
Abstract
Angiotensin-(1-7) (Ang-(1-7)) can regulate vascular inflammation and remodeling, which are processes that have important roles in the pathophysiology of intracranial aneurysms. In this study, we assessed the effects of Ang-(1-7) in the development of intracranial aneurysm rupture using a mouse model of intracranial aneurysms in which aneurysmal rupture (i.e., aneurysmal subarachnoid hemorrhage) occurs spontaneously and causes neurologic symptoms. Treatment with Ang-(1-7) (0.5 mg/kg/day), Mas receptor antagonist (A779 0.5 mg/kg/day or 2.5 mg/kg/day), or angiotensin II type 2 receptor (AT2R) antagonist (PD 123319, 10 mg/kg/day) was started 6 days after aneurysm induction and continued for 2 weeks. Angiotensin-(1-7) significantly reduced the rupture rate of intracranial aneurysms without affecting the overall incidence of aneurysms. The protective effect of Ang-(1-7) was blocked by the AT2R antagonist, but not by the Mas receptor antagonist. In AT2R knockout mice, the protective effect of Ang-(1-7) was absent. While AT2R mRNA was abundantly expressed in the cerebral arteries and aneurysms, Mas receptor mRNA expression was very scarce in these tissues. Angiotensin-(1-7) reduced the expression of tumor necrosis factor-α and interleukin-1β in cerebral arteries. These findings indicate that Ang-(1-7) can protect against the development of aneurysmal rupture in an AT2R-dependent manner.
INTRODUCTION
Unruptured intracranial aneurysms are common, and the prevalence of unruptured intracranial aneurysms is estimated to be between 1% and 5%. 1 Unruptured aneurysms are asymptomatic until they rupture. However, subarachnoid hemorrhage from aneurysmal rupture results in severe morbidity and high mortality. 1 Inflammation is emerging as a key player in the processes that lead to aneurysmal rupture.2–4 Therefore, the pharmacological prevention of aneurysmal rupture that targets inflammation may be an attractive approach for patients with inoperable unruptured aneurysms.
Angiotensin-(1-7) (Ang (1-7)) is emerging as an integral player of the vascular renin-angiotensin system. 5 It is primarily formed from the conversion of angiotensin II by angiotensin-converting enzyme (ACE). 6 The biologic effects of Ang-(1-7) are primarily mediated by a G protein-coupled receptor, the Mas receptor.7–9 In addition to the Mas receptor, angiotensin II type 2 receptor (AT2R), while not having a strong binding capacity to Ang-(1-7), appears to mediate the effects of Ang-(1-7) in selected tissues.10–12 Through complex interactions that involve the Mas receptor, AT2R, and possibly other unknown receptors, Ang-(1-7) exerts protective roles against vascular inflammation and remodeling through processes that have significant roles in the pathophysiology of intracranial aneurysms. Therefore, the modulation of Ang-(1-7) signaling through AT2 or the Mas/ACE2 axis may modulate the processes that lead to aneurysmal subarachnoid hemorrhage.
In this study, we assessed the potential roles of Ang-(1-7) in the development of intracranial aneurysm rupture. We used a mouse model of intracranial aneurysms in which spontaneous aneurysmal rupture (i.e., aneurysmal subarachnoid hemorrhage) causes neurologic symptoms.13,14
MATERIALS AND METHODS
The animal study was approved by the University of California, San Francisco, Institutional Animal Care and Use Committee. The experiments followed the guidelines and regulations set by the University of California, San Francisco, Institutional Animal Care and Use Committee.
Mouse Model of Intracranial Aneurysms
Intracranial aneurysms were induced in 8- to 10-week-old male wild-type mice (WT; C57BL/6J, The Jackson Laboratory, Bar Harbor, ME, USA) or AT2R knockout mice (AT2KO; C57BL/6J background, provided by Dr Tadashi Inagami, Vanderbilt University). 15 We combined systemic hypertension and a single injection of elastase (0.035 units) into the cerebrospinal fluid at the right basal cistern.13,16 Detailed methods are presented in Supplementary information.
Two blinded observers performed daily neurologic examinations as previously described. 13 Neurologic symptoms were scored as follows: 0: normal function; 1: reduced eating or drinking activity showed by a weight loss of >2 g of body weight (approximately 10% weight loss) over 24 hours; 2: flexion of the torso and forelimbs upon lifting the animal by the tail; 3: circling to one side with a normal posture at rest; 4: leaning to one side at rest; 5: no spontaneous activity. 13 Symptomatic mice were euthanized when they developed neurologic symptoms (scores 1 to 5). All asymptomatic mice were euthanized 21 days after aneurysm induction. The brain samples were perfused with phosphate-buffered saline, followed by a gelatin-containing blue dye to visualize cerebral arteries. Two blinded observers assessed aneurysm formation and subarachnoid hemorrhage.
Experimental Protocol
Our previous studies found that aneurysm formation starts to occur approximately 7 days after the aneurysm induction in this model.13,14 By treating the mice with an experimental agent starting from 6 days after aneurysm induction, we were able to test whether the experimental agent could reduce the rupture rate.13,14 Therefore, in this study, the treatments with Ang-(1-7) (0.5 mg/kg/day)17,18 with or without a Mas receptor antagonist (A779 0.5 mg/kg/day or 2.5 mg/kg/day)17,18 or an AT2R antagonist (PD 123319, 10 mg/kg/day)18,19 were initiated 6 days after aneurysm induction and were continued for 2 weeks. Angiotensin-(1-7), A779, and PD 123319 were dissolved in phosphate-buffered saline and filled into osmotic mini pump (model 1002, Alzet, Cupertino, CA, USA). All drugs (including vehicle control: phosphate-buffered saline) were administered continuously by implanting the osmotic mini pump intraperitoneally. Using the tail cuff method, systolic blood pressure was measured in the mice before aneurysm induction and every week after the elastase injection.13,14
Real-Time PCR Analysis
In separate sets of animals, we collected the total RNA samples from cerebral arteries and aneurysms at 14 days after aneurysm induction. We measured mRNA expression levels of AT2R, Mas receptor, and inflammation-related cytokines (monocyte chemoattractant protein-1: MCP-1, tumor necrosis factor-α: TNF-α, interleukin-1β: IL-1β, and inducible nitric oxide synthase: iNOS). The whole cerebral arteries including aneurysms were isolated and the total RNA was extracted using the RNeasy Mini Kit (Qiagen, Valencia, CA, USA). The total RNAs were transcribed to cDNA using the QuantiTect reverse transcription kit (Qiagen). The mRNA expression levels were determined using SYBR Green technology (Applied Biosystems, Foster City, CA, USA). Quantitative values were obtained from the threshold cycle value (Ct) and the data were analyzed by the 2-ΔΔCT method. The transcript amount of GAPDH (glyceraldehyde-3-phosphate dehydrogenase) was quantified as an internal RNA control.
Statistical Analysis
Rupture rate was calculated as the number of mice with ruptured aneurysms/number of mice with ruptured or unruptured aneurysms. We used Fisher's exact test for the analysis of the rupture rate and the incidence of aneurysms. As an exploratory analysis, the survival analysis was performed using the log-rank test. Mice that did not develop aneurysms were excluded from the survival analysis. Blood pressure levels were analyzed using nonparametric ANOVA (Kruskal-Wallis analysis of variance). mRNA levels were analyzed using ANOVA, followed by the Tukey-Kramer post hoc test. All results are expressed as the mean ± s.d. Statistical significance was set at P < 0.05.
RESULTS
Angiotensin-(1-7) is Protective Against Intracranial Aneurysmal Rupture
As a first step, we assessed the effects of Ang-(1-7) in the development of aneurysmal rupture. In all, 25 mice for the vehicle control group and 25 mice for the Ang-(1-7) treatment group underwent the aneurysm induction surgery. Two mice from each group were excluded from the study because of intraoperative mortality. Angiotensin-(1-7) did not affect the overall incidence of intracranial aneurysms (including both ruptured and unruptured aneurysms) (78% versus 78%: n = 23 versus 23, P = 1.00). However, Ang-(1-7) significantly reduced the rupture rate (rupture rate: 83% versus 33%; n = 18 versus 18, P < 0.05) (Figure 1). For the purpose of exploratory analysis, a symptom-free curve (Kaplan-Meier analysis curve) was plotted after excluding mice that did not have aneurysms (Supplementary information) A log-rank test revealed a significant reduction of aneurysmal rupture with Ang-(1-7) (P < 0.05) (Supplementary information).
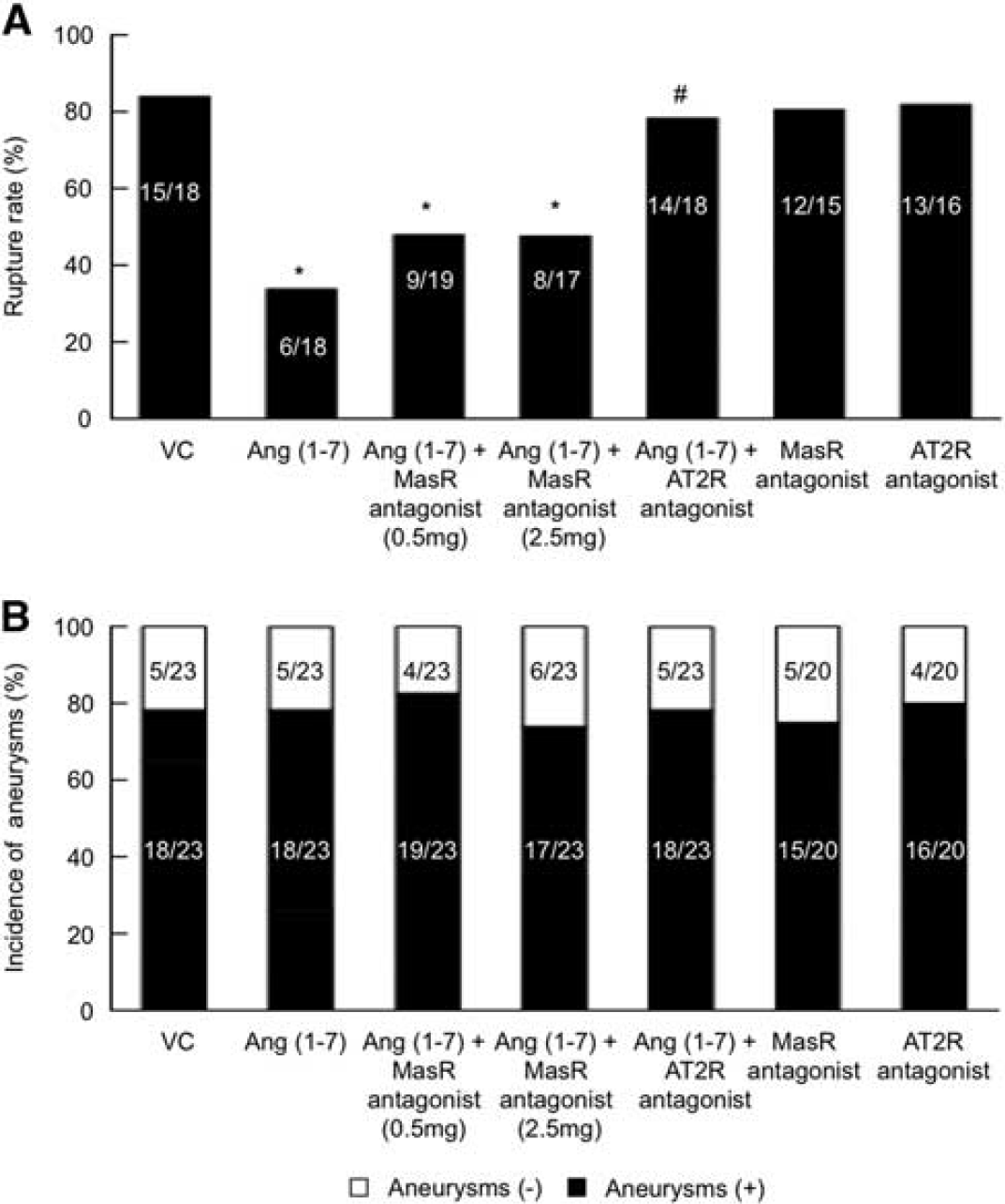
Ang-(1-7) protected against the development of aneurysmal rupture. The protective effect of Ang-(1-7) was abolished by AT2R antagonist, while Mas receptor antagonist only partially reduced the protective effect of Ang-(1-7). (
Figure 2 shows a representative ruptured aneurysm (Figure 2, right panel) from the vehicle-treated group, a representative unruptured aneurysm (Figure, middle panel) from the mice treated with Ang-(1-7) and a control brain sample (Figure 2, left panel). When a mouse was euthanized immediately after it developed neurologic symptoms that are associated aneurysmal rupture (i.e., subarachnoid hemorrhage), we found an aneurysm surrounded by fresh blood clots from a subarachnoid hemorrhage (Figure 2, right panel).
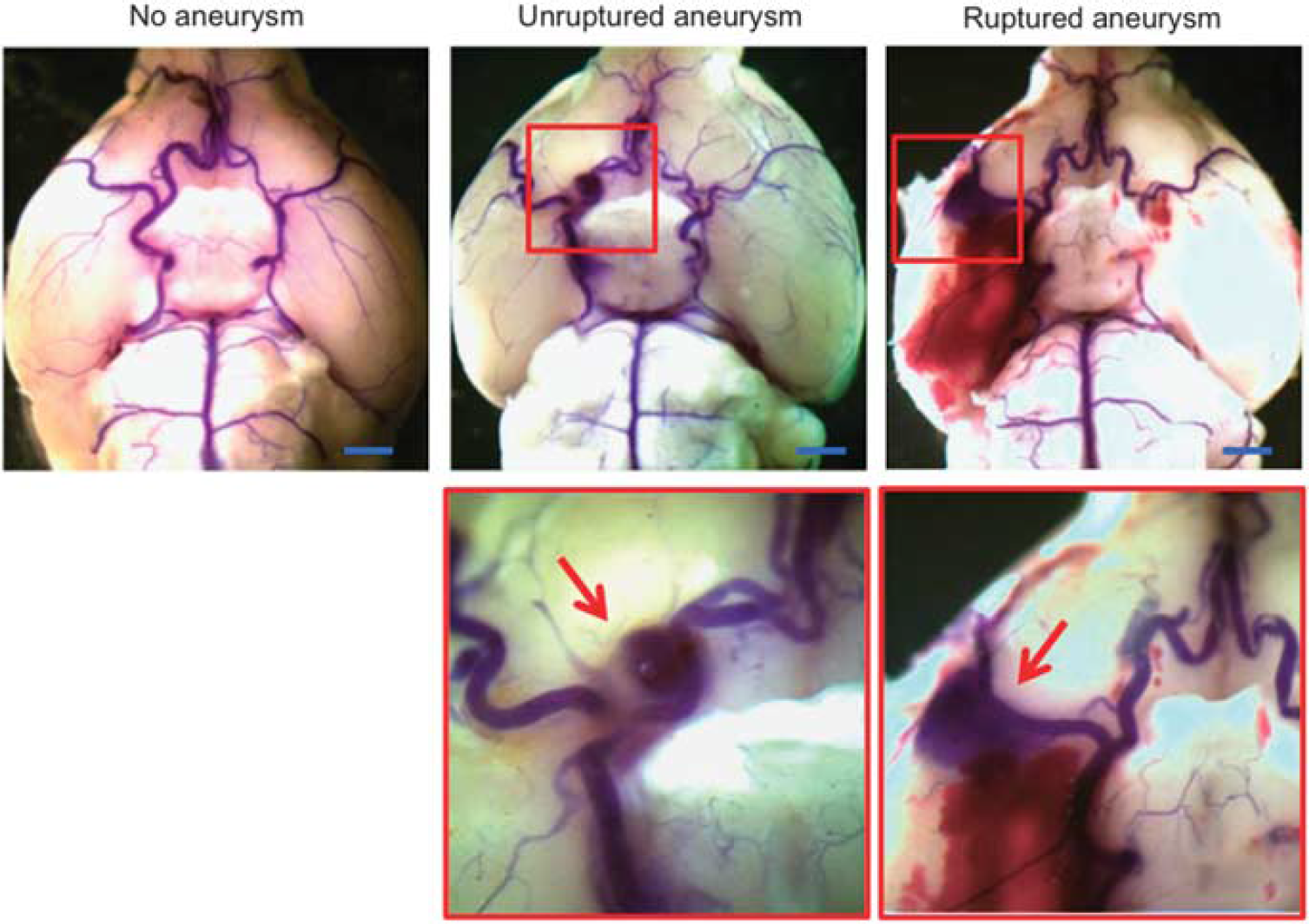
Representative intracranial aneurysms in mice. Left panel: Normal cerebral artery. Middle panel: Unruptured aneurysm in the right anterior cerebral artery. Right panel: Ruptured aneurysm with subarachnoid hemorrhage in the right middle cerebral artery. Cerebral arteries were visualized by blue dyes dissolved in gelatin. Arrows indicate aneurysms. Bar = 2.5 mm.
As a next step, we assessed whether the protective effect of Ang-(1-7) required the Mas receptor and AT2R by treating 25 mice with Ang-(1-7) and a Mas receptor antagonist (A779) at 0.5 mg/kg/day, the dose that has been successfully used in the previous studies,17,18,20,21 and 25 mice with Ang-(1-7) and the Mas receptor antagonist a five-fold higher dose of (2.5 mg/kg/day). Two mice from each group were excluded because of intraoperative mortality.
The addition of the Mas receptor antagonist at 0.5 mg/kg/day or 2.5 mg/kg/day did not affect the overall incidence of aneurysms (Ang-(1-7) versus A779 at 0.5 mg/kg/day versus A779 at 2.5 mg/kg/day: 78% versus 83%. versus74%: n = 23 versus 23 versus 23).
A779 at 0.5 mg/kg/day only partially blocked the protective effects of Ang-(1-7) against the development of aneurysmal rupture (Figure 1A) (rupture rate: 33% versus 47%: n = 18 versus 19, P = 0.51). Even when a five-fold higher dose of A779 (2.5 mg/kg/day) was used, the restoration of the rupture rate remained only partial (Figure 1A) (rupture rate: 33% versus 47%: n = 18 versus 17, P = 0.51). The treatment with A779 alone did not affect the rupture rate or incidence of aneurysms.
However, the AT2R antagonist (PD 123319, 10 mg/kg/day) significantly increased the rupture rate in the Ang-(1-7)-treated mice (78% versus 33%; n = 18 versus 18, P < 0.05) and effectively abolished the protective effect of Ang-(1-7). AT2R alone did not affect the rupture rate (Figure 1A). A log-rank test revealed a significant increase in the rupture risk in the mice that received Ang-(1-7) and PD 123319 compared with the mice that received Ang-(1-7) alone (P < 0.05) (Supplementary information).
Treatments with Ang-(1-7) with or without the Mas receptor antagonist or the AT2R antagonist did not affect the systemic hypertension induced by the deoxycorticosterone acetate (DOCA)-salt treatment (data not shown).
The Protective Effect of Angiotensin-(1-7) Required Angiotensin II Type 2 Receptor
To confirm that the protective effect of Ang-(1-7) requires AT2R, we compared the effect of Ang-(1-7) between the wild-type mice and AT2R knockout mice.
In all, 20 wild-type mice in the Ang-(1-7) group and 20 wild-type mice in the vehicle group underwent the aneurysm induction surgery. There was no intraoperative mortality. In the wild-type mice, there was no difference in the overall incidence of aneurysms (80% versus 85%: n = 20 versus 20; P = 1.00) (Figure 3). However, Ang-(1-7) treatment significantly reduced the rupture rate (31% versus 76%: n =16 versus 17, P < 0.05) (Figure 3).
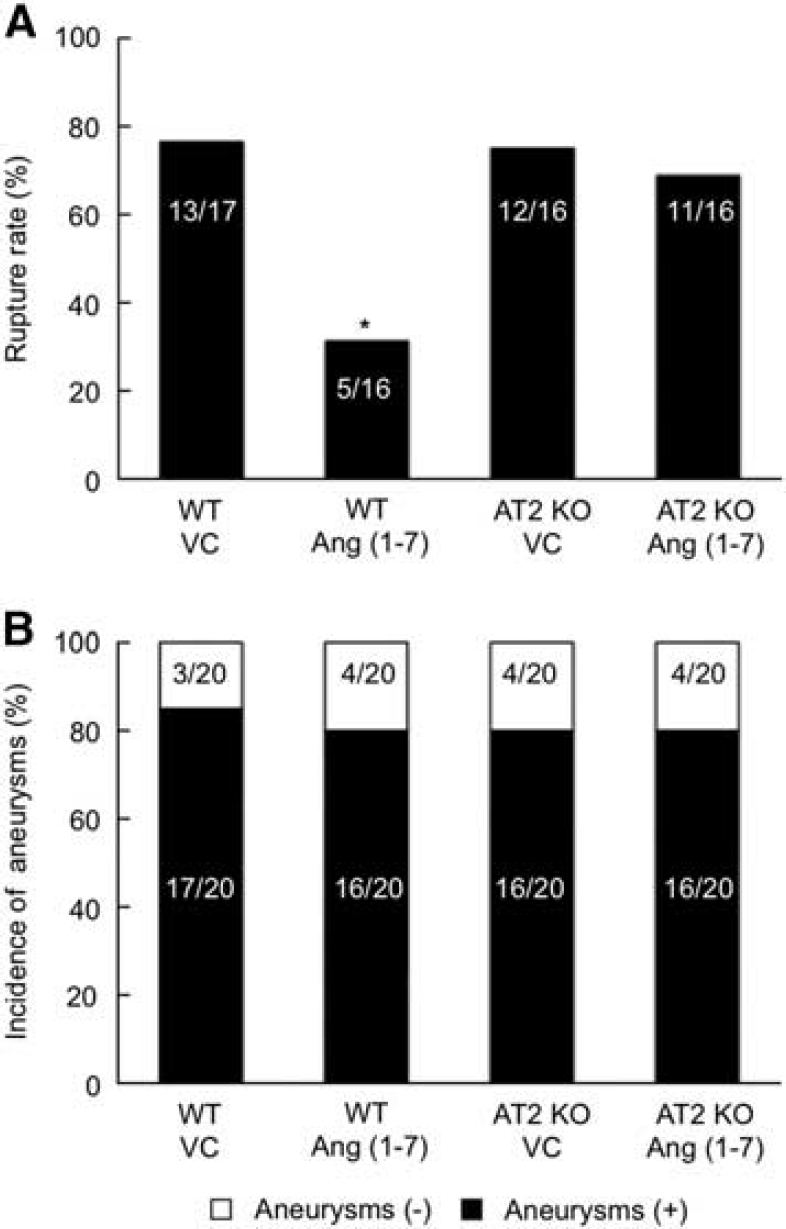
Ang-(1-7) requires AT2R for the protective effects against intracranial aneurysmal rupture. (
In all, 20 AT2R knockout mice in the vehicle group and 20 AT2R knockout mice in the Ang-(1-7) group underwent the aneurysm induction surgery. There was no intraoperative mortality. There was no difference in the overall incidence of aneurysms between the AT2R knockout mice treated with vehicle and AT2R knockout mice treated with Ang-(1-7) (80% versus 80%: n = 20 versus 20; P =1.00) (Figure 3).
The protective effect of Ang-(1-7) was lost in the AT2R knockout mice, as evidenced by no difference in the rupture rate between the AT2R knockout mice treated with vehicle and AT2R knockout mice treated with Ang-(1-7) (75% versus 68%: n = 16 versus 16; P =1.00) (Figure 3).
The Expression of Angiotensin II Type 2 Receptor and the Mas Receptor in Cerebral Vessels or Aneurysms
As a next step, we examined the expression of AT2R and the Mas receptor in the cerebral arteries and intracranial aneurysms in mice. Real-time PCR was performed for eight cerebral arteries and eight aneurysm tissues. As shown in Figure 4, the Mas receptor and AT2R mRNAs were expressed in both control cerebral arteries and aneurysms. In both tissues, the expression levels of AT2R were significantly higher than that of the Mas receptor.
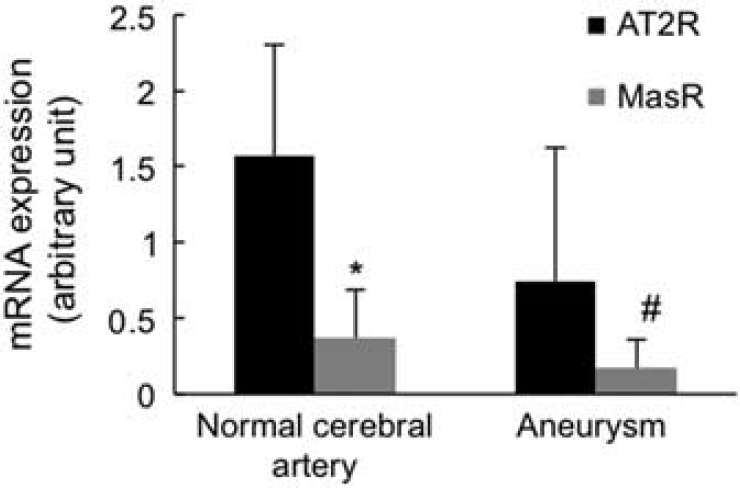
mRNA expression of AT2R and Mas receptor in normal cerebral artery and aneurysm. Real-time PCR: mRNA expression levels of AT2R and Mas receptor in normal cerebral arteries and aneurysms (each group n = 8). AT2R, angiotensin II type 2 receptor. *,#: P < 0.05 compared to AT2R.
Effects of Angiotensin-(1-7) Treatment on Inflammatory Cytokines
To explore the effects of Ang-(1-7) treatment on the inflammatory cytokines, we assessed mRNA expression levels of MCP-1, TNF-α, IL-1β, and iNOS in the cerebral arteries tissues that included surrounding adventitial tissues. We collected the cerebral artery tissues from eight Ang-(1-7)-treated mice and eight vehicle-treated mice. The Ang-(1-7) treatment significantly reduced the expression of IL-1β and TNF-α compared with the vehicle treatment (IL-1β: Ang-(1-7) versus vehicle = 0.7±0.6 versus 4.3±4.6, P < 0.05; TNF-α: 1.5±2.7 versus 3.6±4.3, P < 0.05) (Figure 5). However, there was no significant difference in iNOS or MCP-1 between the vehicle-treated group and Ang-(1-7)-treated group (iNOS: vehicle versus Ang-(1-7) = 2.9±6.1 versus 1.9±2.2, P = 0.35; MCP-1: 3.5±4.4 versus 2.7±1.9, P = 0.32).
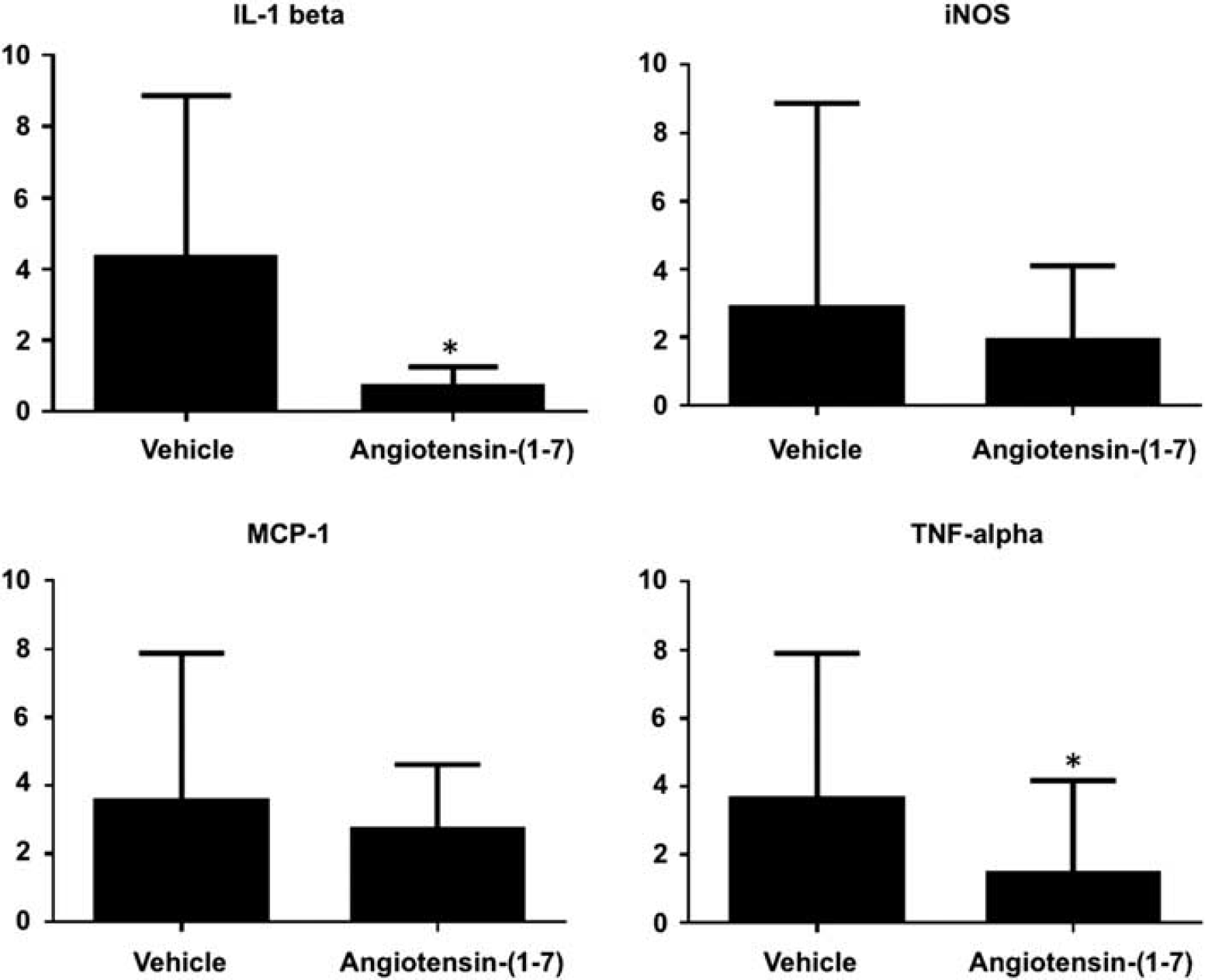
Effects of angiotensin (1-7) (Ang-(1-7)) treatment on inflammatory cytokines. Ang-(1-7) treatment significantly reduced the expression of IL-1β and TNF-α compared with the vehicle treatment. However, there was no significant difference in iNOS or MCP-1 between the vehicle-treated group and Ang-(1-7)-treated group. iNOS, inducible nitric oxide synthase; IL-1β, interleukin-1β; MCP-1, monocyte chemoattractant protein-1; TNF-α, tumor necrosis factor-α.
DISCUSSION
In this study, we found that Ang-(1-7) can protect against the development of intracranial aneurysm rupture (aneurysmal sub-arachnoid hemorrhage) in mice. The protective effect of Ang-(1-7) required AT2R, a receptor that is more abundant than Mas receptor in cerebral arteries and aneurysms. In addition, we found that Ang-(1-7) reduced expression of IL-1β and TNF-α in cerebral arteries, suggesting anti-inflammatory effects of Ang-(1-7). Previously, we showed that the activation of the ACE/AngII/AT1R axis promotes the processes that lead to aneurysmal rupture. 14 Taken together, a balance between the Ang-(1-7) system and the ACE/AngII/AT1R axis may determine the progression of the disease course of intracranial aneurysm to either aneurysmal rupture or stabilization.
Ang-(1-7) exerts both acute and chronic effects on the cardiovascular system. It can reduce vascular inflammation, oxidative stress, cell proliferation, and nitric oxide availability, thereby reducing atherosclerosis and enhancing plaque stability.18,21 It possesses a high binding capacity to Mas receptor, a G protein-coupled receptor that appears to mediate many of the biologic effects of Ang-(1-7).7,9 Although the binding affinity of AT2R with Ang-(1-7) is relatively weak, AT2R appears to mediate some of the vascular effects of Ang-(1-7).10,11,22 In spontaneously hypertensive rats, the vaso-suppressive effect of Ang-(1-7) appears to be dependent on the activation of AT2R. 10 The Mas receptor antagonist (A779) did not block the vaso-supressive effect of Ang-(1-7) in these rats. In a chronic setting, the athero-protective effect of Ang-(1-7) was abrogated by an AT2R antagonist. 18 These findings suggest a functional and possibly physical interaction between Mas receptor and AT2R in the vascular system. 11
We found that Ang-(1-7) reduced expression of IL-1β and TNF-α in cerebral arteries. Both IL-1β and TNF-α have linked with the formation and progression of intracranial aneurysms.3,23 By reducing vascular inflammation, Ang-(1-7) may block the processes that lead to aneurysmal rupture. Excessive inflammation has been suggested as a key mechanism for the development of aneurysmal rupture in previous studies.2,23,24 Although we confirmed the requirement of AT2R in the protective effect of Ang-(1-7) using AT2R knockout mice, the requirement of Mas receptor in this context remains to be further elucidated. Although our pharmacological study using standard and high doses of the Mas receptor antagonist strongly suggested that the activation of Mas receptor is not a prerequisite for the protective role of Ang-(1-7), we did not confirm this finding using Mas receptor knockout mice. Interestingly, we found significantly higher mRNA expression of AT2R than that of Mas receptor in both normal cerebral arteries and aneurysms. In the cerebrovasculature, the activation of AT2R may be the primary regulator of the biologic effect of Ang-(1-7) while Mas receptor has a more permissive role. However, it should be note that we did not verify the differential expression levels of AT2R and Mas receptor at the protein levels because of the potential cross-reactivity of commercially available anti-AT2R antibodies with angiotensin II type 1 receptor.25,26
In our study, Ang-(1-7) did not affect the systemic hypertension induced by the DOCA-salt treatment. The hypotensive effect of Ang-(1-7) appears to be apparent only when the animals are rendered hypertensive by augmenting the endogenous levels of angiotensin II. 5 The DOCA-salt treatment's hypertensive effect bypasses the activation of the systemic renin-angiotensin system; the endogenous angiotensin II levels are reduced in the DOCA-salt hypertension as a result of negative feedback. This explains why the hypotensive effect of Ang-(1-7) was not observed in our study. The protective effect of Ang-(1-7) against the development of aneurysmal rupture appears to be independent of its hypotensive effect.
The DOCA-salt hypertension has been widely used as a model of human essential hypertension, especially when studying the end-organ or vascular changes associated with systemic hypertension. It should be noted that DOCA-salt hypertension is the low renin, volume overload form of hypertension. This particular type of hypertension represents approximately 25% of essential hypertension in humans. 27 However, we have shown that aneurysm can be induced by either DOCA-salt hypertension or angiotensin II-induced hypertension,16,24 indicating that the mode for inducing hypertension is not important for aneurysm formation in this model.
Animal models may not recapitulate all processes involved in aneurysm formation in humans. The exact events inciting the formation of intracranial aneurysms are not known. There are certainly genetic components in the pathophysiology of intracranial aneurysms, but the disease-causing genes have not been identified, making it difficult to develop a genetic model of intracranial aneurysm. In animal models, inciting factors that lead to the disease often need to be exaggerated or accentuated to achieve a severe disease phenotype at high incidence within a practical time frame. However, by doing so, the model may fail to recapitulate or skip events that have key roles in the human disease.
Although aneurysms were induced, but not spontaneously formed, in our model, the phenotype and presentation of intracranial aneurysms in our model closely mimic those of human intracranial aneurysms.16,24 More importantly, human intracranial aneurysms and aneurysms in this model share the common end phenotype, i.e., aneurysmal rupture and associated symptoms.13,14 This model with or without modifications have been successfully used by other groups to study the mechanisms for aneurysmal formation and rupture.3,28 Clinical studies that assess the correlation between aneurysmal rupture and the use of agents that affect the Ang-(1-7) system will be need to validate our findings.
This study focuses on the potential roles of the Ang-(1-7) system in the development of aneurysmal rupture. We did not assess the roles of Ang-(1-7) in the formation of aneurysms. Mechanisms for aneurysmal formation may be fundamentally different from mechanisms for aneurysmal rupture. Future studies may reveal the differential roles of Ang-(1-7) between aneurysmal formation and aneurysmal rupture.
Summary
Our previous study showed the involvement of the vascular renin-angiotensin system, specifically the ACE/Ang II/AT1R axis, in the development of aneurysmal rupture. 14 The activation of the vascular local renin-angiotensin system appears to lead to aneurysmal rupture, independent of the hypertensive effect of the renin-angiotensin system. A complex balance and interaction between the ACE/Ang II/AT1R system and the Ang-(1-7) system may determine the disease course of intracranial aneurysms.
DISCLOSURE/CONFLICT OF INTEREST
The authors declare no conflict of interest.
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.