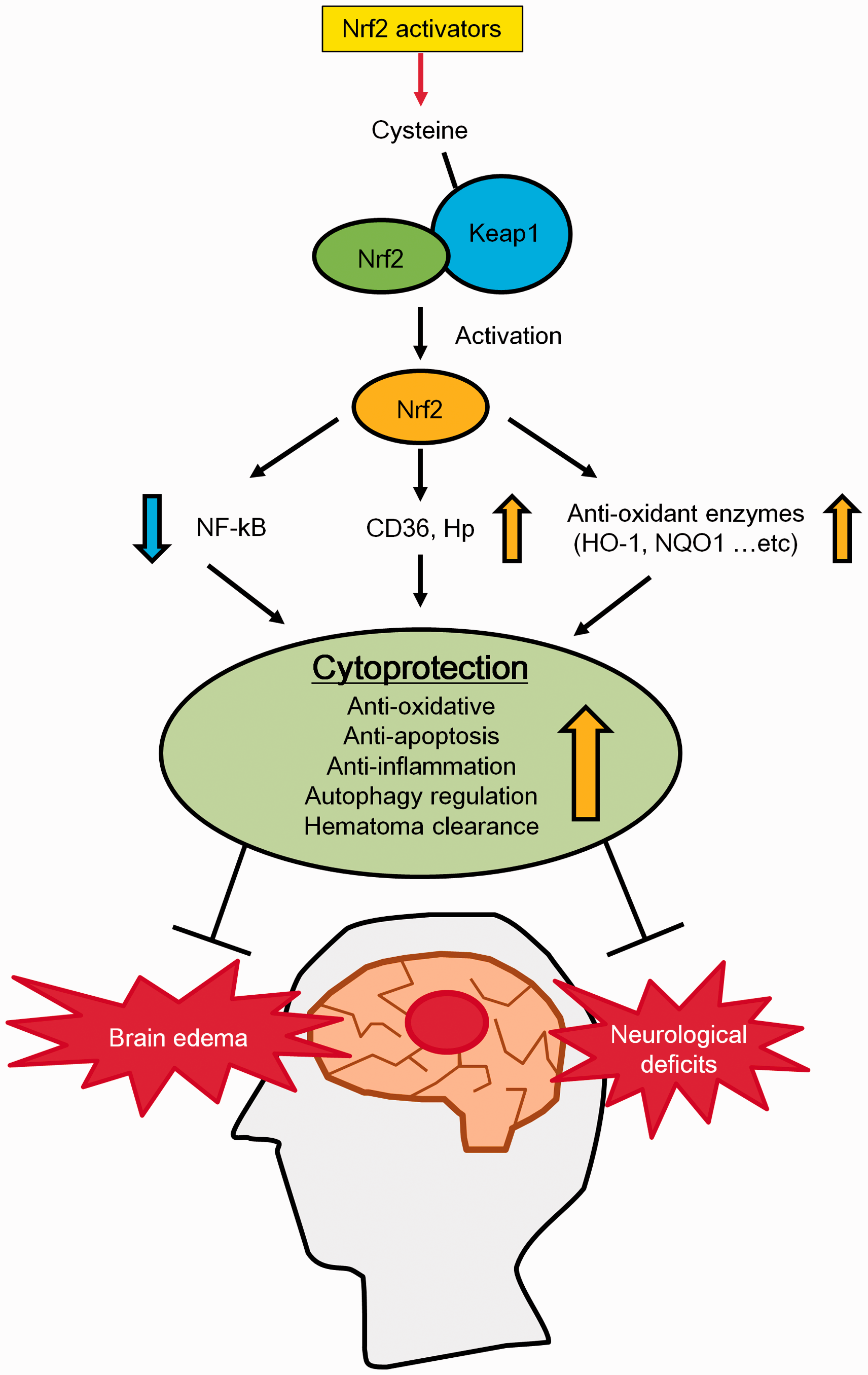
The protective effects of Nrf2 activators on ICH. Summary of the effects of Nrf2 activators on ICH. Nrf2 activation induces the expression of anti-oxidant enzymes and proteins which play roles in hematoma clearance. Nrf2 activation also suppresses NF-κB up-regulation. These events lead to the activation of anti-oxidative, anti-apoptosis and anti-inflammatory pathways, which confer cytoprotection against ICH. As a result, brain oedema and neurological deficits are attenuated after ICH.
In ICH pathology, PBI is mainly attributed to the hematoma physical mass effect. On the other hand, SBI occurs via multiple detrimental processes, especially oxidative and inflammatory stresses. After ICH onset, the mechanism of each detrimental process that contributes to SBI is highly complex and progresses more drastically over time. Therefore, although PBI can be treated with surgical intervention and/or osmotic diuretics, there is no effective therapeutic agent for SBI. Various hematoma components (Hb, heme, iron and immune cells) play important roles in the induction of SBI. These substances induce severe oxidative and inflammatory stresses via stimulating ROS production, autophagy, apoptosis and ferroptosis. Mainly, we summarized ICH pathology with a specific focus on oxidative and inflammatory stresses in this review. Next, we described the efficacy of Nrf2 activators for treating ICH-induced brain damage. These Nrf2 activators have strong anti-oxidative and anti-inflammatory activities that are mediated by targeting multiple factors including Nrf2, NF-κB, p62 and various anti-oxidative enzymes (HO-1, NQO1, etc). Over ten preclinical research articles provided evidence supporting the therapeutic potential of Nrf2 activators for treating ICH-induced SBI. These studies used both collagenase- and blood injection-induced ICH animal models. In particular, the Nrf2 activator DMF has already been approved for the treatment of multiple sclerosis, and clinical trials are currently being conducted to investigate the effects of the Nrf2 activator BARD on kidney disease associated with type II diabetes mellitus. In addition, flavonoids are also Nrf2 activators that may be effective for treating ischemic stroke and neurodegenerative diseases such as Alzheimer’s disease and Parkinson’s disease. Based on these positive results, other Nrf2 activators might also be approved for several diseases including ICH if their efficacy and safety can be confirmed in clinical trials.
Authors' contributions
Takahiko Imai wrote the manuscript. Hirofumi Matsubara and Hideaki Hara gave advice and helped to draft the manuscript. All authors discussed the contents and implications and commented on the manuscript at all stages.
Footnotes
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.