
Abstract
Receptor-interacting protein kinase-1 (RIPK1) is a master regulator of cell death and inflammation, and mediates programmed necrosis (necroptosis) via mixed-lineage kinase like (MLKL) protein. Prior studies in experimental intracerebral hemorrhage (ICH) implicated RIPK1 in the pathogenesis of neuronal death and cognitive outcome, but the relevant cell types involved and potential role of necroptosis remain unexplored. In mice subjected to autologous blood ICH, early RIPK1 activation was observed in neurons, endothelium and pericytes, but not in astrocytes. MLKL activation was detected in astrocytes and neurons but not endothelium or pericytes. Compared with WT controls, RIPK1 kinase-dead (RIPK1D138N/D138N) mice had reduced brain edema (24 h) and blood-brain barrier (BBB) permeability (24 h, 30 d), and improved postinjury rotarod performance. Mice deficient in MLKL (Mlkl-/-) had reduced neuronal death (24 h) and BBB permeability at 24 h but not 30d, and improved post-injury rotarod performance vs. WT. The data support a central role for RIPK1 in the pathogenesis of ICH, including cell death, edema, BBB permeability, and motor deficits. These effects may be mediated in part through the activation of MLKL-dependent necroptosis in neurons. The data support development of RIPK1 kinase inhibitors as therapeutic agents for human ICH.
Keywords
Introduction
Intracerebral hemorrhage (ICH) accounts for approximately 15% of all strokes but has the highest mortality rate among stroke subtypes with ≤50% 30-day survival and poor neurological outcome in half of survivors. 1 Treatment is supportive with no specific therapy to reduce neurological sequelae. The pathogenesis of ICH includes blood vessel rupture, hematoma expansion, disruption of the blood–brain barrier, brain edema formation, apoptotic and necrotic cell death, and induction of a sustained perihematomal inflammatory response. 2 However, molecular drivers of outcome in ICH are only beginning to be elucidated. 3
Receptor-interacting protein kinase-1 (RIPK1) is a serine/threonine kinase that is activated by phosphorylation, dimerization, and assembly of mild detergent-insoluble amyloid complexes with itself and RIPK3. 4 The RIPK1-RIPK3-mixed lineage kinase like (MLKL) “necrosome’ complex (IIb) leads to RIPK3-mediated MLKL phosphorylation, dissociation of phosphorylated MLKL from the necrosome, insertion into the plasma membrane, and cell lysis. 5 RIPK1 can also promote apoptosis as well as cell death-independent inflammation. A growing body of evidence implicates RIPK1 in neuronal death and inflammation in neurodegenerative diseases, traumatic brain injury, traumatic spinal cord injury, and ICH.4,6,7 We previously reported that genetic inhibition of RIPK1-kinase function reduced acute neuronal cell death in hemorrhagic striatum and improved body weight gain and cognitive outcome after autologous blood ICH8 and that mice deficient in RIPK3 had decreased acute cell death vs. WT in a collagenase ICH model. 9 These studies primarily used genetic tools to interrogate effects of RIPKs in ICH models to avoid the confound of off target effects of pharmacological inhibitors. However, specific cell types that activate RIPKs and MLKL after ICH remain to be determined.
In contrast to RIPK1, MLKL has been less studied in CNS injury paradigms. Besides inducing necroptosis, MLKL has RIPK3- and cell death-independent functions in demyelination, 10 macrophage inflammation, 11 and endosomal and extracellular vesicle trafficking. 12 Mlkl-/- (Mlkl gene knock-out) mice show protection in models of systemic ischemia/reperfusion and TNF alpha toxicity,13–15 but MLKL inhibition has not been reported in an ICH model. Theoretically, RIPK1 might be a more important disease driver because it regulates apoptosis (unaffected by MLKL) and multiple inflammation pathways whereas MLKL pathways overlap to a significant degree with RIPK1 (e.g., necroptosis and NFkB dependent inflammation). Indeed, RIPK1 kinase inhibition has been reported to confer greater benefit than MLKL deficiency in mouse models of renal ischemia/reperfusion, TNF alpha toxicity, and pro-survival protein A20 deficiency. 16
Here, we used RIPK1D138N/D138N mice expressing catalytically inactive RIPK1 due to a loss-of-function mutation in the kinase domain of the Ripk1 gene, and Mlkl-/- mice to test the hypothesis that RIPK1 inhibition is more broadly protective in experimental ICH. The results demonstrate a role for MLKL in several important outcomes and support the notion that the RIPK1 kinase domain is an important disease driver in a mouse autologous blood ICH model.
Methods
All experimental protocols were approved by the Massachusetts General Hospital Institutional Animal Care and Use Committee and conducted according to the National Institutes of Health guide for the care and use of laboratory animals and ARRIVE (Animal Research: Reporting of In Vivo Experiments) guidelines. 17 Studies were performed using adult (8–12 weeks, 25–30 g) C57BL/6J (Wild-type) male mice (Jackson Laboratories, Bar Harbor, ME), RIPK1D138N/D138N (RIPK1 kinase-dead knock-in; a kind gift from Dr. Vishva Dixit, Genentech, Inc., San Francisco, CA) 18 and Mlkl−/− (Mlkl knock-out) 19 mouse lines (obtained from Dr. Siddharth Balachandran, Fox Chase Cancer Center, Philadelphia, PA) both derived solely from C57BL/6J. RIPK1D138N/D138N and Mlkl−/− mutations showed no abnormality in development or in adult animals for up to 6 mos. Mice were housed in a pathogen-free environment with 12-hour day/night cycles. Food and water were provided ad libitum. A descriptive table of experimental groups, the timeline of experimentation, and total number of mice is provided in Supplemental Table I. All experiments were performed and analyzed in a fully randomized and blinded design.
Autologous blood injection intracerebral hemorrhage (ICH) model
ICH surgeries were performed as previously described. 8 Briefly, nonheparinized autologous blood (30 μL for all experiments except edema, for which we used 60 μl) collected from the facial vein was injected into the right striatum over 2 minutes using a 26-gauge Hamilton syringe and syringe pump (Harvard Apparatus). Stereotactic co-ordinates were 3.5 mm deep, 0.5 mm anterior, and 2.0 mm lateral to bregma. Sham injured mice were administered an equal volume of sterile saline instead of autologous blood.
Cell isolation by magnetic-activated cell sorting (MACS) and western blotting
Mice were transcardially perfused with cold phosphate buffered saline (PBS). The brains were removed, and hemispheres without cerebellum were separated as contralateral (non-injured) and ipsilateral (injured). MACS was performed for astrocyte, neuron, endothelium and pericyte cell isolation according to manufacturers’ instructions. Western blotting methods are described in Supplemental Methods).
Preparation of brain tissues and quantification of Fluoro-Jade B-positive cells
At 48 hours after ICH, brains were frozen on dry ice and kept at −80°C until use. The brain sections were cut, stained with Fluoro-Jade B (FJB, Millipore), and FJB positive cell counts were performed as described previously. 8
Immunohistochemistry
Neuronal nuclei (NeuN, Millipore), astrocyte (GFAP, Abcam), and endothelium (CD31, BD Pharmingen) histochemistry was performed as described in Supplemental Methods.
Quantitation of endothelial cell loss and GFAP+ astrocytes
Frozen brain sections were stained with anti-mouse CD31 (1:100, BD Pharmingen) or GFAP (1:100, Abcam), and photomicrographs were obtained from 8 hemorrhagic x100 fields and corresponding contralateral hemispheric striatal brain regions. CD31+ and GFAP+ cells were counted as described in Supplemental Methods.
Assessment of blood-brain barrier (BBB) permeability
Evans blue (EB, Sigma, 2% wt/vol in saline) was given by tail vein injection (4 cc/kg) to mice immediately after ICH and allowed to circulate for 24 hours to capture dynamic changes in BBB permeability. At 24 hours or 30 days after ICH, mice were transcardially perfused with PBS, and EB extravasation measured either with spectrophotometry (for 24h ICH group) or from photomicrographs (for 30d ICH group, to increase sensitivity of EB detection) as described in Supplemental Methods.
Assessment of brain edema
At 24 h after ICH, mice were deeply anesthetized with isoflurane and decapitated. The brains were removed immediately and dissected into contralateral and ipsilateral striatum and cortex. Brain water content was expressed as a percentage of the wet weight: (wet weight – dry weight)/(wet weight) × 100.
Assessment of hemorrhage volume
Separate cohorts of mice were used to assess hemorrhage volume in WT, RIPK1D138N/D138N, and MLKL−/− at 24 and 72 h after ICH. Mice were anesthetized and subjected to ICH (30 ul blood) and killed at 24 or 72 h by transcardial perfusion with 4% PFA. Brains were post-fixed in 4% PFA for 24 h and then were cryoprotected in 30% sucrose for 24 h. Brains were frozen in nitrogen vapor, stored at −80°C and cut on a cryostat (20 um thick sections) every 400 um from anterior to posterior lesion. White light microscope images of the brain slices were taken under a 1x objective. The hemorrhage area in each section was measured by ImageJ software and hemorrhage volume obtained by summing the areas and multiplying the sum by 0.4mm
Behavior testing
The rotarod test was performed as previously described. 20
Statistical analyses
Data are mean ± standard deviation of the mean, and data collected at a single time point are presented in dot plots. Densitometry, cell counts, Evans blue extravasation, and brain water content were analyzed by t-test. Weight loss and rotarod data were analyzed by repeated measures ANOVA (group × time) using GraphPad Prism 8. Behavioral tests were powered based on our previous publications. All other experiments were powered for >50% differences between group means and estimated standard deviation 25% to achieve power = 0.8. When pilot studies indicated that apparent differences between means might be significantly less than 50%, the projected sample size was appropriately increased. Normality of data sets was established by the Shapiro-Wilk normality test and Q-Q plot analysis. Data that were not normally distributed were analyzed by the Mann-Whitney U-test for non-parametric data (brain edema and western blot data) or the Kruskal-Wallis test for mortality data.
Results
A total of 314/329 mice survived ICH and the experimental protocols (Supplemental Table I). Of the mice that died (all within 24 h of ICH), 6/161 were WT, 7/85 were RIPK1D138N/D138N, and 2/83 were Mlkl-/-. No differences were observed in mortality rates among the three groups (p = 0.15, Kruskal-Wallis test).
Autologous blood intracerebral hemorrhage (ICH) induces cell-specific activation of RIPK1 and MLKL
To determine cell-specific RIPK1 and MLKL responses after ICH, mice were subjected to autologous blood ICH and specific brain cell types were isolated by immunopanning. Purity of the isolated cells
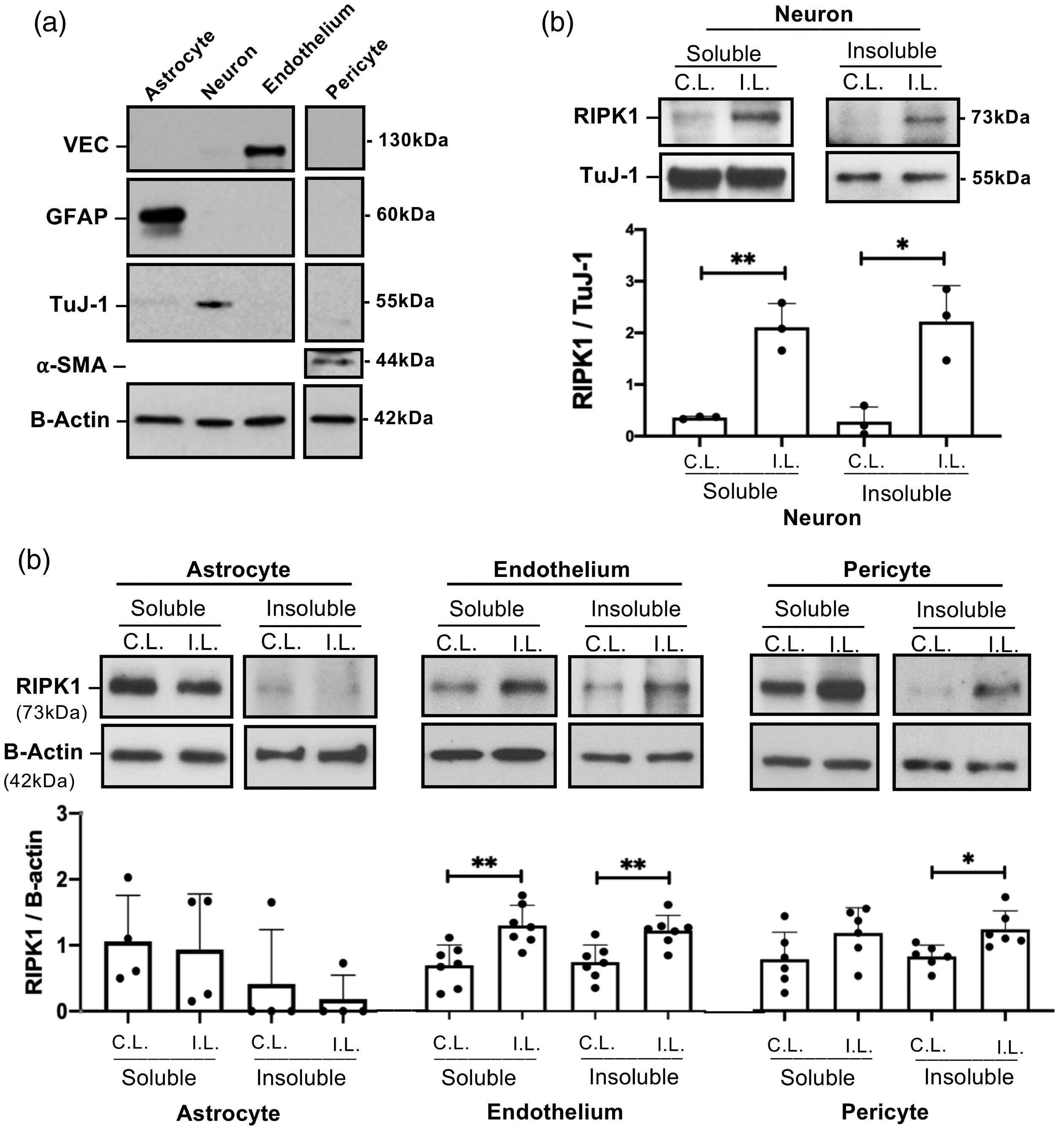
Cell-specific activation of RIPK1 after intracerebral hemorrhage (ICH). (a) Purity of isolated cell populations assessed by western blot using antibodies to glial fibrillary acidic protein (GFAP), neuron-specific class III beta-tubulin (TuJ-1), vascular endothelial cadherin (VEC), and alpha-smooth muscle actin (a-SMA). (b) RIPK1 was increased in both soluble and insoluble cell fractions in neurons isolated from ipsilateral (I.L.) hemispheres of wild-type (WT) mice 12 hours after ICH (**p < 0.005, *p < 0.05 vs. contralateral (C.L.) hemispheres, n = 3/group). (c) Increased RIPK1 in the detergent-insoluble fraction was detected in endothelium and pericytes isolated from ipsilateral hemispheres at 24 hours after ICH (**p < 0.005 vs. contralateral hemisphere, n = 7/group and *p < 0.05 vs. contralateral hemisphere, n = 6/group), but not in astrocytes (n = 3/group, p = ns, Mann Whitney U-test).
In whole cell lysates, phospho-MLKL (pMLKL) was increased in ipsilateral hemispheric neurons and astrocytes but not endothelium from injured WT mice (Figure 2(a)) and was not detected at all in isolated pericytes (not shown). In contrast, total MLKL protein expression decreased in isolated neurons and pericytes but increased in astrocytes and endothelium of ipsilateral compared to contralateral hemispheres
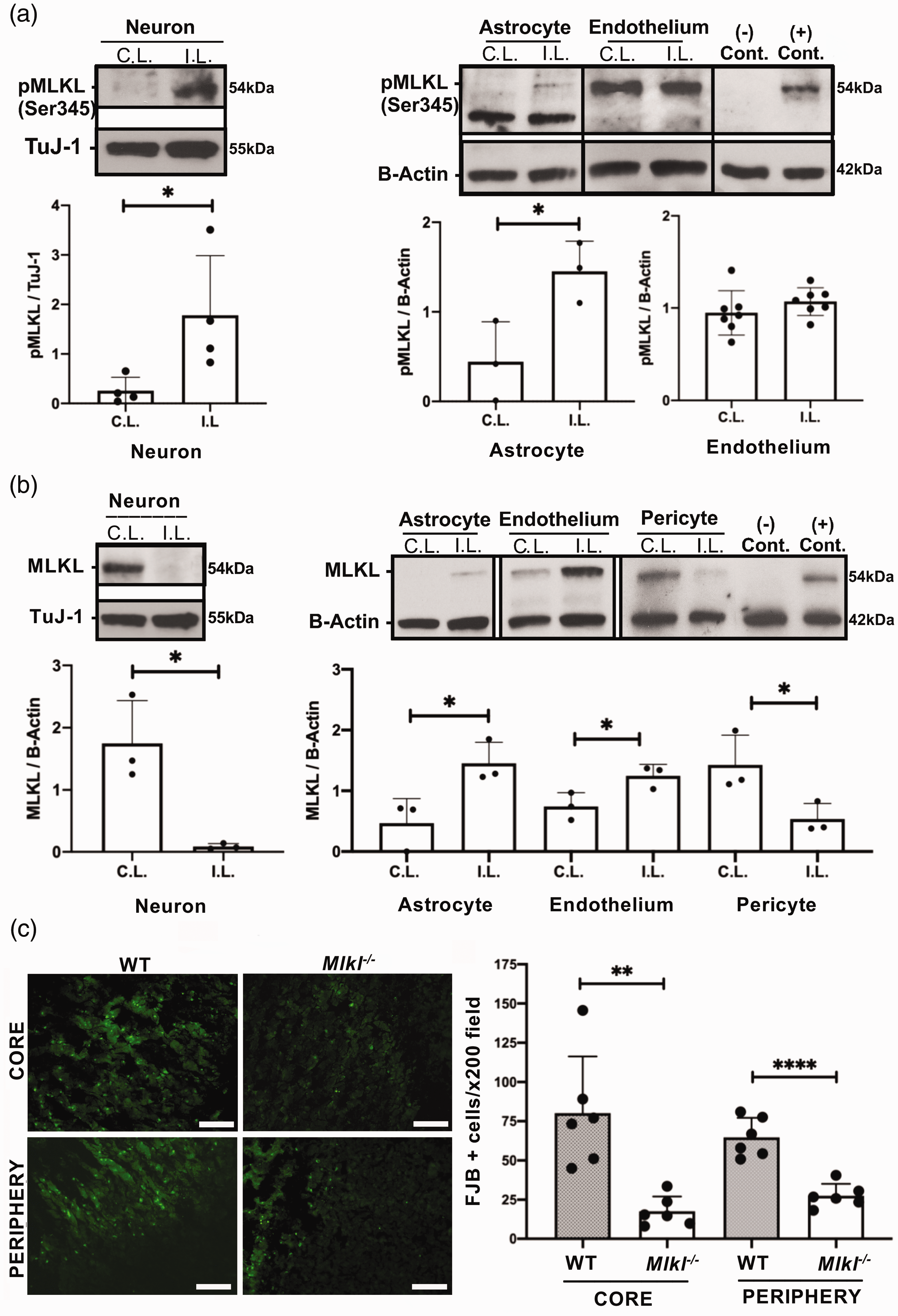
Cell-specific activation of MLKL after intracerebral hemorrhage (ICH). (a) MLKL phosphorylation (Ser345) was increased in neurons at 12 h and astrocytes at 24 h after ICH (*p < 0.05, n = 3/group) but not in endothelium (p = 0.28, n = 7/group). In isolated pericytes, pMLKL was not detected at 24 h (n = 3, not shown). (–) control: CD31+ cells from ipsilateral hemisphere of Mlkl-/- mouse at 24 h; (+) control, RAW 264.7 cells treated with SMAC mimetics to induce necroptosis. (b) MLKL was decreased in neurons isolated from the ipsilateral hemisphere of WT mice 12 hours after ICH and in pericytes isolated at 24 h (*p < 0.05, n = 3-4/group). MLKL expression was increased in astrocytes and endothelium from ipsilateral hemispheres (*p < 0.05, n = 3/group). (−) and (+) controls same as in panel a. (c) Left panel: Representative photomicrograph of fluoro-Jade B-positive cells in core and periphery of the hemorrhagic lesion in wild type (WT) and Mlkl−/− mice. Right panel: Fluoro-Jade B (FJB) positive cells were significantly reduced in core and peripheral hemorrhage regions of Mlkl−/− mice compared to WT at 48 hours after ICH (**p < 0.005, ****p < 0.0005 vs. WT, n = 6/group). Scale bars, 100 µm.
Mlkl deficiency protects against acute neuronal cell death after ICH
We previously reported reduced acute neuronal cell death after ICH in RIPK1 kinase-dead knock-in mice. 8 We next sought a functional role for MLKL in the autologous blood ICH model. Compared to WT, Mlkl-/- mice had significantly reduced Fluoro-Jade B+ (FJB+) cells in core and peripheral hemorrhagic regions at 48 hours after ICH (Figure 2(c)). No differences in neuron numbers or Hoechst+ cell counts were observed in uninjured striatum between WT and Mlkl-/- mice (Supplemental Figure II). In addition, endothelial cell loss within the hemorrhagic lesion and astrocyte coverage around the lesion were similar between WT and Mlkl−/− at 48 h after ICH (Supplemental Figure III, IV).
RIPK1 kinase inactivation and Mlkl deficiency reduce blood-brain barrier (BBB) permeability after ICH
At 24 h, ICH caused significant BBB permeability to Evans blue (EB) (Figure 3(a) and (b)
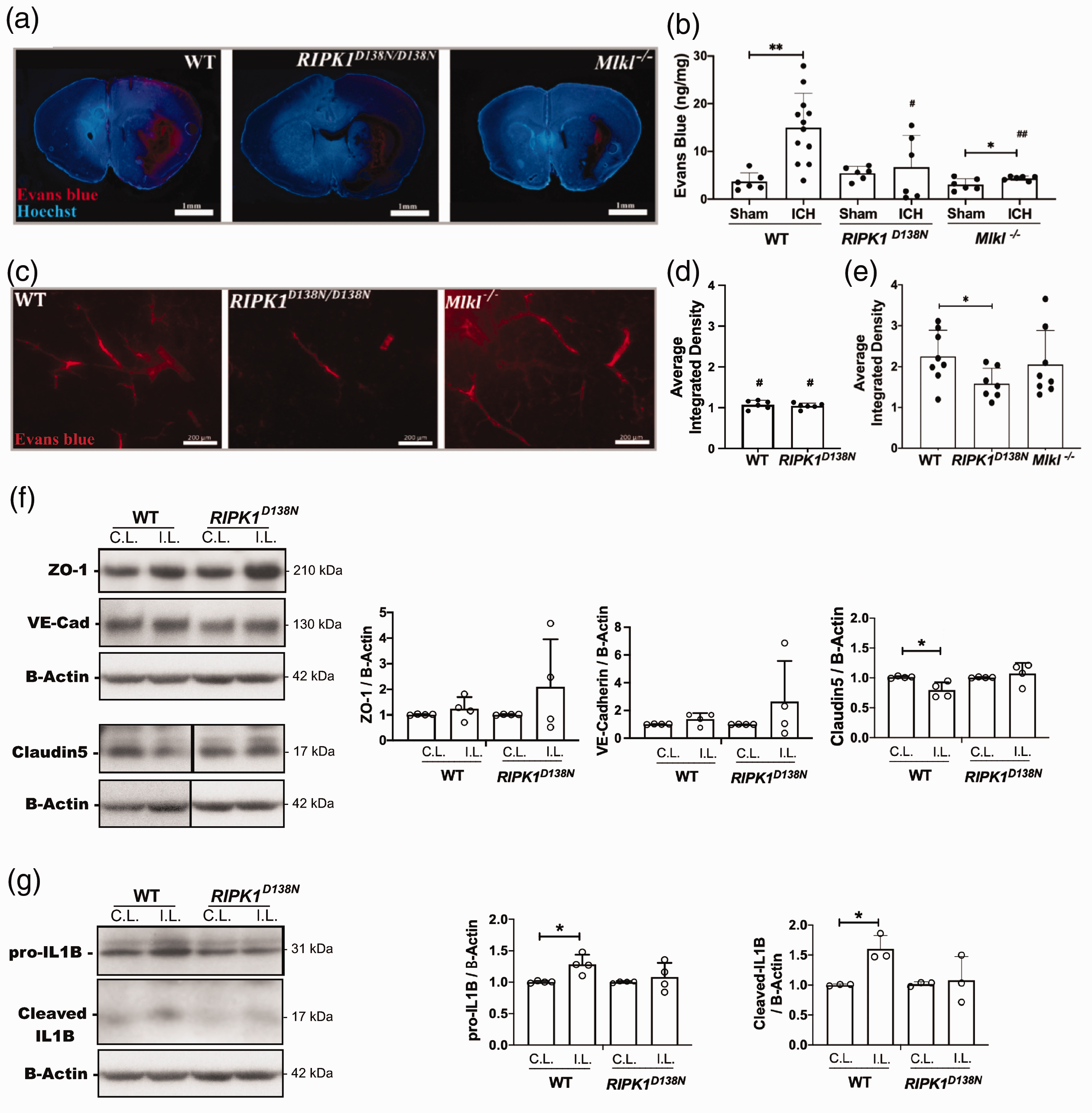
RIPK1 and Mlkl contribute to blood-brain barrier damage after intracerebral hemorrhage (ICH). (a) Representative photomicrographs (1X) of the ICH lesion at 24 h showing Evans Blue extravasation (red). (b) Spectrophotometric quantification of extravasated Evans blue (EB) in ipsilateral hemispheres of wild-type (WT), RIPK1D138N/D138N, and Mlkl−/− mice. (**p < 0.005 and *p < 0.05 vs. sham; #p < 0.05 and ##p < 0.005 vs. WT ICH, n = 6 sham and 6-12 ICH/group). (c) Left panel: Representative photomicrographs of injured striatum at 30 d after ICH showing Evans blue dye
RIPK1 kinase inactivation, but not Mlkl deficiency, reduces brain edema after ICH
Sham ICH did not increase brain water content in ipsilateral cortex or striatum, however at 24 h post ICH, brain water content was significantly increased in ipsilateral cortex and striatum of WT and Mlkl-/- mice (Figure 4(a) and (b)). In contrast, compared to contralateral, brain water content was not increased in ipsilateral cortex or striatum of RIPK1D138N/D138N mice (Figure 4(b)). Decreased brain edema in RIPK1D138N/D138N mice was not explained by differences in hematoma volume at 24 or 72 h (Figure 4(c)).
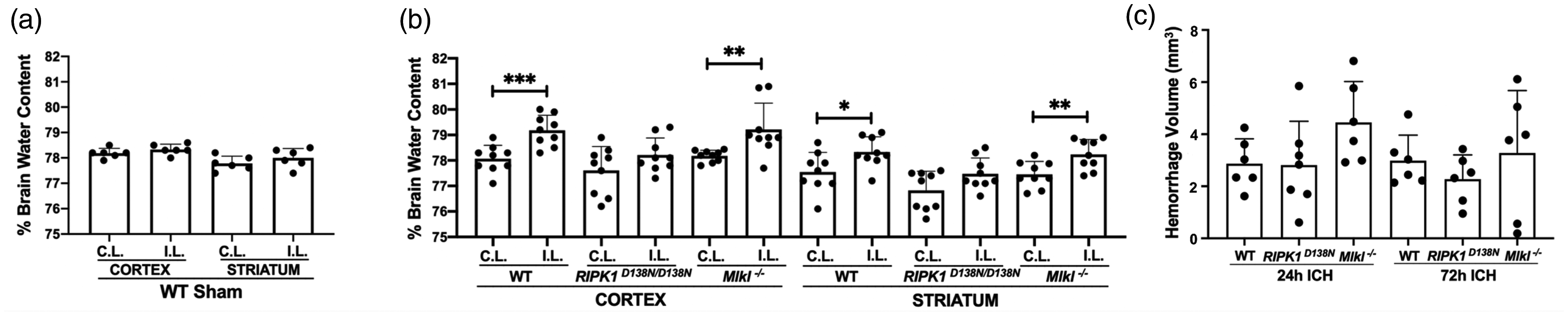
Reduced brain edema in RIPK1D138N/D138N mice at 24 h after intracerebral hemorrhage (ICH). (a) Sham ICH did not induce brain edema in WT mice. (b) Brain water content differed significantly between contralateral (C.L.) and ipsilateral (I.L.) hemispheres in cortical and striatal brain regions of wild-type (WT) and Mlkl knock-out (Mlkl-/-) but not RIPK1 kinase dead (RIPK1D138N/D138N) mice at 24 hours after ICH (***p < 0.005, **p < 0.01, and *p < 0.05, n = 9/group; RIPK1 D138N/D138N data analyzed by Mann Whitney U-test). (c) Hemorrhage volume did not differ among groups at 24 or 72 h after ICH (n = 6-7/group, p = ns, ANOVA).
RIPK1 kinase inactivation and Mlkl deficiency improve physiological and behavioral outcomes after ICH
As expected, 8 weight loss differed in sham (p < 0.01 for group) and injured (p < 0.01 for group) RIPK1D138N/D138N mice compared to WT (Figure 5(a) and (b)), however relative weight loss did not differ after sham or ICH in Mlkl-/- vs. WT (Figure 5(a) and (c)). In rotarod analyses, an overall group effect was detected by RM ANOVA that included all sham and ICH groups (p < 0.02 for group). No differences were observed among sham groups in rotarod performance (p = 0.12, Figure 5(d)), and no differences in rotarod performance were observed between sham and their corresponding injured WT, RIPK1 D138N/D138N , or Mlkl-/- groups (p > 0.1 for all comparisons). However, injured RIPK1 D138N/D138N mice performed significantly better on the rotarod test (Figure 5(e)) compared to injured WT (p < 0.01 for group). Similar to RIPK1 D138N/D138N mice, injured Mlkl-/- mice performed significantly better on rotarod testing compared to injured WT (p < 0.05 for group, Figure 5(f)), but performance seemed to return to baseline on days 21 and 28.
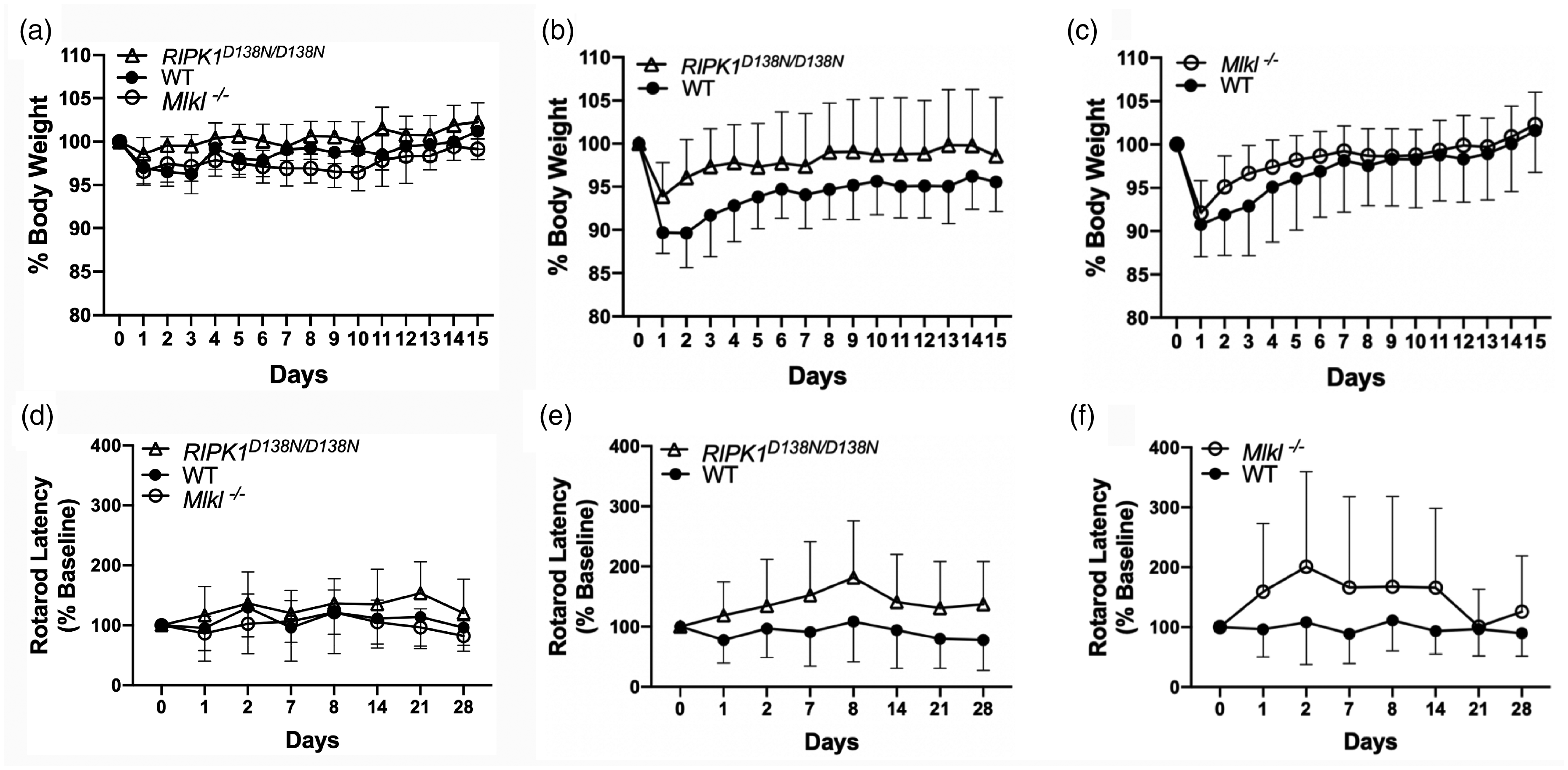
Physiological and behavioral outcome of RIPK1 kinase dead (RIPK1D138N/D138N) and Mlkl knock-out (Mlkl-/-) mice after intracerebral hemorrhage (ICH). (a) Changes in body weight as a percentage of baseline in sham injured mice. RIPK1D138N/D138N mice had less weight loss after sham injury compared to wild type (WT) (p < 0.01 for group), whereas no difference between Mlkl-/- and WT was observed (n = 11–12/group). (b) Injured RIPK1 D138N/D138N had reduced weight loss and faster weight recovery after ICH compared to WT (p < 0.05 for group, RM ANOVA). (c) No differences between WT and Mlkl−/− groups were observed for weight loss and weight recovery after ICH (n = 20–22/group). (d) Rotarod performance in sham injured mice did not differ among groups (n = 11–12/group). (e) Rotarod performance was improved after ICH in RIPK1 D138N/D138N (p < 0.01 for group, n = 20/group) and in (f) Mlkl−/− (p < 0.05 for group, n = 20/group) vs. WT.
Discussion
To our knowledge, this is the first study to assess cell specific RIPK1 and MLKL activation and to compare genetic inhibition of RIPK1 kinase activity to MLKL deficiency in an ICH model. RIPK1 and MLKL were activated in specific cell types acutely after ICH, with RIPK1 activation detected in neurons, endothelial cells, and pericytes but not in astrocytes, and MLKL activation detected in neurons and astrocytes. Compared to WT, mice deficient in Mlkl (Mlkl-/-) and RIPK1 kinase activity (RIPK1D138N/D138N) had improved blood brain barrier (BBB) permeability and post-injury rotarod performance, and reduced acute neuronal death, 8 whereas RIPK1D138N/D138N mice additionally had more days of reduced weight loss, reduced brain edema, and more durable BBB protection in the chronic period after ICH. The results confirm a significant contribution of MLKL, and an important role for RIPK1 kinase function, as disease drivers in an autologous blood ICH model.
Western blot analysis of neurons isolated from ICH hemispheres showed significant RIPK1 activation by 12 h, defined as increased RIPK1 expression in mild detergent-insoluble fractions, 4 and MLKL activation, defined as phosphorylation of MLKL at Ser 345. 21 In support for a functional role for MLKL in neuronal death, FJB+ neurons were significantly decreased in Mlkl-/- vs. WT mice at 48 h, a time of peak cell death in our ICH model. 8 These data are the first that we know of to demonstrate in vivo evidence for neuronal necroptosis in a preclinical ICH model using genetic tools. In contrast, we did not observe increased pMLKL in injured endothelium or reduced endothelial cell loss at 48 h in Mlkl-/- vs. WT mice, arguing against necroptosis as a predominant acute death mechanism in endothelium after ICH.
RIPK3/MLKL-mediated necroptosis of astrocytes influences inflammatory phenotypes in cerebral ischemia and spinal cord injury models.22,23 pMLKL was increased in astrocytes after ICH but GFAP+ astrocyte coverage did not differ between Mlkl-/- and WT at 48 h. It is possible that in astrocytes, necroptosis is redundant to other modes of cell death such as ferroptosis. 24 Alternatively, phosphorylation of MLKL may be required but not sufficient for astrocyte death. Phosphorylated MLKL can be exported in extracellular vesicles or degraded by lysosomes- mechanisms that may ensure that only signals of sufficient intensity induce necroptosis. 12 Thus, cells with active MLKL may not commit to death but can be resuscitated in some conditions, which suggests the existence of additional regulators. 25 It is also possible that astrocytes may proliferate and die at different rates after ICH in Mlkl-/- vs. WT mice, making interpretation of astrocyte coverage data at a single time point challenging. Thus, our data do not rule out a role for necroptosis in astrocyte death, rather further studies are needed to definitively resolve this question.
Interestingly, MLKL but not RIPK1 was activated in astrocytes isolated from ipsilateral hemispheres at 24 hours after ICH. Lack of detection of increased RIPK1 in the detergent-insoluble fraction of astrocytes could mean that RIPK1 is not required for MLKL phosphorylation. Indeed, RIPK3 phosphorylation of MLKL, a prerequisite for necroptosis, does not necessarily require RIPK1 in all contexts. 26
While pMLKL was increased, total MLKL was decreased in ipsilateral vs. contralateral hemispheric neurons early after ICH. In an ischemic stroke model, decreased brain MLKL expression was associated with increased MLKL ubiquitination, however functional analysis of proteasomal degradation of MLKL was not reported in that study. 27 HSP90 is required for MLKL stability, and loss of HSP90 function results in MLKL destabilization and rapid degradation by the ubiquitin-proteasome pathway. 28 It is possible that MLKL undergoes proteasomal degradation in injured neurons in an attempt to limit necrotic death after ICH. Other mechanisms that might contribute to loss of MLKL include cleavage by caspase-8 29 and transcriptional regulation.
BBB damage was robust in WT and was significantly reduced at 24 h in RIPK1D138N/D138N and Mlkl-/- mice. Previous studies using pharmacological inhibitors suggested a role for RIPK1 in BBB damage in subarachnoid hemorrhage, ischemic stroke, and multiple sclerosis models.30–34 Though our data do not support a role for reduced endothelial necroptosis, cell death-independent mechanisms of RIPK1 could also mediate BBB damage. RIPK1 promotes vascular permeability enabling tumor cell metastasis in a lung cancer model 35 and RIPK1 kinase dead (K45A) mice had decreased BBB damage attributed to reduction in blood monocytes in an experimental autoimmune encephalitis model. 34
We detected RIPK1 but not MLKL activation in injured pericytes, suggesting the possibility that RIPK1 signaling in this cell type might also contribute to BBB damage independent of necroptosis. BBB integrity could be disrupted after ICH by dissociation of pericytes from the endothelial membrane.
36
Other possible BBB mechanisms that might be influenced by RIPK1 and MLKL include maintenance of tight and adherens junction proteins and production of pro-inflammatory cytokines.
36
RIPK1D138N/D138N mice had preserved claudin-5 and reduced activation of interleukin-1 beta in brain homogenates at 24 h after ICH. These data support the premise that blood-brain barrier proteins and interleukin-1 beta are targets of RIPK1 in the setting of ICH, but more work is needed to provide a fuller understanding of the underlying mechanisms
BBB disruption is a prequel to dementia in Alzheimer’s disease patients, 37 and recent studies have shown an increased risk of cognitive impairment and early dementia in survivors of ICH.38–42 We therefore examined BBB damage at early and chronic time points in the current study. Interestingly, BBB damage was similar in Mlkl-/- and WT mice at 30 d, whereas RIPK1D138N/D138N mice remained protected even as late as 30d after injury, again arguing against reduced necroptosis as a key mechanism at this later time point. The finding that RIPK1 kinase dead mice had lasting BBB protection may be relevant to the increased risk of dementia in ICH survivors. 39 Indeed, few studies have examined BBB damage in the chronic period after experimental stroke, 43 and no other published studies that we know of have directly examined a role for MLKL in BBB damage after acute brain injury. Further studies are needed to elucidate the mechanisms of RIPK1 mediated BBB damage in the chronic period of ICH.
Of note, protection against early BBB damage in Mlkl-/- mice did not equate to reduced brain edema, as injured Mlkl-/- mice had increased brain water content in cortex and striatum similar to WT. These findings suggest that BBB damage may not contribute significantly to brain edema at 24 h in our ICH model, or that RIPK1 and MLKL may have differential effects on other mechanisms contributing to brain edema after ICH. Importantly, hematoma volume did not differ among groups at 1 or 3 days, suggesting that differences in clot retraction or coagulation, known to be influenced by MLKL and RIPK3,44–46 do not contribute to outcome or explain reduced brain edema in RIPK1D138N/D138N mice. Since brain edema contributes to life-threatening intracranial hypertension in ICH patients, targeting RIPK1 therapeutically could conceivably help reduce morbidity (e.g., brain ischemia) and mortality (due to brain herniation) in patients with ICH.
Mlkl-/- mice were protected against post-injury rotarod deficits similar to RIPK1 kinase dead mice, however unlike RIPK1 D138N/D138N , performance in injured Mlkl-/- seemed to return to that of WT at 21 and 28 days (Figure 5). Mlkl deficiency delays demyelination in a peripheral nerve injury model. 10 If rotarod deficits induced by ICH are related to demyelination, Mlkl-/- may exert only temporary protection because demyelination of striatal axons may eventually occur later after ICH via other mechanisms. It is also possible that protection in Mlkl-/- mice might be related to differences in brain inflammation. Inhibition of necroptosis by Mlkl-/- may reduce inflammation that is induced by release of damage associated molecular patterns by necrotic cells (e.g., necroinflammation), or by inhibition of necroptosis-independent inflammation pathways induced by toll-like receptors. 11 However, inhibition of MLKL is pro-inflammatory in some contexts such as intestinal inflammation models. 47 Further studies are needed to examine the contribution of MLKL to functional outcome in ICH models.
Our study has several important limitations. First, developmental effects in genetically modified mice lacking RIPK1 kinase activity or MLKL may confound interpretation of ICH injury phenotypes. This caveat is only partially addressed by the lack of passenger DNA in these strains, and similar striatal neuron and total cell counts in Mlkl-/- and WT mice (and as previously reported in RIPK1D138N/D138N mice 8 ). We also addressed possible strain-related differences by indexing weight loss and rotarod data to pre-injury baseline measurements. To date, no inducible Mlkl-/- mice have been reported, and inducible RIPK1 deletion in intestinal epithelium causes spontaneous gut inflammation, caspase-8 mediated epithelial cell apoptosis, and death in mice, a phenotype not observed in RIPK1 kinase dead mice. 48 These data suggest that, like constitutive global RIPK1 knockout, even inducible RIPK1 knockout might be incompatible with life. Second, autologous blood injection does not model blood vessel disruption that occurs in human ICH, and autologous blood injection may cause less robust rotarod deficits compared to collagenase ICH. 49 In the current study, autologous blood ICH did not induce motor deficits below baseline in WT or transgenic mice, but WT mice were unable to further improve their scores after sham injury and ICH whereas RIPK1D138N/D138N and Mlkl-/- mice did improve after ICH. We speculate that the absence of functional RIPK1 or MLKL may lead to induction of neurotrophic factors that allow for better recovery after ICH, but further studies are needed to examine this possibility. Third, our exploratory studies did not include a comprehensive time course of cellular RIPK1 and MLKL activation, or examination of their potential interactions with age or sex in ICH. These questions need to be addressed in future studies. Fourth, we did not assess the degree of BBB damage due to the blood component of the ICH model at 30 days in Mlkl-/- mice as we did not have a sham injured group, however no protection vs. WT was observed at this later time in injured Mlkl-/-.
Conclusions
Our data reveal wide-spread RIPK1 activation in multiple CNS cell lineages following ICH. Activation of RIPK1 plays a central role in the pathogenesis of ICH, including cell death, BBB permeability, brain edema, and motor outcome. These effects may be mediated in part through the activation of MLKL-dependent necroptosis in neurons. The data support development of RIPK1 kinase inhibitors as therapeutic agents for human ICH.
Supplemental Material
sj-pdf-1-jcb-10.1177_0271678X20973609 - Supplemental material for Cell-specific activation of RIPK1 and MLKL after intracerebral hemorrhage in mice
Supplemental material, sj-pdf-1-jcb-10.1177_0271678X20973609 for Cell-specific activation of RIPK1 and MLKL after intracerebral hemorrhage in mice by Sevda Lule, Limin Wu, Aliyah Sarro-Schwartz, William J Edmiston III, Saef Izzy, Tanya Songtachalert, So Hee Ahn, Neil D Fernandes, Gina Jin, Joon Yong Chung, Siddharth Balachandran, Eng H Lo, David Kaplan, Alexei Degterev and Michael J Whalen in Journal of Cerebral Blood Flow & Metabolism
Supplemental Material
sj-pdf-2-jcb-10.1177_0271678X20973609 - Supplemental material for Cell-specific activation of RIPK1 and MLKL after intracerebral hemorrhage in mice
Supplemental material, sj-pdf-2-jcb-10.1177_0271678X20973609 for Cell-specific activation of RIPK1 and MLKL after intracerebral hemorrhage in mice by Sevda Lule, Limin Wu, Aliyah Sarro-Schwartz, William J Edmiston III, Saef Izzy, Tanya Songtachalert, So Hee Ahn, Neil D Fernandes, Gina Jin, Joon Yong Chung, Siddharth Balachandran, Eng H Lo, David Kaplan, Alexei Degterev and Michael J Whalen in Journal of Cerebral Blood Flow & Metabolism
Footnotes
Authors’ contributions
Dr. Lule, Dr. Wu, Dr. Whalen, Dr. Degterev, Dr. Lo, Dr. Balachandran, Dr. Izzy, and Dr. Kaplan planned the experiments and interpreted the data; Dr. Whalen wrote the manuscript; Dr. Lule, Dr. Wu, Aliyah Sarro-Schwartz, and Joon Chung performed Western blot and Dr. Wu performed additional immunohistochemistry experiments; Dr. Lule performed the in vivo ICH model, immunopanning, immunohistochemistry experiments and histological studies including brain edema, clot volume, and BBB; So Hee Ahn, and Neil Fernandes performed microscopy and cell counting; Tanya Songtachalert, Gina Jin and William Edmiston performed rotarod experiments. Dr. Whalen and Dr. Lule performed statistical analyses.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by National Institute of Neurological Disorders and Stroke RO1NS071072 (Dr. Whalen), R21AI124049 and R21NS111395 (Dr. Degterev), 1R01CA190542 (Dr. Degterev and Dr. Balachandran), and R01NS092847 (Dr. Kaplan).
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.