
Abstract
Necroptosis is a newly identified type of programmed necrosis initiated by the activation of tumor necrosis factor alpha (TNFα)/Fas. Necrostatin-1 is a specific inhibitor of necroptosis that reduces ischemic tissue damage in experimental stroke models. We previously reported decreased tissue damage and improved functional outcome after controlled cortical impact (CCI) in mice deficient in TNFα and Fas. Hence, we hypothesized that necrostatin-1 would reduce histopathology and improve functional outcome after CCI in mice. Compared with vehicle-/inactive analog-treated controls, mice administered necrostatin-1 before CCI had decreased propidium iodide-positive cells in the injured cortex and dentate gyrus (6 h), decreased brain tissue damage (days 14, 35), improved motor (days 1 to 7), and Morris water maze performance (days 8 to 14) after CCI. Improved spatial memory was observed even when drug was administered 15 mins after CCI. Necrostatin-1 treatment did not reduce caspase-3-positive cells in the dentate gyrus or cortex, consistent with a known caspase-independent mechanism of necrostatin-1. However, necrostatin-1 reduced brain neutrophil influx and microglial activation at 48 h, suggesting a novel anti-inflammatory effect in traumatic brain injury (TBI). The data suggest that necroptosis plays a significant role in the pathogenesis of cell death and functional outcome after TBI and that necrostatin-1 may have therapeutic potential for patients with TBI.
Introduction
Necroptosis is a recently discovered, regulated form of programmed necrosis initiated by the activation of tumor necrosis factor alpha (TNFα) and/or Fas that is distinct from caspase-dependent apoptotic cell death (Degterev et al, 2005). In susceptible cell types such as Jurkat cells and U937 monocytes, TNFα- or Fas-induced cell death occurs by caspase-mediated apoptosis. However, in the presence of caspase inhibitors, these cell types die in response to TNFα or Fas activation with ultrastructural features of necrosis (Denecker et al, 2001a, b ; Holler et al, 2000; Matsumura et al, 2000; Vanden Berghe et al, 2002, 2004a, b; Vercammen et al, 1997, 1998a, b ). This alternative, death-receptor-mediated, necrosis-like mode of cell death has been termed necroptosis (Degterev et al, 2005).
Using an
Here, we tested the hypothesis that necroptosis contributes to cell death after traumatic brain injury (TBI) and that treatment with necrostatin-1 would reduce histopathology and functional deficits after controlled cortical impact (CCI) in mice. Necrostatin-1 reduced acute cellular plasmalemma permeability, short- and long-term tissue damage, and motor and cognitive deficits in mice subjected to CCI. The data suggest an important role for necroptosis in the pathogenesis of TBI, and suggest that necrostatin-1 may be a novel therapeutic agent for the treatment of patients with head injury and perhaps other acute central nervous system disorders that feature necroptosis as a mode of cell death.
Materials and methods
For all studies, investigators were masked to treatment during surgery, data acquisition, and analysis.
Controlled Cortical Impact
The CCI model was used as previously described with minor modifications (Bermpohl et al, 2007). The trauma protocol was approved by the Massachusetts General Hospital Institutional Animal Care and Use Committee and complied with the NIH Guide for the Care and Use of Laboratory Animals. Mice (6 to 12 weeks of age) were anesthetized with 4% isoflurane (Anaquest, Memphis, TN, USA) in 70% N2O and 30% O2 using a Fluotec 3 vaporizer (Colonial Medical, Amherst, NH, USA) and positioned in a stereotaxic frame. Anesthesia was maintained using 2% to 3% isoflurane. A 5-mm craniotomy was made using a portable drill and trephine over the left parietotemporal cortex (the center of the coordinates of craniotomy relative to bregma: 1.5 mm posterior, 2.5 mm lateral), and the bone flap was removed. Mice were then subjected to CCI using a pneumatic cylinder with a 3-mm flat-tip impounder, velocity 6 m/secs, set depth of 0.6 (for immunohistochemical studies requiring preserved cortical tissue) or 1.0 mm (for histopathological and behavioral studies), and 100 ms impact duration. The scalp was sutured closed and mice returned to their cages to recover from anesthesia.
Administration of Necrostatins and Related Compounds
Necrostatin-1 (5-[(7-chloro-l
Assessment of Lesion Volume
Morphometric image analysis (MCID, Imaging Research Inc, St Catherines, ON, Canada) was used to determine lesion volume and brain tissue loss after CCI. Mice were deeply anesthetized with isoflurane, decapitated, and brains were removed and frozen in nitrogen vapor. Coronal brain sections (12 μm) were cut on a cryostat at 0.5 mm intervals from anterior to the posterior and mounted on poly-
Evaluation of Motor and Morris Water Maze Performance
Vestibulomotor function was assessed using a wire grip test. Mice were placed on a metal wire (45 cm long) suspended 45 cm above a foam pad and allowed to traverse the wire for 60 secs. The latency that a mouse remained on the wire within a 60-sec interval was measured, and wire grip scores were quantitated using a 5-point scale (Bermpohl et al, 2006).
The Morris water maze (MWM) task was used to evaluate spatial memory performance as previously described (Bermpohl et al, 2007) with minor modifications, including the use of a 10 cm platform and the inclusion of a 60-sec probe trial after the visible platform trials. Performance in the MWM was quantitated by latency to the platform.
Administration of Propidium Iodide and Detection of Propidium Iodide-Positive Cells
Propidium iodide (PI; 10 mg/mL; Sigma) was diluted in 0.9% NaCl and 1 mg/kg was administered 1 h before killing by intraperitoneal injection in a total volume of not more than 100 μL. Mice were killed at 6 h after CCI, the brains frozen in nitrogen vapor, and cryostat brain sections (12 μm) were cut at 150 to 200 μm intervals from the anterior to posterior hippocampus. The cryostat sections were placed on poly-
Caspase-3, Neutrophil, and Astrocyte Immunohistochemistry
At 48 h after CCI, mice were anesthetized with isoflurane and the brain was rapidly removed intact and frozen at −80°C. Coronal brain sections (12 μm) were placed on poly-
Assessment of Propidium Iodide-Positive, Neutrophil, and Caspase-3-Positive Cell Counts
Cell counts were determined as previously described (Bermpohl et al, 2007). The regions of interest in the injured cortex (x 200 microscopic fields, 1,100 μm × 1,100 μm) for PI- and caspase-3-positive cell counts were a cortical field at the medial edge and a cortical field at the lateral edge of the contusion. Three coronal brain sections separated by at least 150 μm were randomly sampled from brain regions between bregma −1.90 and −2.70 for a total of six × 200 cortical fields assessed per animal. Because the depth of the × 200 fields was always 1,100 μm, comparable cortical fields were analyzed in each of the three coronal brain sections. For cell counts in the dentate gyrus, two adjacent × 200 fields (1,100 μm × 1,100 μm) encompassing the upper and lower blades of the dentate gyrus were evaluated in the three brain sections selected for cortical cell counts (above). Cell count data for each mouse were reported as the average of the cortical or hippocampal fields. In all cases, image acquisition and analyses were performed by a blinded observer.
For the estimation of neutrophils in the injured cortex, cortical brain fields (x 200; 1,100 μm × 1,100 μm) from three brain sections separated by at least 150 mm and within the approximate center of the contusion were selected for analysis. Neutrophils were photographed and quantitated in three × 200 cortical fields randomly chosen from each brain section. Thus, a total of nine × 200 fields were analyzed per mouse. From these data, an average neutrophil count per × 200 field was obtained for each animal.
Microglia Immunohistochemistry
At 48 h after CCI, mice were anesthetized with isoflurane and transcardially perfused with 4% paraformaldehyde. Coronal brain sections (12 μm) were cut at 150 to 200 mm interval. Cryostat-fixed brain sections were treated with 1% hydrogen peroxide in PBS. The slides were blocked in PBS with 3% normal goat serum for 1 h and incubated overnight at 4°C with rabbit anti-Iba-1 antibody (1:200; Wako Pure Chemical Industries Ltd, Osaka, Japan). The sections were then washed in PBS with 1% normal goat serum and incubated with biotinylated goat anti-rabbit immunoglobulin G (1:200; Vector Laboratories, Burlingame, CA, USA) antibody for 1 h. After washing, the signal was detected using an ABC kit (Vector Laboratories) and diaminobenzidine (Vector Laboratories).
Quantitation of Astrocytes and Activated Microglia in Mouse Cortex after Controlled Cortical Impact
For the analysis of glial fibrillary acidic protein-positive astrocytes or Iba-1-positive microglia, photomicrographs of × 200 fields from the medial and lateral edges of the contusion were analyzed from three brain sections as described above (cell counts). Using MCID image analysis software, activated glial fibrillary acidic protein-positive astrocytes or Iba-1-positive microglia were quantified as previously described (Bermpohl et al, 2007).
Statistical Analyses
Data are mean ± s.e.m. Cell count data were analyzed by rank sum test. Lesion volume data and probe trial data were analyzed by analysis of variance on ranks and rank sum tests. Motor and MWM test data (hidden and visible platform acquisition latencies) were analyzed by two-factor repeated measures analysis of variance (group-time). For all comparisons,
Results
All animals survived CCI and the experimental period. No overt toxic effects of necrostatin-1, inactive analog, or vehicle treatment were observed in any of the mice studied.
Necrostatin-1 Inhibits Brain Tissue Damage after Controlled Cortical Impact
In animals administered necrostatin-1 immediately before CCI (pretreated), lesion size was significantly reduced by approximately 30% to 40% compared with pretreatment with vehicle or inactive analog at 14 days (Figure 1A). Pretreatment with necrostatin-1 also reduced lesion size assessed at 35 days after CCI (Figure 1A), suggesting that protection against brain tissue damage is a long-term effect of necrostatin-1 in our CCI model. To determine the therapeutic window for necrostatin-1, drug or vehicle was administered to mice before or at various times after CCI and lesion size was assessed at 2 weeks. Figure 1B shows that administration of necrostatin-1 preinjury or at 15 but not 30 mins after CCI significantly reduced brain lesion size.
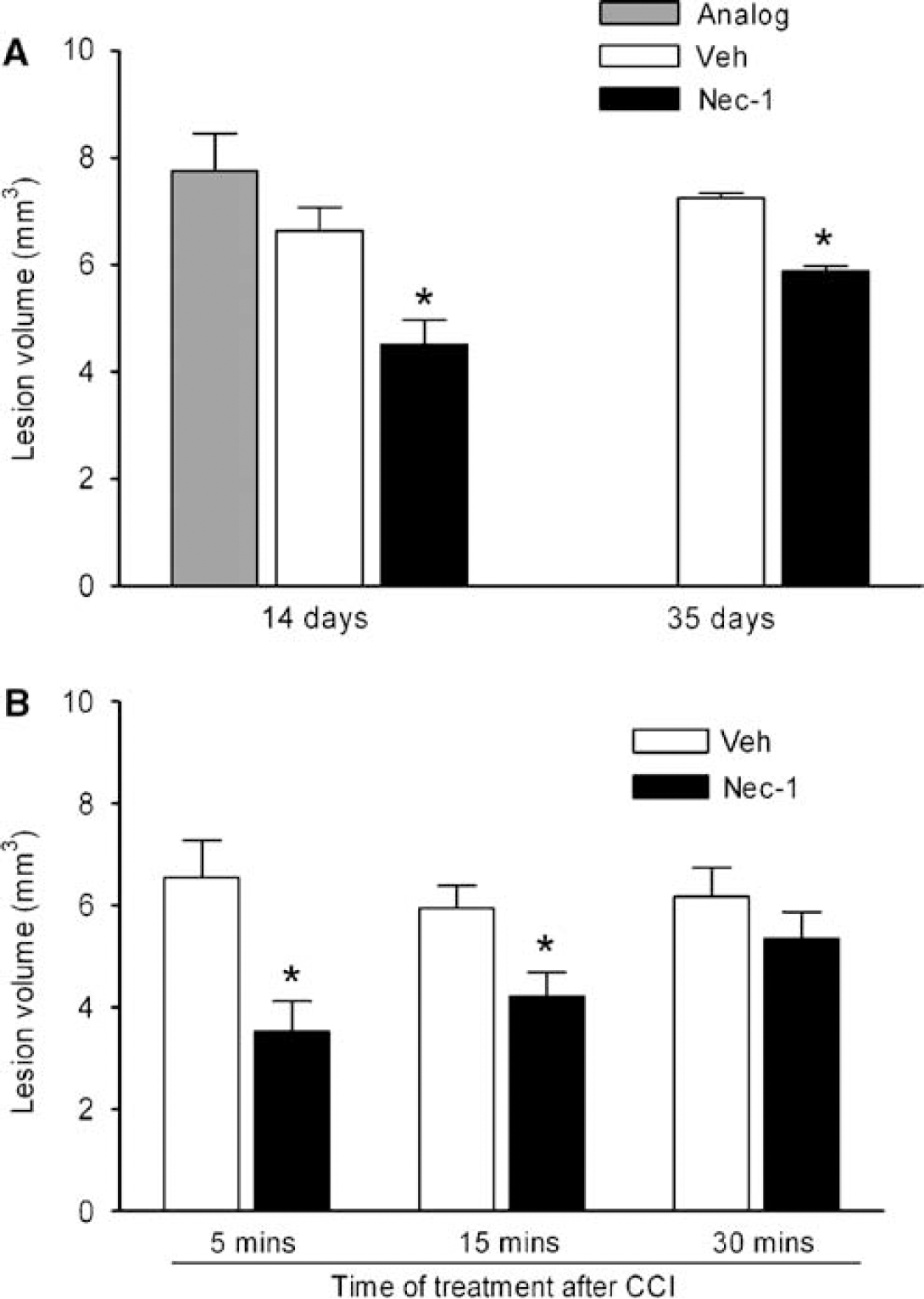
Brain lesion volume is reduced in necrostatin-1 (Nec-1)-treated mice after controlled cortical impact (CCI). (
Necrostatin-1 Improves Recovery of Motor Performance after Controlled Cortical Impact
No difference in baseline motor function before CCI was observed between groups of mice administered drug, vehicle, or inactive analog (Figure 2). In addition, sham-injured mice administered necrostatin-1 or vehicle performed similar to naive (untreated) C57B1/6 mice in pretests of motor function before MWM testing (data not shown). After CCI, recovery of motor function was significantly improved in necrostatin-1-pretreated mice versus mice treated with inactive analog or vehicle (Figure 2A,
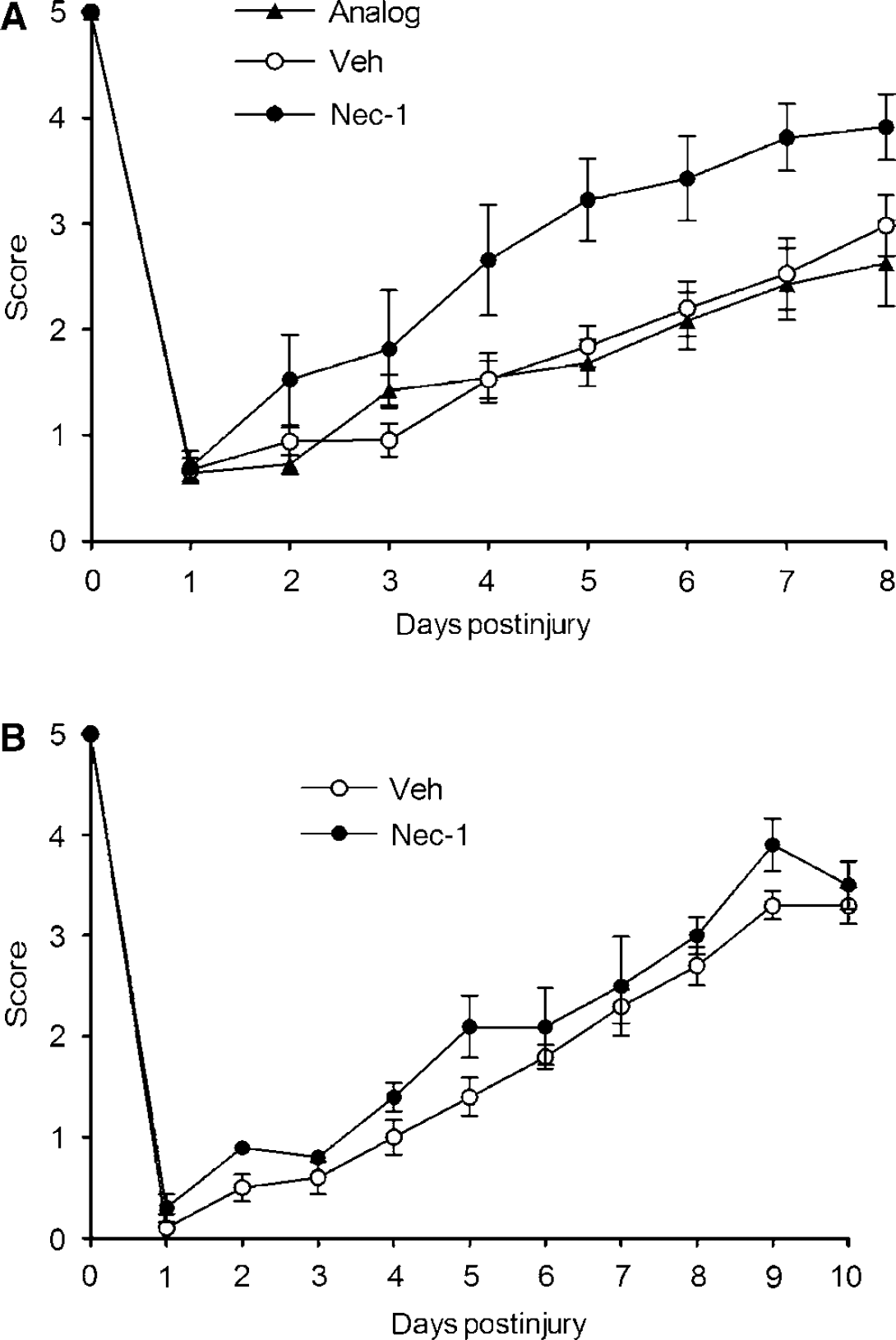
Reduced motor deficits after controlled cortical impact (CCI) in necrostatin-1 (Nec-1)-treated mice. (
Necrosatin-1 Improves Spatial Memory Acquisition after Controlled Cortical Impact
To examine the effect of necrostatin-1 on postinjury cognitive function, performance of injured mice pretreated with necrostatin-1, inactive analog, or vehicle was assessed in a MWM paradigm. Mice administered necrostatin-1 before CCI had significantly improved latency to the hidden platform compared with vehicle- or inactive analog-treated mice, whereas all three groups had similar performance in visible platform tests (Figure 3A). The protective effect of necrostatin-1 in injured mice could not be attributed to the effects on baseline MWM performance as sham-injured mice administered necrostatin-1 or vehicle did not differ in acquisition of hidden or visible platform tests (Figure 3B). Results of probe trial testing are shown in Figure 3E. The injured mice pretreated with necrostatin-1 performed similar to the sham-injured mice (administered vehicle or necrostatin-1) and significantly better compared with injured vehicle-or inactive analog-treated mice. No difference in probe trials was observed between the sham-injured vehicle- or drug-treated mice. In a separate set of experiments, mice were treated with necrostatin-1 or vehicle at various times after CCI and MWM performance was assessed as above. Although no significant differences between groups were observed in hidden platform trials with limited numbers of mice per group (Figures 3C and 3D), mice administered necrostatin-1 at 5 or 15 mins after injury had significantly improved probe trial performance versus vehicle-treated animals (
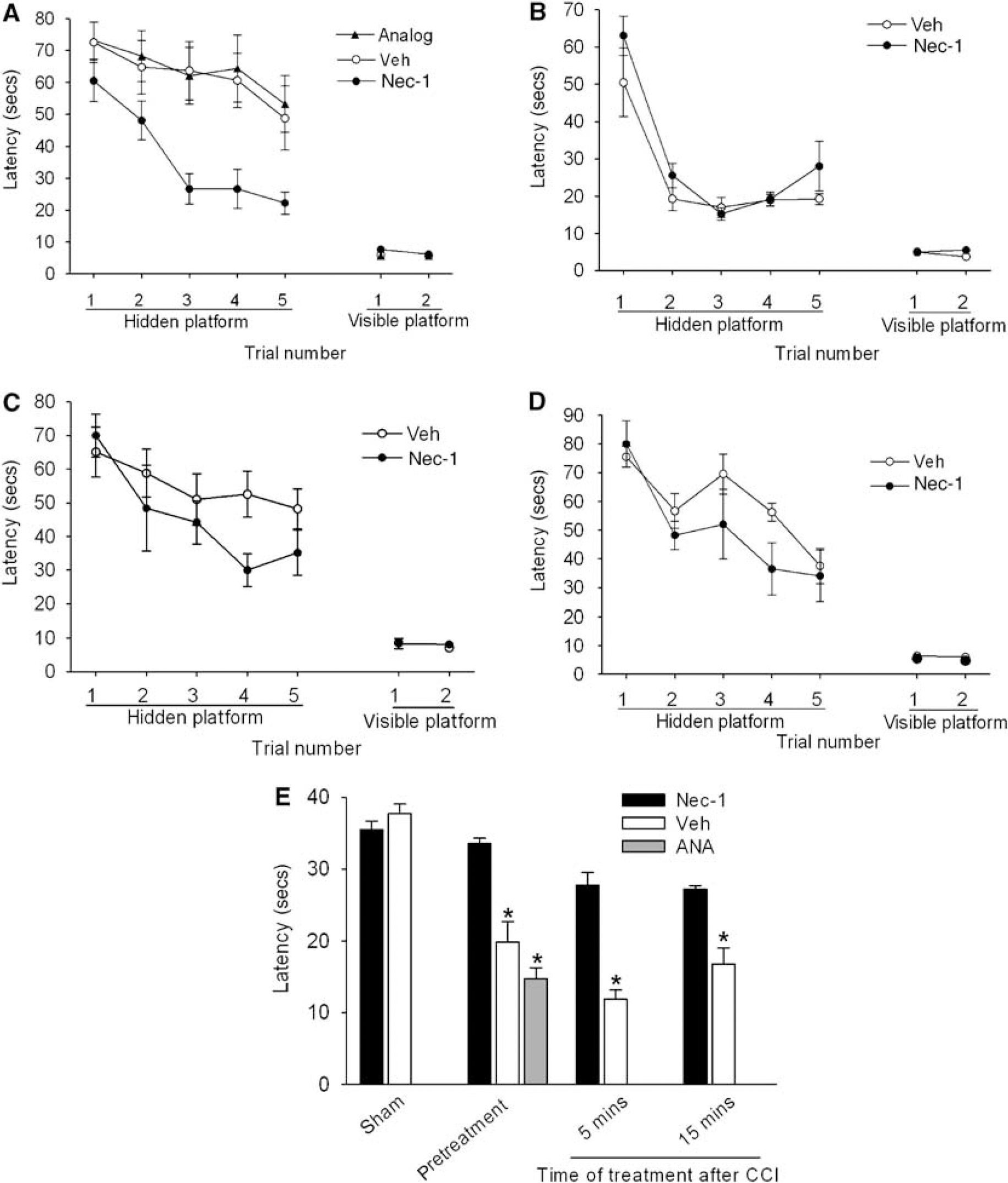
Necrostatin-1 (Nec-1) treatment improves Morris water maze (MWM) performance after controlled cortical impact (CCI). (
Necrostatin-1 Reduces Acute Plasmalemma Permeability in Injured Cells after Controlled Cortical Impact
Plasmalemma permeability is a hallmark of necrotic, including necroptotic, cell death. To begin to address cellular mechanisms of reduced postinjury tissue damage, we assessed the effect of necrostatin-1 treatment on loss of plasmalemma integrity in the cortical and hippocampal brain regions using
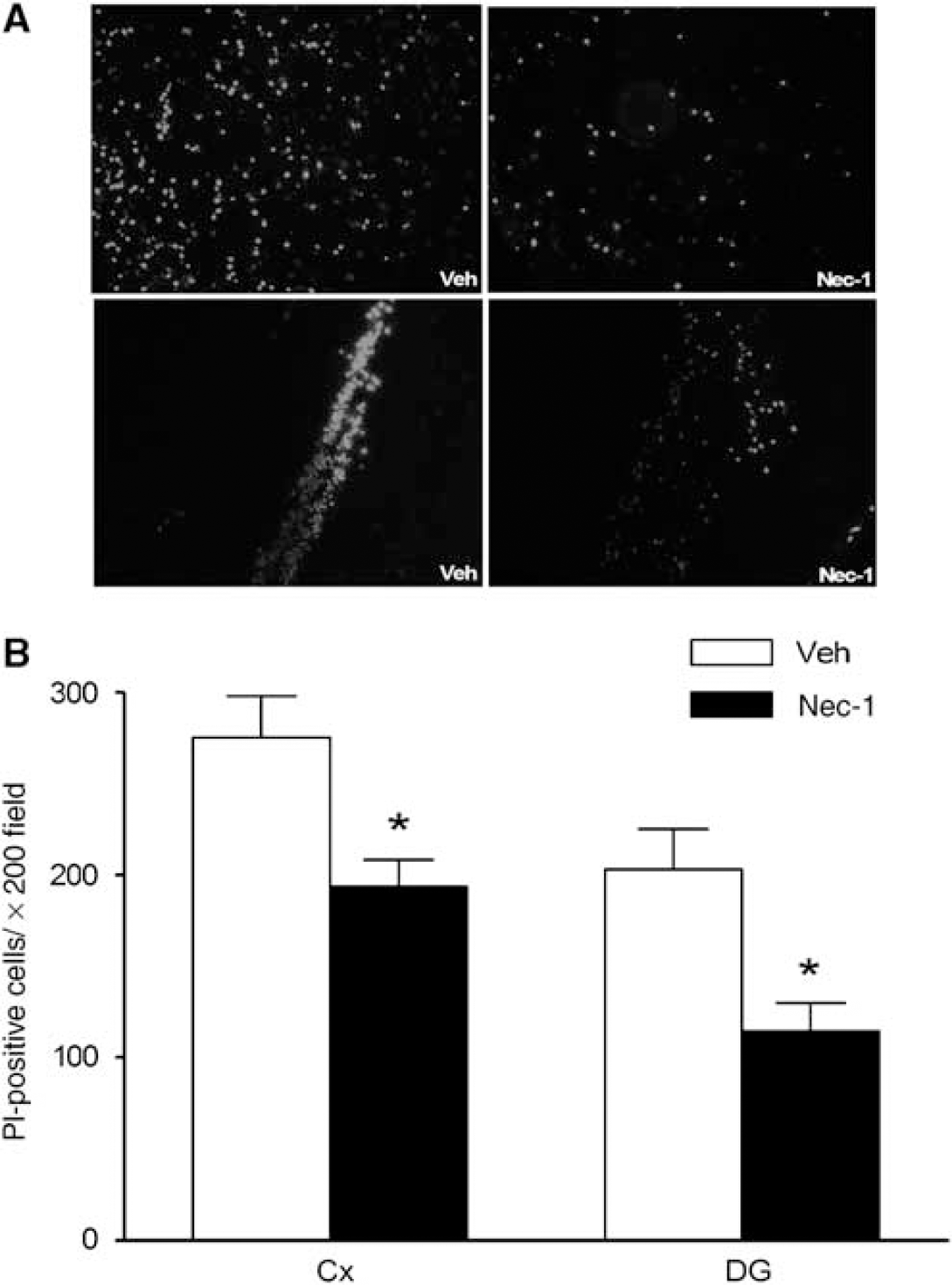
Pretreatment with necrostatin-1 (Nec-1) reduces propidium iodide (PI)-positive cells at 6 h after controlled cortical impact (CCI) in the injured cortex and hippocampus. (
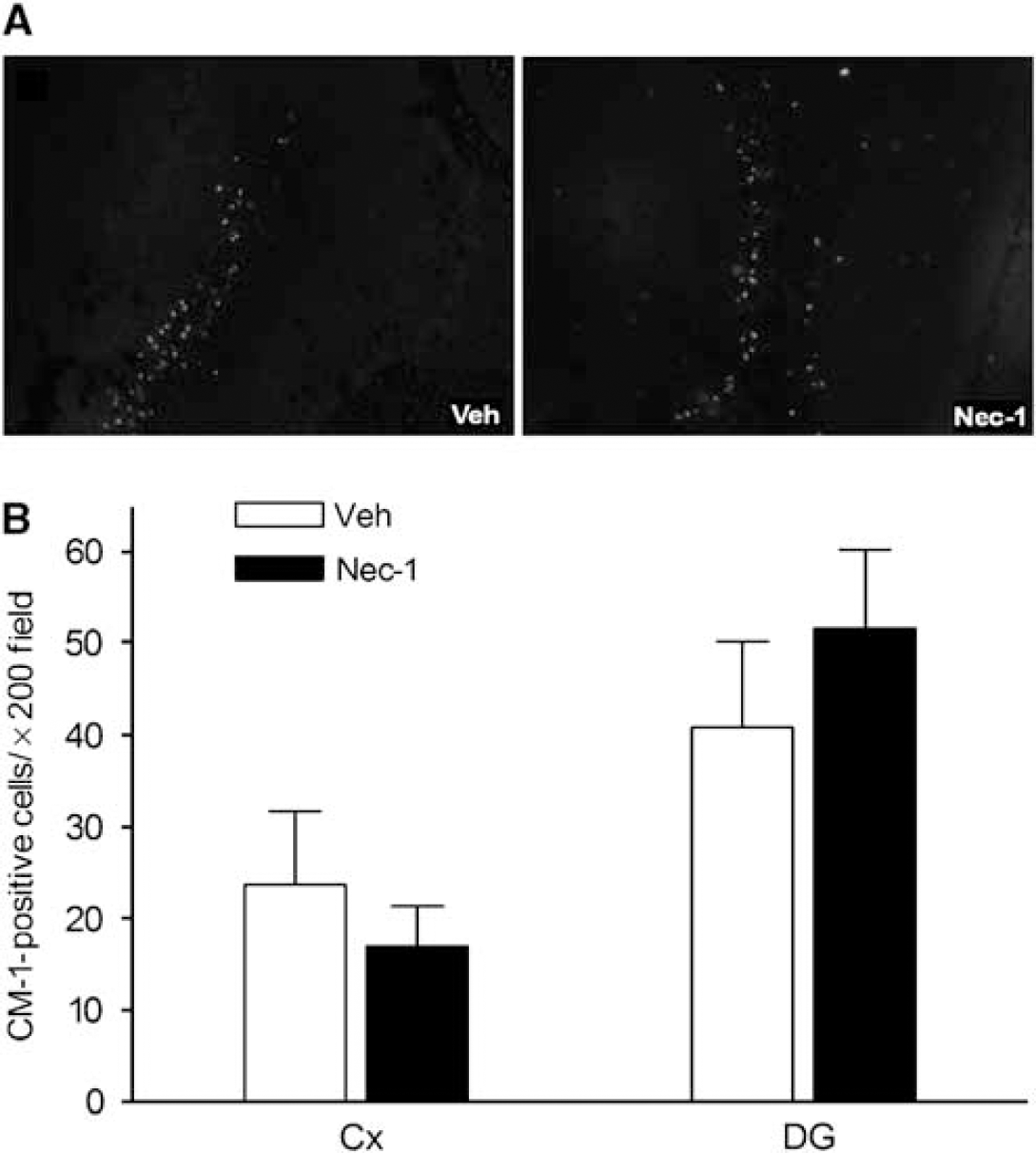
No effect of necrostatin-1 (Nec-1) on caspase-3-positive cells after controlled cortical impact (CCI). Necrostatin-1 or vehicle (Veh) was administered to mice (
Necrostatin-1 Inhibits Cellular Neuroinflammation after Controlled Cortical Impact
To assess the potential effect of necrostatin-1 on neuroinflammation after CCI, brain neutrophil counts, astrocytosis, and the microglial response to CCI was assessed in the injured mouse cortex at 48 h in mice pretreated with necrostatin-1 or vehicle. This time point was chosen because all the three of these inflammatory markers are robust in the injured brain at 48 h (Bermpohl et al, 2007). Figure 6A shows that brain neutrophil counts in the injured cortical brain regions were decreased by approximately 50% in necrostatin-1-treated mice. Similarly, necrostatin-1 markedly inhibited the staining intensity of activated microglia detectable with Iba-1 antibody (Figure 6B). In contrast, immunohistochemical density analysis of astrocytes showed no differences between groups (data not shown).
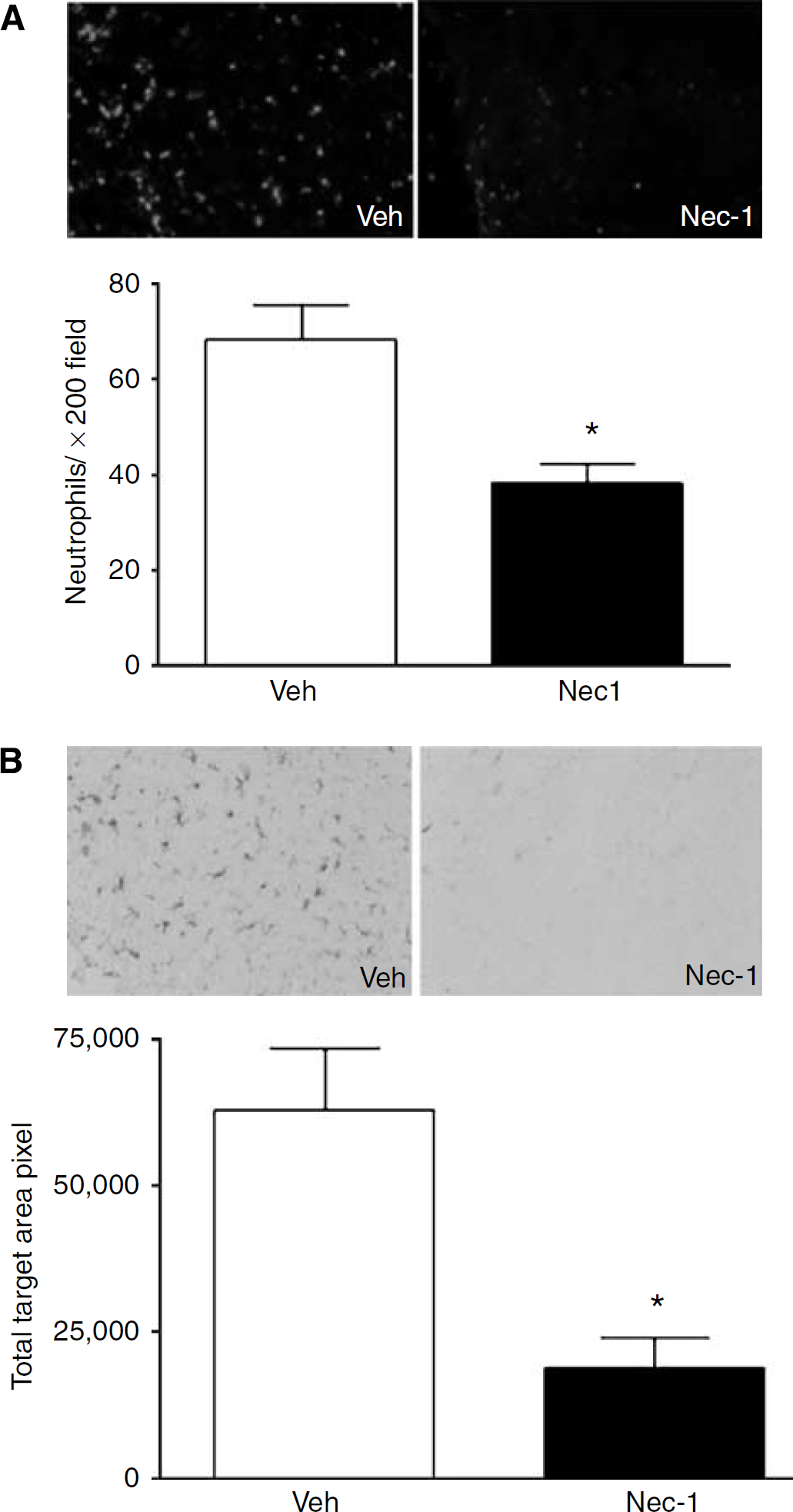
Pretreatment with necrostatin-1 (Nec-1) reduces neuroinflammation assessed at 48 h after controlled cortical impact. (
Discussion
This study suggests a possible role for necroptosis in the pathogenesis of cell death and neurologic dysfunction after TBI. Necrostatin-1, a specific inhibitor of necroptosis (Degterev et al, 2005), reduced acute cellular injury, brain tissue damage, motor dysfunction, and spatial learning deficits after CCI in mice. Indeed, necrostatin treatment recapitulated all of the protective effects of TNFα/Fas knockout in our CCI model, consistent with the known mechanism of action of necrostatins in cultured cells (Degterev et al, 2005). Beneficial effects of necrostatin-1 on learning and memory deficits were observed even when necrostatin-1 was administered 15 mins after CCI. Necrostatin-1 treatment also markedly suppressed the neutrophil and microglial responses to CCI, suggesting a heretofore-unreported anti-inflammatory effect in acute brain injury.
Two lines of evidence suggest that necrostatin-1 may inhibit necroptosis in our CCI model. First, pretreatment with necrostatin-1 reduced the numbers of cells with the loss of plasmalemma integrity, a hallmark of necroptosis, in the cortex and dentate gyrus 6 h after CCI. Second, necrostatin-1 did not reduce the number of caspase-3-positive cells in cortex or dentate gyrus at 48 h after CCI, a time of maximal cleaved caspase-3 expression in our CCI model. These findings are consistent with
On the basis of prior studies showing a beneficial effect of necrostatin-1 in ischemic brain injury and our previous report of reduced tissue damage in mice lacking functional TNFα and Fas (Bermpohl et al, 2007), we anticipated that necrostatin-1 would reduce posttraumatic lesion size after CCI. Pretreatment with necrostatin-1 significantly reduced cavitary lesion size with a magnitude similar to that obtained in rodents using cysteine protease inhibitors or apoptosis gene knockout mice (Clark et al, 2000, 2007; Bermpohl et al, 2006, 2007). Moreover, necrostatin-1 treatment conferred long-lasting reduction in postinjury brain tissue damage after CCI, an effect not seen with strategies targeting caspases or Bid (Bermpohl et al, 2006; Clark et al, 2007). These data suggest that necroptotic mechanisms contribute to short- and long-term brain tissue loss after CCI. Moreover, necrostatin-1 produced functional improvement after CCI (discussed below), a beneficial effect that is not necessarily attributable to reduced tissue damage in this model, as tissue damage does not always correspond to behavioral outcome after CCI (Bermpohl et al, 2006; Clark et al, 2007).
The therapeutic window for necrostatin-1 to reduce lesion size was 5 to 15 mins under the conditions of our intracerebroventricular (ICV) administration protocol. In contrast, ICV administration of necrostatin-1 significantly reduced ischemic infarct size even when administered as late as 6 h after experimental MCAO and reperfusion in mice (Degterev et al, 2005). The different therapeutic window for necrostatin-1 in MCAO versus CCI may reflect different kinetics of necroptosis in the two brain injury models. In support of this possibility, the onset of loss of cellular plasmalemma integrity, a hallmark of necroptosis, peaks at 60 mins after CCI in the injured cortex and hippocampus (Whalen et al, 2008). Early loss of plasmalemma integrity was also reported in another (closed head injury) TBI model (Farkas et al, 2006). In contrast, significant plasmalemma damage was not detected until 6 h after reperfusion in mice subjected to MCAO (Unal Cevik and Dalkara, 2003; Unal-Cevik et al, 2004). The short therapeutic window for necrostatin-1 suggests that necroptosis is rapidly initiated after CCI, and implicate necroptotic mechanisms as one possible cause of traumatic membrane poration in the CCI model (Farkas et al, 2006; Kristensen et al, 2003; Lee et al, 1999). In support of this notion, induction of TNFα and Fas receptor and assembly of TNFR1 and Fas DISC are rapid events measured in minutes to hours after experimental TBI (Beer et al, 2000; Lotocki et al, 2004; Qiu et al, 2002).
Mice pretreated with necrostatin-1 had modest but significant improvement in posttraumatic motor dysfunction. Necrostatin-1 also reduced sensorimotor deficits after MCAO in mice, although with a much longer therapeutic window compared with CCI (Degterev et al, 2005). In this study, necrostatin-1 administration at 5 mins after CCI was not effective in reducing postinjury motor deficits. Thus, necroptotic pathways that influence posttraumatic motor deficits may have a shorter therapeutic window for necrostatin-1 treatment compared with mechanisms responsible for cell death and cognitive outcome in the CCI model.
An important finding in our study was that necrostatin-1 treatment partially reduced cognitive deficits induced by CCI. Compared with vehicle or inactive analog, necrostatin-1 led to a significant improvement in injured mice in hidden platform and probe trials (the latter being a sensitive and specific test of spatial learning). The finding that necrostatin-1-treated and control groups performed equally well on visible platform trials suggests that improved MWM performance is not attributable to differences in motivation, swim speed, sensorimotor function, or other nonspatial brain functions, but likely represents improved spatial memory acquisition and retention. Necrostatin-1 improved probe trial performance in injured mice even when administered at 15 mins after CCI, suggesting that necrostatin-1 might be useful for the treatment of postinjury cognitive deficits in humans with TBI. Even with a narrow therapeutic window, rapid postinjury administration of necrostatins using an intravenous protocol would be feasible in patients with witnessed TBI.
Pretreatment with necrostatin-1 markedly reduced brain neutrophil influx and microglial activation at 48 h after CCI. Little is known regarding the molecular pathways that activate microglia after TBI; however, a previous study showed that small molecule inhibitors of microglial activation can reduce spatial learning deficits associated with neurodegenerative disease in mice (Ralay Ranaivo et al, 2006). This report notwithstanding, reduced inflammation is not a prerequisite for improved MWM performance mediated by the TNFα/Fas signaling pathways (Bermpohl et al, 2007). Our data suggest that necroptotic signaling pathways contribute to cellular inflammation in injured brain, and raise the possibility that necrostatin-1 may inhibit proinflammatory gene expression after TBI. This hypothesis is currently under investigation in our laboratory.
The present study adds necroptosis to the list of cell death mechanisms that operate in brain after TBI. Other TBI-related cell death mechanisms include caspase-mediated apoptosis and caspase-independent programmed cell death (apoptosis inducing factor), as well as oxidative stress (Aoyama et al, 2002; Zhang et al, 2002, 2005). Our data suggest that necroptosis is also relevant to TBI and that further work is justified to identify necroptosis signaling pathways after TBI, and to identify the molecular target(s) that mediate protection by necrostatin-1. In addition, other necrostatins with increased activity and serum half-life should be tested in brain injury models (Jagtap et al, 2007; Wang et al, 2007).
In conclusion, the data suggest that necroptosis, a novel form of programmed necrosis triggered by death receptors, may play a significant role in cell death and neurologic dysfunction after CCI in mice. This study provides proof of principle that necrostatin-1 and perhaps other necrostatins are promising therapeutic agents to reduce the histopathological and functional neurologic sequelae of TBI.
Footnotes
The authors declare that they have no competing financial interests. Brigham and Women's Hospital and Harvard Medical School have filed patents for necrostatins.