
Abstract
Brain injury after intracerebral hemorrhage (ICH) occurs in cortex and white matter and may be mediated by blood breakdown products, including hemoglobin and heme. Effects of blood breakdown products, bilirubin and bilirubin oxidation products, have not been widely investigated in adult brain. Here, we first determined the effect of bilirubin and its oxidation products on the structure and function of white matter in vitro using brain slices. Subsequently, we determined whether these compounds have an effect on the structure and function of white matter in vivo. In all, 0.5 mmol/L bilirubin treatment significantly damaged both the function and the structure of myelinated axons but not the unmyelinated axons in brain slices. Toxicity of bilirubin in vitro was prevented by dimethyl sulfoxide. Bilirubin oxidation products (BOXes) may be responsible for the toxicity of bilirubin. In in vivo experiments, unmyelinated axons were found more susceptible to damage from bilirubin injection. These results suggest that unmyelinated axons may have a major role in white-matter damage in vivo. Since bilirubin and BOXes appear in a delayed manner after ICH, preventing their toxic effects may be worth investigating therapeutically. Dimethyl sulfoxide or its structurally related derivatives may have a potential therapeutic value at antagonizing axonal damage after hemorrhagic stroke.
INTRODUCTION
Spontaneous intracerebral hemorrhage (ICH) accounts for only 15% of all strokes 1 but it has the highest mortality of the major types of stroke, with only about 41% of affected individuals surviving for a year. 2 It is also an important contributor to poor outcome after traumatic brain injury. 3 Survivors are often left with severe disabilities, with only 20% of them regaining full independence. 4 To date, translational research and clinical trials have not been successful in finding effective pharmacological therapies which can prevent secondary injury, or decrease morbidity and mortality. 5
The pathogenesis of brain damage after ICH includes direct destruction of brain tissue by the physical mass of blood. Additionally, there can be increased intracranial pressure, expansion of the hematoma, edema, breakdown of the blood-brain barrier, inflammation, oxidative stress, hydrocephalus, apoptosis, necrosis, activation of the complement system, spreading depolarization, and possibly ischemia around the hematoma. 6 , 7 Toxicity of blood breakdown products has been recognized as a contributor to secondary injury that occurs after ICH. Much work has been performed to understand the effects of hemoglobin and iron, as well as whether decreasing the quantities of these molecules after ICH improves functional outcome.8–10 However, the effects of the intermediate breakdown products of hemoglobin, such as bilirubin, have largely been overlooked in the context of ICH, despite the fact that free (i.e., unconjugated) bilirubin is known to cause brain injury in newborn infants, and less commonly in older children and adults. 11
After ICH, hemoglobin breaks down and releases heme, which is further broken down into biliverdin and iron by heme oxygenases. Biliverdin is then further reduced to bilirubin by biliverdin reductase. Since bilirubin is sparingly soluble in water if it is found in the plasma, it binds tightly to albumin in order to be transported to the liver where it is conjugated by glucuronyl transferase by breaking the internal hydrogen bonds and forming, most commonly, bilirubin IXα-diglucuronide. 11 Conjugated bilirubin is water soluble and is the form excreted in bile and urine. However, when the concentration of bilirubin exceeds the binding capacity of albumin, the concentration of free unconjugated bilirubin increases. Free unconjugated bilirubin may have toxic effects. In addition, under the oxidative stress environment of ICH, bilirubin may be degraded into bilirubin oxidation products (BOXes). 12 Bilirubin oxidation products can be produced by nucleophilic attack of biliverdin and bilirubin with hydrogen peroxide, superoxide, nitric oxide, or peroxynitrite radicals. Additionally free iron, released by heme degradation, interacts with hydrogen peroxide to produce hydroxyl-free radicals, which can also interact with bilirubin to generate BOXes. 12
Compared with ischemic stroke and other cerebral small vessel pathologies, patients with ICH show considerably larger volumes of white-matter hyperintensity based on the analyses of volumetric magnetic resonance images taken within 2 weeks of ICH. 13 Furthermore, the extent of white-matter damage is predictive of both early and long-term outcome, correlates well with the Glasgow coma scale at admission, and with both 1 month and long-term mortality. 14 , 15 This suggests that the white-matter damage makes the brain more vulnerable to further insult as well as higher risk of mortality. White-matter damage is also associated with poorer cognitive outcome and a higher risk of stroke reoccurrence and poststroke dementia. 14 , 15 Additionally, white-matter damage is of prognostic value as higher volumes of white-matter hyperintensity correlate with greater ICH volumes and are associated with greater degrees of hematoma expansion. 16 Despite clinical evidence that white matter is damaged after ICH, few studies have investigated effects of ICH and blood breakdown products on white matter.
The aims of this work were to determine whether bilirubin and its oxidation products cause white-matter injury after ICH. We first determined the effect of bilirubin and its oxidation products on the structure and function of white matter in vitro using acutely isolated brain slices. Subsequently, we determined whether these compounds have an effect on the structure and function of white matter in vivo. On the basis of the previous knowledge about the effects of these molecules, we hypothesized that bilirubin and its oxidation products would cause functional loss in white-matter tracts through direct or indirect structural disruption of myelin and axonal integrity.
MATERIALS AND METHODS
Animals
All animal protocols were approved by the Animal Care Committee at St Michael's Hospital and complied with the regulations of the Canadian Counsel on Animal Care. We used CD-1 male mice (25 to 35 g, Charles River, Sherbrooke, Quebec, Canada). The animals were kept in a 12-hour light-dark cycle and had access to food and water ad libitum. All bilirubin treatment experiments in vitro were performed on acutely isolated brain slices from naïve mice.
Bilirubin Treatment In vivo
Animals were anesthetized with an intraperitoneal injection of ketamine/ xylazine (100/20 mg/kg), and the scalp at the incision site was infiltrated with a subcutaneous injection of 0.5% bupivacaine (5 mL/kg). The head of the mouse was fixed in a stereotactic frame (David Kopf Instruments, Tujunga, CA, USA). Body temperature was maintained at 37°C with a heating pad (Homeothermic Systems, Harvard Apparatus, Holliston, MA, USA). A 2-cm midline incision was made in the scalp and the skull exposed. A burr hole was made with a 1-mm drill 1mm posterior to the bregma and 1mm lateral to the midline. A previously published protocol was used with slight modifications. 17 Briefly, a 50-μL glass syringe with an attached needle was lowered into the cranium through the burr hole at a 15° angle with the following coordinates (1mm posterior to bregma, 1mm lateral to the midline, and 1.8mm depth) A microinjection pump was used to inject saline or fresh 0.5 mmol/L bilirubin intracerebrally. During the initial 3 minutes 5 μL was injected, which was followed by a 7-minute pause. Subsequently, 10 μL was injected over 5 minutes, and the needle removed 10 minutes later. Some previous studies have used smaller amounts of bilirubin (1 μL of a 4.5-mmol/L solution) but this was accompanied by 10 μL of whole blood. 6 Others have used higher amounts of pure bilirubin at 12 mg/mL (205 mmol/L) at a volume of 30 μL. 9 All animals received subcutaneous buprenorphine (0.2 mg/kg) and 0.5 mL subcutaneous 0.9% NaCl (saline) twice daily for 48 hours.
Electrophysiologic Recording of Brain Slices In Vitro
Procedures were similar to those we published (for details, see Supplementary Methods). 18 In brief, coronal sections of brain slices were cut using a vibratome (Leica, 1200, Leica Microsystem, Richmond Hill, ON, Canada). Slices were placed in artificial cerebrospinal fluid (aCSF) aerated with 95% O2 and 5% CO2 for at least 1 hour before recording.
Unless otherwise specified, all brain slices were incubated for 7 hours in an incubation chamber without perfusion, and all solutions were made in aCSF and continuously bubbled with 95% O2 and 5% CO2. Concentrated bilirubin stock solution was made by dissolving powdered bilirubin (Sigma-Aldrich, St Louis, MO, USA) in distilled water titrated with 2 mol/L NaOH until fully dissolved (the final concentration of NaOH is ∼ 0.1 mol/L). The pH of the solution was then adjusted to 7.4 using HCl. The solution was either immediately diluted to the appropriate concentration in aCSF and used (fresh bilirubin), or placed in a tightly sealed glass vial and kept in the dark at 4°C (stored/oxidized bilirubin). The final solution of aCSF containing bilirubin (0.1 or 0.5 mmol/L) had a similar pH and osmolarity to normal aCSF. The pH of aCSF and aCSF with 0.1 and 0.5 mmol/L bilirubin was 7.37, 7.43, and 7.39, respectively (n = 3 measurements). The osmolarity of normal aCSF and aCSF with 0.1 and 0.5 mmol/L bilirubin was 314, 322, and 321, respectively (n = 3 measurements). In preliminary experiments, addition of solutions of 2 mol/L NaOH with pH adjusted to 7.4 with HCl had no effect on compound action potentials (CAPs).
Electrophysiologic recordings were obtained within 8 hours after sectioning. Compound action potentials were recorded extracellularly from the corpus callosum using a pulse range from 30 to 1,500 μA, at a rate of 0.05 Hz. The tract was stimulated with a bipolar concentric tungsten electrode (FHC, Bowdoinham, ME, USA). Signals were amplified with an amplifier (Multiclamp 700A, Molecular Devices, Foster City, CA, USA), digitized (Digidata, 1320, Molecular Devices) and collected through a computer running Clampex 8.0 (Molecular Devices). A digital stimulator was used and was controlled by a computer program (STG 1001, Multichannel System, Reutlingen, Germany).
Transmission Electron Microscopy
Coronal brain sections, fresh or incubated for 7 hours in various conditions (normal aCSF, 0.1 mmol/L bilirubin, 0.5 mmol/L bilirubin, 0.1mmol/L bilirubin +dimethyl sulfoxide (DMSO), normal aCSF+DMSO) were immersed in 4% formaldehyde and 1% glutaraldehyde in phosphate buffer at pH 7.3 for 1 day. They were postfixed in 1% osmium tetroxide for 1 hour, after which they were dehydrated in a graded series of acetone solutions. The sections were then cut sagittally through the midline and the area containing the corpus callosum embedded in epon-araldite epoxy resin. Sagittal brain sections were cut using a diamond knife on a microtome (Reichert Ultracut E, Leica Inc., Vienna, Austria), and stained with uranyl acetate and lead citrate. The sections were examined using a transmission electron microscope (TEM, JEM-1011, JEOL Corp., Peabody, MA, USA). The images were acquired with a 1024 × 1024 pixel charge coupled device camera (AMT Corp., Denver, MA, USA).
Immunohistochemistry
Procedures were similar to those we published (see Supplementary Methods for details).18 Animals used for histology were perfused by 4% paraformaldehyde. Brain tissues were processed and blocks were embedded in paraffin. Some brains were prepared for cryosectioning with optimal cutting temperature medium and stored at −80°C.
Staining was performed according to the ABC protocol using Vectastain Elite ABC kit (Vector Laboratories, Burlingame, CA, USA). All sections were counterstained in methyl green solution for 5 minutes then dehydrated quickly through increasing concentrations of ethyl alcohol. Images of the slides were taken using the Olympus DP72 microscope (Center Valley, PA, USA) and the Olympus cellSense software (Olympus, Center Valley, PA, USA), and qualitatively assessed for the presence of positive staining.
Mass Spectrometry
Mass spectrometry analysis of oxidized bilirubin was performed using an AB Sciex QSTAR XL ESI-Qq-TOF mass spectrometer in negative mode with a nano flow electrospray source (AB Sciex, Framingham, MA, USA). The samples were mixed with an equal volume of 5% NH4OH before loading to a glass capillary tip (Econotip, New Objective, Woburn, MA, USA) for static infusion.
UV Spectrophotometry
Solutions were prepared in aCSF at serially diluted concentrations and were placed in triplicate into a 96-well plate (200 μL/well). The absorbance of BOXes at 320 nm6 was detected by spectroscopy using a microplate reader (SpectraMax M5e, Molecular Devices) and analyzed using the SoftMax Pro 5.4.1 software (Molecular Devices).
Data Analysis and Statistics
Analysis of transmission electron microscope images. The G-ratio, a commonly used index of structural and functional myelination, was calculated using the equation G-ratio =(A1/A2) where A1 is the area within the inner lumen of the axon and A2 is the area of the entire crosssection of the axon including the myelin sheath. 19 The axon density was calculated by averaging the number of axons per squared area.
Analysis of electrophysiologic data. Raw electrophysiologic data were analyzed using the Clampfit 9.0 software (Molecular Devices, Sunnyvale, CA, USA). Amplitude was calculated by measuring the distance between the trough and the average of the two peaks. Latency was measured as the distance between onset of the stimulation and the trough of the curve, whereas the area under the curve was calculated using the formula area under the curve = (a × b)/2 where a is the amplitude and b is the time between the first and second peak of the curve (Supplementary Figure 1).
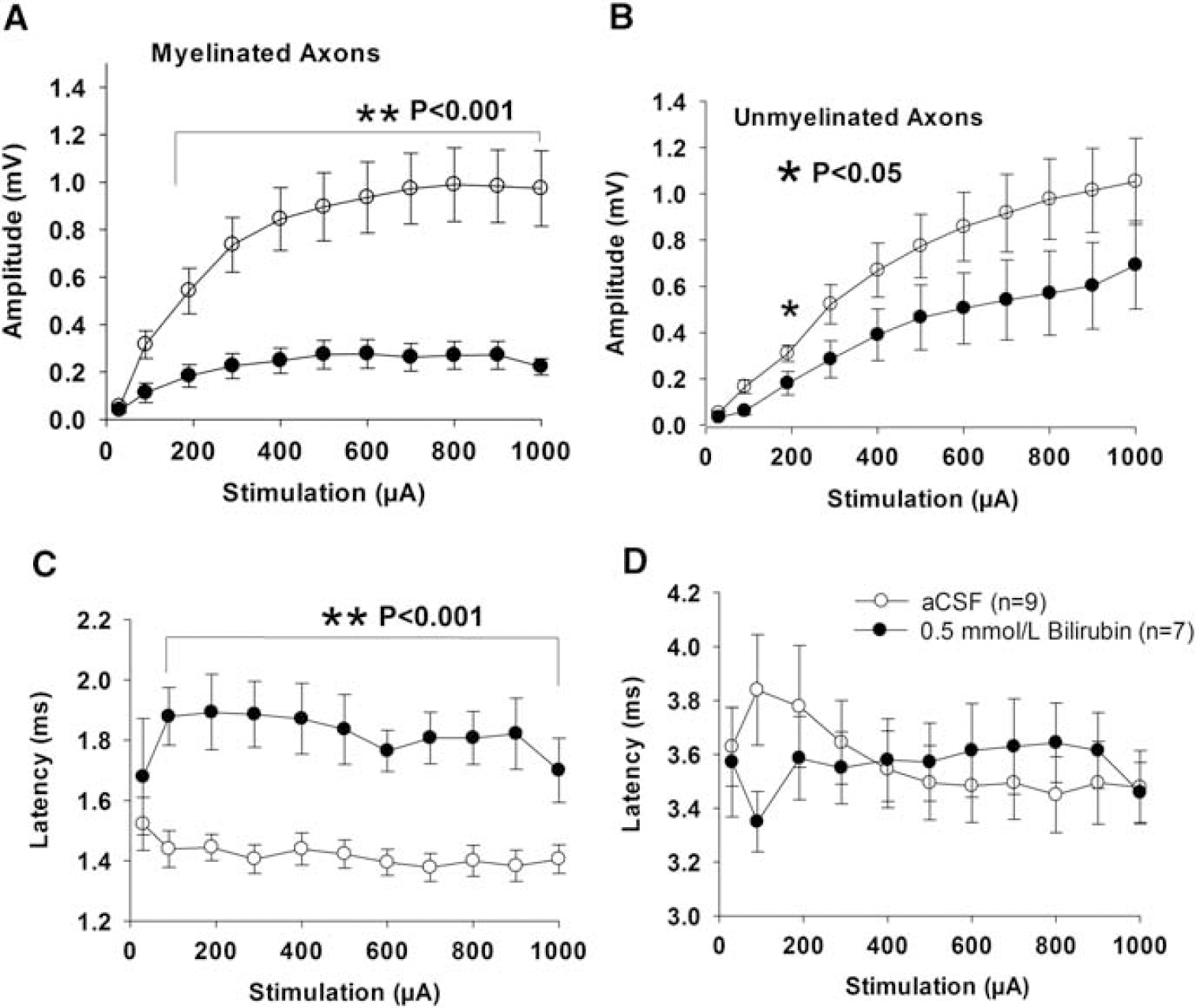
Effect of bilirubin on compound action potential (CAP) of axons in brain slices. Incubation with bilirubin, 0.5 mmol/L, for 7 hours resulted in a significantly decreased amplitude
All statistical analyses were performed using the SigmaStat 3.1 program (Systat Software, San Jose, CA, USA). All data are presented as means ± standard error of mean (s.e.m.), and were compared between groups by analysis of variance. If significant variance was found, then a post hoc test was performed using the Holm-Sidak method. Student's t-test was used for comparision between two groups. P<0.05 was considered as significant.
RESULTS
We first confirmed that the CAPs recorded were consistent with prior descriptions. 18 As shown in Supplementary Figure 1A, the typical CAP comprises two downward peaks in response to electrical stimulation. They are corresponding to CAPs from myelinated (N1, the fast conducting fibers) and unmyelinated (N2, the slower conducting fibers) axons in the corpus callosum. We next quantified the amplitude, latency, and area under the curve of the two peaks. The amplitude of CAP is proportional to the total number of axons. The latency is an inverse function of axonal conductance and their structural integrity. Both peaks were diminished by treatment of Na+-free aCSF (Supplementary Figure 1B) or 0.5 μmol/L tetrodotoxin (Supplementary Figure 1C), indicating that CAPs are mainly generated by Na+ channels. This is consistent with the previous reports showing that these CAPs are of axonal origin and not synaptic. 20
Bilirubin Diminishes Compound Action Potential of Myelinated Axons In Vitro
We next studied the effect of incubation of brain slices with varying concentrations of stored/oxidized bilirubin solutions. Incubation of the brain tissue for 7 hours in 0.02 or 0.1 mmol/L stored/oxidized bilirubin did not result in any difference in the latency of CAPs compared with recordings from tissue incubated in aCSF. However, the amplitude of both N1 and N2 is significantly diminished by bilirubin at some of the stimulation points in the range of 50 to 1,000 μA.
When the slices were treated for 7 hours with 0.5 mmol/L bilirubin, the amplitude of myelinated axons was significantly lower than that in aCSF-treated controls (P<0.001, stimulations from 190 to 1,000 μA, Figure 1A). Meanwhile, the latency of myelinated axons was significantly increased (P<0.001, stimulations from 90 to 1,000 μA, Figure 1B). This suggests that bilirubin caused a decreased number of total myelinated axons and compromised function of the remaining myelinated axons. In addition, there was a trend for decreased amplitude of unmyelinated axons from slices treated with 0.5 mmol/L bilirubin (Figure 1B). This was not statistically significant as compared with aCSF-treated controls except at one stimulation point (Figure 1B). There was no difference in latency of unmyelinated axons between the two conditions (Figure 1D). In all, 0.5 mmol/L bilirubin affected myelinated axons to a greater extent than unmyelinated axons (Figure 1).
Since the effects of bilirubin may resemble those of hyoxia, the oxygen saturation of applied solutions was measured. In normal aCSF and aCSF with 0.1 and 0.5 mmol/L bilirubin, oxygen saturation was 99.9% for all the solutions (n = 3 for each measurement). Therefore, the toxic effect of bilirubin is probably not due to reduced diffusion of oxygen.
Bilirubin Disrupts Structural Integrity of Axons In Vitro
The effects of stored/oxidized bilirubin solution on axonal structure were studied using TEM. There was a decreasing number of myelinated axons with increasing bilirubin concentration (Figure 2). There were empty spaces between axons in bilirubintreated slices but not in the normal controls. The remaining axons had increased diameter but decreased thickness of the myelin sheaths, suggesting the presence of cytotoxic or ionic edema. 21 Furthermore, the axons were less compact with more debris between them in the bilirubin-treated slices. We quantified the density of axons and observed a trend toward decreased density with increasing concentration of bilirubin (P<0.05 to 0.001 for 0.5 mmol/L bilirubin group compared with the three aCSF control groups, Figure 2B). Dimethyl sulfoxide treatment significantly increased density of axons as compared with bilirubin-treated groups and aCSF controls (P<0.05 to 0.001, analysis of variance, Figure 2B).
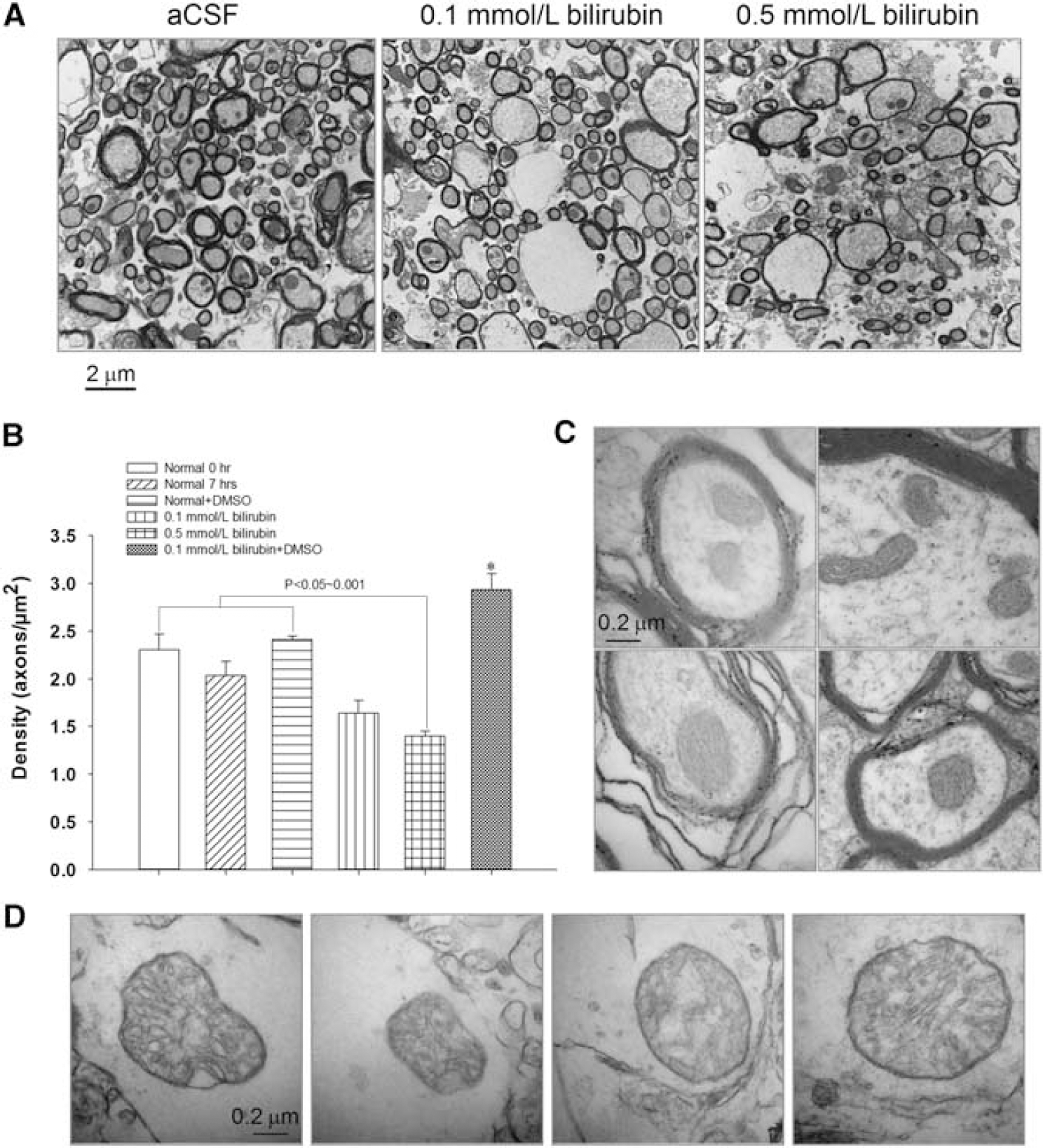
Effect of bilirubin on ultrastructure of axons in brain slices.
Images taken at a higher magnification showed differences in mitochondria (Figures 2C and 2D). Axons from tissue incubated in aCSF had a well-defined outer edge surrounded by myelin, and small mitochondria with compact, well-defined cristae (Figure 2C). However, axons incubated in 0.5 mmol/L bilirubin for 7 hours had significantly enlarged mitochondria, and vacuole-like bubbling, as well as loss of definition of the cristae (Figure 2D). In addition, the outer membranes of the remaining axons appeared to have lost their integrity after bilirubin treatment. This suggests that bilirubin, at a concentration of 0.5 mmol/L, disrupted the integrity of both axonal membranes and mitochondria within the axons. This structural damage may result in disrupted function shown as increased latency of axonal response to electrical stimulation.
Antagonists of N-methyl-D-aspartate, α-Amino-3-hydroxy-5- methyl-4-isoxazolepropionic acid receptors, and L-type Ca2+ channels Have No Effect of Bilirubin Toxicity on Compound Action Potentials
We next determined what could reverse the toxic effect of stored/ oxidized bilirubin on axons. Incubation of brain tissue for 7 hours in 0.1 mmol/L bilirubin or 0.1 mmol/L bilirubin with 0.1 mmol/L amlodipine besylate, an L-type calcium (Ca2+) channel antagonist, did not have statistically different effects on amplitude or latency in either myelinated or unmyelinated axons. Electrophysiologically, there was no difference between the tissue incubated with bilirubin alone or with 0.2 mmol/L N-methyl-D-aspartate (NMDA) receptor antagonist D-AP5 or 30 μmol/L α-amino-3-hydroxy-5- methyl-4-isoxazolepropionic acid (AMPA) receptor antagonist NBQX. This suggests that NMDA and AMPA receptors are not involved in the effects of bilirubin on the structure and function of white matter.
Dimethyl Sulfoxide Reverses the Toxic Effect of Stored/oxidized Bilirubin on Axons in Corpus Callosum In Vitro
A comparison of electrophysiologic recordings from tissues incubated in various solutions (aCSF, aCSF with DMSO, 0.1 mmol/L bilirubin and 0.1 mmol/L bilirubin with DMSO) indicates that there were no differences between latencies recorded in these groups (Figures 3C and 3D). However, there were significant differences between the amplitudes of these groups in both myelinated and unmyelinated axons (Figures 3A and 3B, P<0.05, one-way ANOVA). In myelinated axons, treatment with 0.1 mmol/L bilirubin and DMSO alone slightly but statistically insignificantly reduced the amplitude of CAP (Figure 3A). Dimethyl sulfoxide treatment completely reversed bilirubin's effect and significantly increased CAP amplitude (Figure 3A). Similarly, in unmyelinated axons, DMSO treatment significantly reversed the diminished amplitude back to normal control level at several stimulation points (Figure 3B).
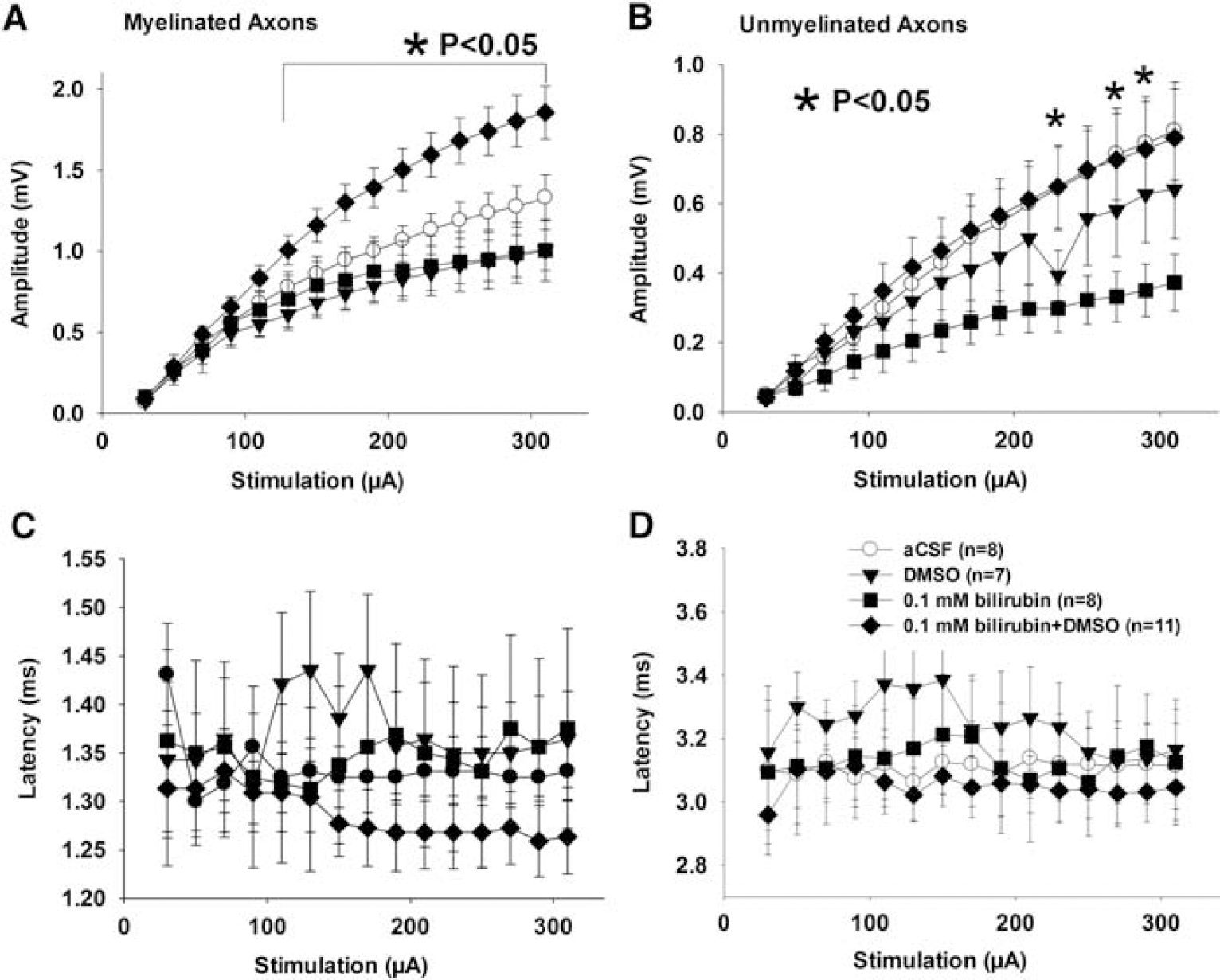
Dimethyl sulfoxide (DMSO) protects axons from bilirubin toxicity. DMSO alone has a moderate toxic effect on axons; however, it completely prevented the effect of bilirubin on compound action potential (CAP) amplitude of both myelinated
Figure 4A shows representative TEM images of cross-sections of the corpus callosum. The images of normal controls showed a dense population of round myelinated axons of varying sizes, with few unmyelinated axons filling in the gaps between the myelinated ones. The tissue incubated in DMSO alone or together with bilirubin looked similar to the normal control, with slightly more space not occupied by any axons. The tissue incubated in 0.1 mmol/L bilirubin had a number of oddly shaped axons as well as significantly more space where there were no axons present, similar to that in Figure 2A.
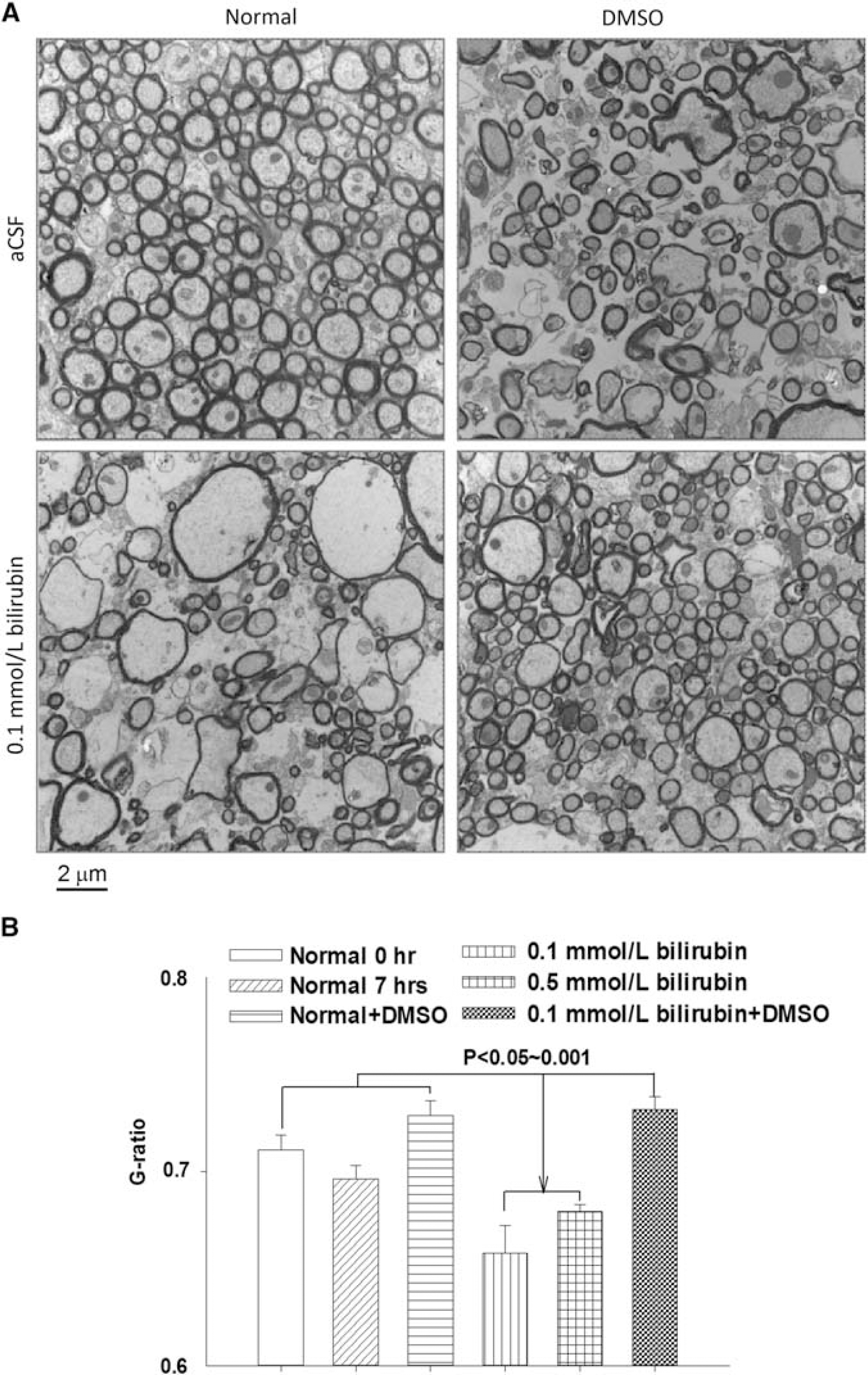
Dimethyl sulfoxide (DMSO) prevented bilirubin-induced effects on structure of axons. Representative transmission electron microscope (TEM) images showing DMSO preserves axonal structure after exposure to bilirubin (0.1 mmol/L for 7 hours)
We calculated the G-ratio to quantitatively evaluate the effect of DMSO and bilirubin on the structural integrity of axons. Each condition had a sample size of 103 axons, which were measured from images taken at the same magnification. Except the normal control incubated with aCSF for 0 hour, all other samples were incubated for 7 hours. The G-ratio values for each condition were 0.71 ± 0.008 for normal control at 0 hour, 0.70 ± 0.007 for normal control incubated in aCSF for 7 hours, 0.73 ± 0.008 for tissue incubated in aCSF and DMSO, 0.66 ± 0.01 for tissue incubated in 0.1 mmol/L bilirubin, 0.068 ± 0.004 for tissue incubated in 0.5 mmol/L bilirubin, and 0.73 ± 0.007 for tissue incubated in 0.1 mmol/L bilirubin+DMSO (Figure 4B). There was no statistical difference among the three normal control groups. However, bilirubin (0.1 or 0.5 mmol/L) significantly decreased the G-ratio as compared with the controls (Figures 4B, P<0.05 to 0.001, ANOVA). DMSO treatment reversed the decreased G-ratio of 0.1 mmol/L bilirubin-treated tissue back to normal level. Although DMSO reversed the toxic effect of 0.1 mmol/L bilirubin, it did not reverse that of 0.5 mmol/L bilirubin (Supplementary Figure 4).
Storage of Bilirubin Is Related to its Toxic Effect In Vitro
To investigate the effect of storage on the toxic effect of bilirubin on brain white matter, we compared the effects of bilirubin solution made immediately before the experiment (fresh bilirubin) and solution made 7 days prior and kept at 4°C (stored/oxidized bilirubin) on CAPs. There was no difference between fresh or oxidized bilirubin on CAP at a concentration of 0.1 mmol/L. However, at a concentration of 0.5 mmol/L, tissue treated with stored/oxidized bilirubin showed a lower amplitude in myelinated axons (Supplementary Figure 2A), which reached statistical significance (P<0.05) at stimulations of 90, 190, and 900 μA. Similarly, there was a longer latency of myelinated axons incubated in stored/oxidized bilirubin, which was statistical significant (Supplementary Figures 2C, P<0.05) at most stimulation currents. Neither the amplitude nor the latency of unmyelinated axons was different between the two conditions at 0.5 mmol/L bilirubin (Supplementary Figures 2B and 2D).
To investigate whether the effect of oxidized bilirubin was time dependent, we performed a series of experiments with brain slices incubated with stored/oxidized bilirubin for increasing times. Compound action potentials were recorded after every hour of incubation in 0.5 mmol/L of oxidized bilirubin from 1 to 5 hours. There were no differences in the unmyelinated axons across the time points (Supplementary Figures 3B and 3D). Myelinated axons did not show a decrease in CAP amplitude until 5 hours of incubation (Supplementary Figures 3A, P<0.05 as compared with other time point groups). There was no statistically significant difference in the latency of the myelinated axon CAP (Supplementary Figure 3C).
Bilirubin Oxidation Products Are Formed from Incubated Bilirubin
Bilirubin oxidation products are organic compounds, which form as a result of bilirubin oxidation. 22 They can be quantified by assessing the absorbance at a wavelength of 320 nm. 6 The 7-day stock solution of bilirubin either stored ot oxidized at 4°C or 37°C had significantly higher absorbance at 320 nm compared with freshly-made solution (Figuress 5A and 5B, P<0.05 to 0.001 across different concentrations).
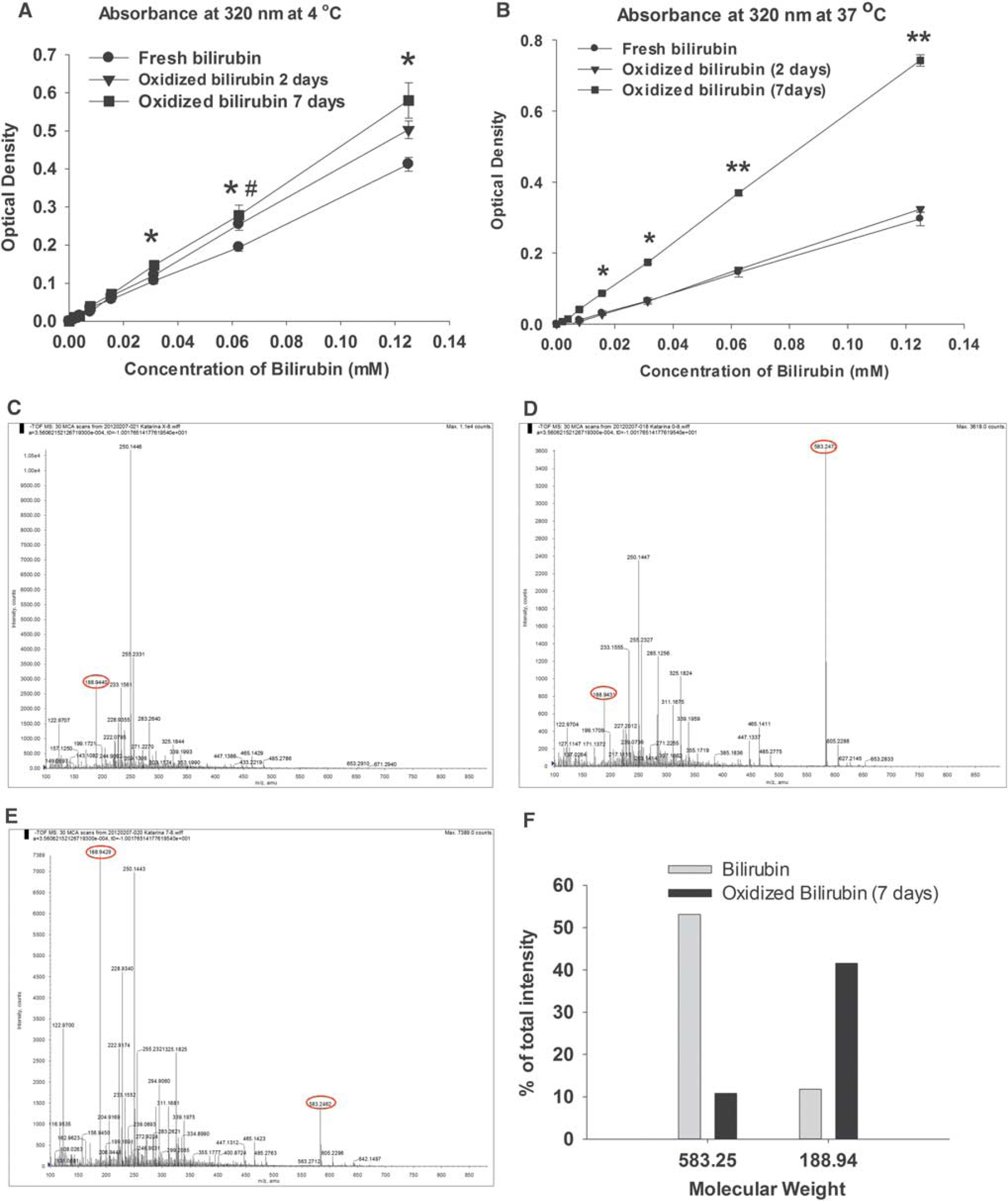
Detection of bilirubin oxidation products (BOXes) in oxidized bilirubin solutions. Ultraviolet spectrophotometry detection of BOXes at 320 nm in stored/oxidized bilirubin solutions at 4°C
We further investigated the identity of compounds in the oxidized bilirubin solution with mass spectrometry. Pure BOXes (donated by Dr JF Clark) showed two major peaks, with the 188.94 m/z (mass-to-charge ratio) being the closest to the previously reported weight of 179.2 m/z (Figure 5C). 23 Analysis of the fresh bilirubin solution showed a large peak at 583.25 m/z, which is very close to the 584.66 m/z molecular weight of bilirubin. Fresh bilirubin solution also showed the presence of a small peak at 188.94 m/z (Figure 5D). The analysis of 7-day stock bilirubin solution showed the opposite, with a small peak at 583.25 m/z and a large peak at 188.94 (Figures 5E and 5F). Interestingly, there was also a peak at 250.14 m/z that was present in the solution of BOXes. This suggests an intermediate compound in the metabolism of bilirubin to BOXes. Thus, mass spectrometry showed a decrease in bilirubin accompanied by an increase in BOXes after 7 days of solution storage. This was similar to the findings on UV absorbance.
Bilirubin Affects the Structure and Compound Action Potential of the Corpus Callosum in vivo
The effects of bilirubin on the corpus callosum in vivo were assessed by stereotactic injection of 15 μL of 0.5 mmol/L of bilirubin or saline (0.9% NaCl). When the CAP of brain slices was assessed 2 days after injection, we did not find any difference between bilirubin- and saline-injected animals. When brain slices from bilirubin- or saline-injected animals were assessed 7 days after the injection, there were no effects on amplitude or latency of myelinated axons (Figures 6A, 6C, and 6E). In contrast, the amplitude of unmyelinated axons from brain slices of animals injected with 0.5 mmol/L bilirubin was significantly lower than the saline-injected controls (Figure 6B). There was no effect on latency of the unmyelinated axons (Figure 6D). However, the area under the curve of umyelinated axons from tissue injected with bilirubin was significantly affected at some stimulation points (Figure 6F). Immunohistochemical staining showed no positive staining for β- APP, MBP, or CD68 in saline-injected mice (Figure 7). However, all mice injected with bilirubin had positive staining for axonal damage (β-APP), degraded myelin (dMBP), and activated microglia (CD68). The contralateral side of the bilirubin-injected brains was also positive for β-APP and CD68 (Figure 7).
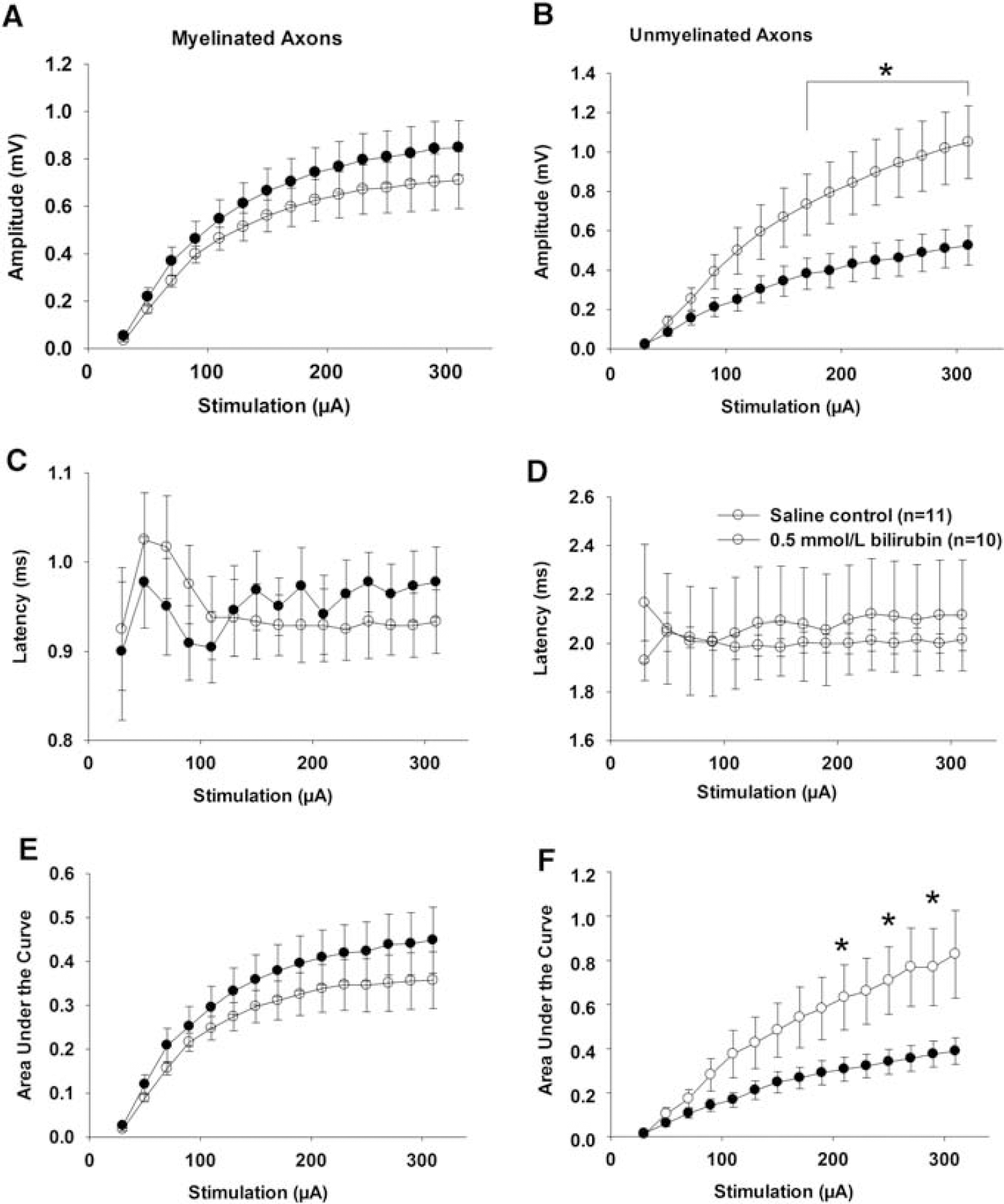
Effect of bilirubin injected into the corpus callosum in vivo on the compound action potential (CAP) of axons in brain slices. Seven days after injection of 15 μL of 0.5 mmol/L bilirubin into the corpus callosum there was significantly decreased CAP amplitude and area under the CAP curve of unmyelinated axons (
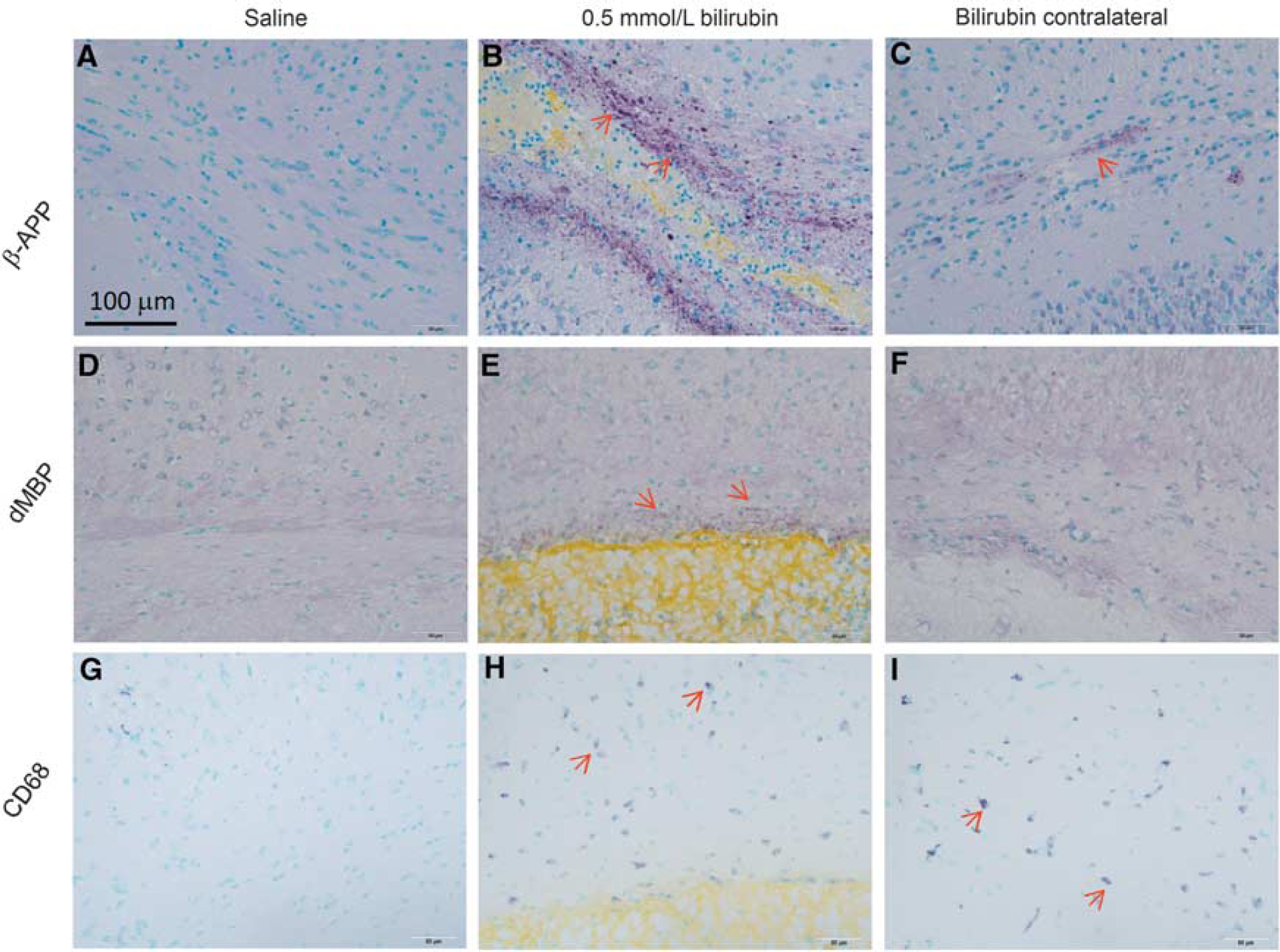
Effect of bilirubin injected into the corpus callosum in vivo on axonal integrity. Immunohistochemical staining 7 days after injections showed no immunoreactivity for β-APP, dMBP, or CD68 in saline-injected control mice (
DISCUSSION
Major Findings of the Study
This study found that bilirubin affects the structural integrity and function of white-matter tracts of the corpus callosum both in vitro and in vivo. The toxic effect of bilirubin may be due to its oxidation products such as BOXes. Dimethyl sulfoxide effectively ameliorates toxicity of bilirubin and confers neuroprotection of axons in the white matter. Finally, unmyelinated axons may be more vulnerable than myelinated axons to bilirubin toxicity in vivo.
Although it is known that neurons and white-matter tracts within and around an ICH are damaged, most experimental work focused on neuronal injury. Wasserman and Schlichter 24 studied white-matter damage around ICH in mice, reporting that there was axonal damage with or without demyelination. They did not investigate which blood components caused white-matter injury. The current work shows the initial cause of damage to axons and myelin may be due to effects of hematoma breakdown products including bilirubin and BOXes.
Toxicity of Bilirubin
Toxicity of bilirubin has been studied in animals. Bilirubin treatment leads to death of neurons, 25 oligodendrocytes, 26 and astrocytes. 27 Mechanisms may include activation of NMDA (but not AMPA) receptors, apoptosis, increase in free intracellular Ca2+, and activation of caspase-3. 25 The toxic effects of bilirubin on oligodendrocytes and astrocytes were suggested to be to induce apoptosis by activation of caspase-3. 26 , 27
Unconjugated bilirubin disrupted hippocampal function and memory by impairment of long-term potentiation and long-term depression in rat hippocampi. 28 These effects may be caused by calpain-mediated cleavage of NMDA receptors that are crucial for synaptic changes associated with long-term potentiation and depression. 29
The current experiments show direct effects of bilirubin and BOXes on white-matter integrity. Other studies focused on in vivo models of ICH support a toxic effect of hemoglobin breakdown products including bilirubin and BOXes, but have mainly investigated inflammatory mechanisms. 30 Unconjugated bilirubin caused excessive glutamate release, as well as an increase in proinflammatory cytokines interleukin-1β, interleukin-6 and tumor necrosis factor-α in rat cortical astrocytes. 31 Loftspring et al 6 , 7 hypothesized that bilirubin and BOXes activate microglia and astrocytes which release proinflammatory cytokines and chemokines, and recruit peripheral leukocytes. Inflammation increases oxidative stress. Microglial and astrocyte activation also causes release of proapoptotic factors such as tumor necrosis factor-α and interleukin-1β, and the death of these cells causes further leukocyte influx, which is facilitated by the breakdown of the BBB. The current results suggest bilirubin can be toxic in concentrations similar to those found around ICH. Clark and Sharp12 and Clark et al 32 reported bilirubin concentrations reached as high as 0.70 mmol/L (41 ± 3 mg/dL) within the hematoma, and ∼ 0.18 mmol/L in the perihematoma white matter in an ICH model in pigs. Use of 0.5 mmol/L in our study is consistent with the concentration used by Loftspring et al. 6 , 33 They used 0.45 mmol/L unconjugated bilirubin combined with 10 μL blood injection to study the role of unconjugated bilirubin in mediating secondary injury after ICH. In the study by Huang et al, 9 20 mmol/L bilirubin (12 mg/mL in saline) was used to study its effect on brain edema. Lower concentrations of bilirubin (20 to 150 μmol/L) may have antioxidant effects.34–37 It is likely that these effects are outweighed by toxicity caused by high concentrations present in pathologic conditions such as in ICH, especially if such large increases are not accompanied by increases in biliverdin reductase.
Potential Mechanisms Underlying Bilirubin Toxicity On Axons
The amplitude of the CAP is proportional in individual axons to the inward driving force for Na+ as well as overall to the number of conducting axons whereas latency is related to axonal function. Our data showed that bilirubin decreased the amplitude of the CAP and increased its latency in a concentration-dependent manner, although the effects were most marked on amplitude and latency of myelinated axons after 7 hours incubation in vitro. The effects were different 7 days after injection of bilirubin in vivo, where the greatest effect was on unmyelinated axon amplitude. This suggests that bilirubin affects the electrophysiologic properties of white-matter tracts by interacting with myelin and axonal membranes but that the effects depend on duration of exposure and probably on other effects that may occur in vivo but not in vitro, perhaps including inflammation. Furthermore, breakdown of bilirubin, probably into BOXes, increases toxicity. The TEM data support the CAP measurements in that they showed decreased axonal density in bilirubin-treated brain slices. The nonsignificant effect on the unmyelinated axons, however, may suggest that bilirubin might affect unmyelinated axons of a particular size, or to a varying degree based on the size. The averaged CAP may mask this effect on a specific group of axons. Reeves et al 20 found the CAP amplitude of myelinated axons decreased within 3 days of traumatic brain injury in rats and recovered by 7 days after injury. In contrast, CAP of the unmyelinated axons was suppressed for 7 days after injury. Although we did not find an effect of bilirubin on the CAP 2 days after injection (data not shown), there was a nonsignificant trend for decreased CAP amplitude of myelinated axons at 2 days after injection, which resolved 7 days after injection suggesting a recovery of function of the myelinated axons. There was no effect of bilirubin on the latency of either type of axon 7 days after injection of bilirubin, suggesting loss of conducting axons, not a decrease in conduction speed. The histologic data are consistent with the functional data in that β-APP immunoreactivity was more prominent than degraded MBP immunoreactivity.
We tested whether effects of bilirubin were mediated by activation of NMDA, AMPA, or L-type Ca2+ channels. There is evidence that these channels are located on axons. 38 Antagonizing these channels did not abrogate bilirubin toxicity.
Dimethyl Sulfoxide Protective Effect
Dimethyl sulfoxide is a sulfur containing compound, which is commonly used as a drug delivery vehicle because of its ability to enhance permeability of biologic membranes. 39 However, DMSO is also a free radical scavenger and has been shown to have neuroprotective properties when administered after experimental traumatic brain injury and cerebral ischemia. 40 , 41 The mechanism by which DMSO prevented bilirubin toxicity was not fully determined here but might include antioxidant effects or ability to promote membrane resealing. 42 In hippocampal neurons, DMSO was shown to suppress Ca2+ influx induced by glutamate, reduced NMDA and AMPA receptor activation and protected against glutamate excitotoxicity. 43 However, it is unlikely these effects were involved in white-matter injury, since we did not find that antagonizing these glutamate receptors reduced bilirubin toxicity.
Bilirubin Oxidation Products Are Responsible for Bilirubin Toxicity on Axons
Toxicity of bilirubin in vitro required at least 5 hours of incubation. The increased effects of stored/oxidized bilirubin solutions and the presence of BOXes, as determined by mass and ultraviolet spectrometry, in the stored/oxidized solutions suggest that oxidation occurred during storage and that the BOXes are responsible for the toxic effect of bilirubin on axons. Although direct effects of BOXes on white matter were not assessed here, the importance of BOXes is supported by studies detecting BOXes in brain tissue around experimental ICH. 12 , 32 Compounds with other molecular weights were identified and their contribution to toxicity needs to be determined.
CONCLUSION
We have shown that bilirubin is toxic to both myelinated and unmyelinated axons in vitro and in vivo. Under the experimental conditions used, myelinated axons were more susceptible to exposure to bilirubin in vitro. On the contrary, unmyelinated axons were more susceptible to damage from bilirubin injection in vivo. The toxic effect of bilirubin may be due to its oxidation products such as BOXes. Dimethyl sulfoxide effectively attenuated the toxic effect of bilirubin on axons. These results suggest that unmyelinated axons may have a major role in white-matter damage in vivo. The delayed onset of effect of bilirubin and time required for it to form in vivo after ICH make antagonizing its toxicity of interest for investigation. DMSO or its structurally related derivatives may have a potential therapeutic value at antagonizing axonal damage after hemorrhagic stroke.
Footnotes
Dr RL Macdonald receives grant support from the Physicians Services Incorporated Foundation, Brain Aneurysm Foundation, Canadian Stroke Network, and the Heart and Stroke Foundation of Ontario. RLM is a Chief Scientific Officer of Edge Therapeutics, Inc. KL was supported by an Ontario Graduate Scholarship Program grant.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.