
Abstract
In eukaryotes, the polyamine pathway generates spermidine that activates the hypusination of the translation factor eukaryotic initiation factor 5A (eIF5A). Hypusinated-eIF5A modulates translation, elongation, termination and mitochondrial function. Evidence in model organisms like drosophila suggests that targeting polyamines synthesis might be of interest against ischemia. However, the potential of targeting eIF5A hypusination in stroke, the major therapeutic challenge specific to ischemia, is currently unknown. Using in vitro models of ischemic-related stress, we documented that GC7, a specific inhibitor of a key enzyme in the eIF5A activation pathway, affords neuronal protection. We identified the preservation of mitochondrial function and thereby the prevention of toxic ROS generation as major processes of GC7 protection. To represent a thoughtful opportunity of clinical translation, we explored whether GC7 administration reduces the infarct volume and functional deficits in an in vivo transient focal cerebral ischemia (tFCI) model in mice. A single GC7 pre- or post-treatment significantly reduces the infarct volume post-stroke. Moreover, GC7-post-treatment significantly improves mouse performance in the rotarod and Morris water-maze, highlighting beneficial effects on motor and cognitive post-stroke deficits. Our results identify the targeting of the polyamine-eIF5A-hypusine axis as a new therapeutic opportunity and new paradigm of research in stroke and ischemic diseases.
Introduction
Stroke is undoubtedly one of the worst devastating diseases worldwide with an enormous economic and societal burden. 1 Although tremendous progress has been made in understanding the complex spatial and temporal mechanisms of major drivers of neuronal death in ischemic stroke – primarily excitotoxicity and oxidative stress – neuroprotective drug therapy targeting these and other mechanisms has been a dismal failure in clinical trials. 2 While acknowledging several undesirable issues inherent in experimental stroke models and clinical trial designs, learning from efforts to save other organs from ischemic death can provide novel insight and therapeutic targets if applicable to the brain and ischemic stroke. 3 For instance, in many experimental conditioning setups, ischemic tolerance has been achieved in multiple organs including the heart, kidney and brain and their respective cell types4,5 and highlighted new putative targets.
In kidney ischemic injury, we recently reported the therapeutic potential of eukaryotic initiation factor 5A (eIF5A) inhibition to protect against transient unilateral renal artery occlusion in rats and against anoxia in immortalized mouse renal proximal cells. 6 Originally described for its role in initiating translation, hypusinated eIF5A not only promotes general translation elongation and termination,7,8 but also modulates mitochondrial function and oxygen consumption rates. 9 eIF5A activation by hypusination is achieved by post-translational transfer and hydroxylation of the 4-amino butyl moiety from spermidine to a specific lysine residue 10 by two key enzymes, known as deoxyhypusine synthase (DHS) and deoxyhypusine hydroxylase (DOHH). 11 eIF-5A is the only known cellular substrate for DHS and the spermidine analog N1-guanyl-1,7-diaminoheptane (GC7), a diaminoheptane derivative known as the most efficient inhibitor of DHS proved useful in protecting again renal ischemia in vivo and in vitro. Therefore, we investigated whether GC7 and underpinning DHS inhibition may lead to novel therapeutic opportunity in ischemic stroke.
Material and methods
In vitro models of neuronal death
Primary neuronal cultures containing less than 5% glia were prepared from embryonic day 13.5 C57BL/12 mouse embryos. Dissociated cortical neurons were diluted in Neurobasal medium (Gibco) supplemented with 2% B-27, 1% glutamax, 50 U/mL penicillin and 50 µg/mL streptomycin and seeded in 24-well plates. Addition of 5-fluoro-2ʹ-deoxyuridin (2 µM) was added at DIV1 to limit glia proliferation. The study by Melis et al. 6 previously showed that 30 µM was the lowest dose of GC7, which provided a maximum effect on anoxia-induced renal cell death. Therefore, neuronal cultures were treated with 30 µM GC7 between 13 and 14 days in vitro (AtlanChim Pharma) for 12 h before the toxic insult.
Oxygen and glucose deprivation (OGD)
To model ischemia in vitro, neuronal cultures were exposed to transient OGD for 60 min (lethal) to 120 min (supra-lethal).12,13 OGD was performed by placing the neuronal cultures in glucose-free medium (116 mmol/L NaCl, 5.4 mmol/L KCl, 0.8 mmol/L MgSO4, 1 mmol/L NaH2PO4, 26.2 mmol/L NaHCO3, 0.01 mmol/L glycine, 1.8 mmol/L CaCl2 pH 7.4) balanced with 1.2% O2 (N2 replacing O2) for 60 or 120 min (37°C, 5% CO2). OGD was stopped by returning to normoxic conditions in neurobasal medium.
NMDA, H2O2 and KCl toxicity
Stroke-related in vitro neuronal injuries were induced by either excitotoxicity with the glutamate agonist NMDA (100 µM) for 30 or 60 min or superoxide production/oxidative stress by application of 300 µM H2O2 for 30 min. 14 Neurotoxicity was also induced by chemical depolarization with an exposure to 50 µM KCL for 30 min. 15 Cultures were washed and returned to standard neurobasal medium. Neuronal death was estimated 24 h after the onset of the insult.
Assessment of neuronal death
Death of cultured neurons was quantified by trypan blue staining 24 h after exposure with an automatic counter (Life technologies) or using PI/Hoechst staining. PI-positive neurons were quantified with ImageJ software and expressed as a ratio of PI+ neurons/Hoechst+ nuclei.
Mitochondrial imaging
To monitor the mitochondrial membrane potential (ΔΨm), vehicle or GC7-treated neurons seeded on fluorodishes were loaded with the cationic lipophilic dye tetramethylrhodamine ethyl ester (TMRE, Life Technologies) at 25 nM for 20 min. After washout of the dye and application of vehicle or NMDA (100 µM), time-lapse imaging with a laser confocal LSM780 microscope (Carl Zeiss) was started with 5-min intervals for 60 min. The mitochondrial superoxide production was measured using MitoSOX™ Red (Life technologies). Vehicle and GC7-treated neuronal cultures submitted or not to 60 min OGD were loaded with 3 μM MitoSOX™ Red and 2 µg/mL Hoechst (Sigma Aldrich) 24 h after the onset of the insult. Acquisitions at 488 nm and 568 nm wavelengths were performed using videomicroscope (Carl Zeiss). TMRE and MitoSOX™ fluorescence intensity in individual neuron were analyzed with an image analysis software (n = 12 neurons and 3 separate neuronal cultures). Briefly, the area in each confocal micrograph occupied by each neuron was defined using the ImageJ ‘threshold’ function based on Hoechst staining, and the average fluorescence intensity derived from the indicator dyes (TMRM or MitoSOX Red) was measured within this area.
Calcium imaging
Intracellular calcium concentration [Ca2+]i was determined by monitoring the fluorescence intensity of a calcium indicator Fura-2 AM (Molecular probe). Acquisition was performed after vehicle and GC7 treatment, and 24 h after an OGD challenge of neurons seeded at a density of 1,400,000 cells/35-mm fluorodish. Following a 45-min dye loading with fluorescent probe Fura2-AM (10 µM, Life technologies) at 37°C in an atmosphere of 95% air/5% CO2, media was replaced by calcium imaging media containing (in mM) 116 NaCl, 5.6 KCl, 1.2 MgCl2, 2 CaCl2, 20 HEPES, 5 NaHCO3, 1 NaH2PO4. Imaging was performed on fluorodishes mounted on an inverted fluorescent microscope (Zeiss) using a plan fluor 20X0.75 oil/water immersion fluorescent objective. For each condition, at least 50 neurons were recorded from three independent assays. The ratiometric imaging of Fura-2 fluorescence intensity reflecting the [Ca2+]i was obtained using probe excitation with alternative wavelengths of 340 and 380 nm and expressed as percentage of the respective control condition.
Measurement of cellular ATP content
Intracellular ATP level was measured using a luciferin/luciferase-based assay (ATPlite, PerkinElmer) according to the manufacturer’s instructions. Briefly, after vehicle and GC7 treatment, and 24 h after an OGD challenge, cortical neurons were gently swirled in lysis buffer before being incubated for 5 min with the substrate buffer. The emitted light was then detected with a Luminoskan™ Ascent from Thermo. Total ATP was normalized against total protein determined by the BCA method (Thermo Fisher Scientific) to account for any difference in cell density and expressed as percentage of the respective control condition.
Electrophysiology
Membrane potentials were measured on cortical neurons. Cells were incubated during 12 h in the presence of vehicle (neurobasal) or GC7 (30 µM), with or without KATP channel blocker glibenclamide (10 µM), the mitochondrial KATP blocker 5-hydroxydecaoate (5-HD, 10 µM), apamin (100 nM), or charybdotoxin (100 nM). After the incubation period, cells were patched and membrane potentials were immediately measured using the whole cell patch clamp configuration (Hamill et al., 1981). 16 Each membrane potential was evaluated by using a RK 400 patch clamp amplifier (Axon Instrument), low-pass filtered at 3 kHz and digitized at 10 kHz using a 12-bit analog-to digital converter digidata (1322 series, Axon Instrument, USA). Patch clamp pipettes were pulled using vertical puller (PC-10, Narishige) from borosilicate glass capillaries and had a resistance of 3–5 MΩ. The pipette solution contained (in mM) 155 KCl, 3 MgCl2, 2.5 Na2-ATP, 5 EGTA, 2.1 CaCl2, and 10 HEPES adjusted to pH 7.2 with KOH. All experiments were performed at room temperature (21–22°C). Stimulation protocols and data acquisition were carried out using a microcomputer (Dell Pentium) which used a commercial software and hardware (pClamp 8.2).
In vivo model of ischemic stroke
Animals
Male eight-week-old C57BL/6J mice (Janvier France Breeding) were housed in a temperature- and humidity-controlled animal facility with a 12-h light–dark cycle. Food and water were available ad libitum. All efforts were made to minimize animal suffering and the number of animals used. All studies involving animal followed the guidelines outlined in European Community Directive 86/609/EEC, were approved by the Institutional Animal Care and Use Committee (CIEPAL-Azur, approval n°586-1560112863) and were completed in compliance with the ARRIVE guidelines for how to report animal experiments.
Model of transient focal cerebral ischemia
tFCI was induced in adult male mice (8–10-weeks-old, 25–30 g) by intraluminal occlusion of the right middle cerebral artery for 30 or 60 min with a 6–0 coated filament (Doccol, USA) as previously described. 17 Experimental procedures were performed following criteria derived from Stroke Therapy Academic Industry Roundtable group guidelines for preclinical evaluation of stroke therapeutics, starting by treatments and all outcome assessments that were performed by investigators blinded to experimental group assignments. 18 The regional cerebral blood flow was monitored by laser-Doppler flowmetry (Perimed, France) to standardize tFCI severity and confirm restoration of blood flow during reperfusion. Animals that did not show both criteria, CBF reduction of at least 85% of baseline level and reperfusion within 30 min after the filament withdrawal were excluded from the study.
Treatment with the eIFA5 hypusination inhibitor GC7
Animals were randomly assigned to vehicle or GC7 groups. Vehicle and GC7 were administrated intraperitoneally (3 mg/kg). 6 The pretreatment protocols consisted in three injections 48, 24, 2 h 6 or a single injection 2 h before 60 min of tFCI. Post-treatment consisted in sequential injections 2 h, 2d, 4d, 7d, 10d, 14d and 17d after 30 min tFCI which is the recommended model for studying long-term functional outcomes in mice. 19
Brain ischemic infarction
The infarct volume was assessed 24 h and 4 days after tFCI on 12 equally spaced coronal brain sections stained with cresyl violet. Tissue lesion was measured using a computer image analysis system and corrected for brain edema. Infarct volume (in mm3) was calculated by a linear integration of the corrected lesion areas.17,20
Neurobehavioral performance
Motor recovery
Sensorimotor deficits were evaluated by the rotarod test as previously described. 17 Each mouse was subjected to four sessions: at 24 h before tFCI to determine the baseline (pre-operative score), and two, three and four days after tFCI. In each session, mice were trained on the rotarod for 5 min at a constant speed (4 r/min), after 30 min rest, they were then subjected to a single trial to measure their latency to fall from the rotating rod (diameter, 30 mm) that was accelerated from 4 to 40 r/min over 5 min, the trial being stopped after 10 min.
Cognitive recovery
Long-term cognitive deficits were evaluated in the Morris water maze at 14–18 days after tFCI, in a 120-cm-diameter, 60-cm-high circular swimming pool filled to a depth of 32 cm with 21 ± 2°C opaque water, as previously described. 17 Learning deficits were evaluated measuring the time needed for the animal to locate the submerged platform (escape latency) from 14 to 17 days after tFCI. Spatial memory deficits were evaluated at D18 after tFCI by measuring the time spent in the target quadrant when the platform was removed. Swim pattern of the probe test at D18 was recorded by ANY-maze software (Version 5.6; Stoelting Co., Wood Dale, Il).
Statistical analyses
Statistical analysis was performed using GraphPad Prism 6.0 (GraphPad Software, La Jolla, CA, USA). Results are presented as means ± 95% confidence intervals (CI) for the in vitro experiments and as mean ± SD for the in vivo experiments. Unpaired t test and Mann–Whitney test were performed to assess the difference between groups as appropriate. Two-way ANOVA was performed when two groups (first factor) were compared over the time (second factor). This test was used to compare the GC7 effect on motor recovery and cognitive recovery. Statistically significant value was set to p < 0.05, *p < 0.05, **p < 0.01, ***p < 0.001.
Results
GC7 decreases neuronal death induced in vitro by oxygen–glucose deprivation and ischemic-related stress
A potential anti-ischemic effect of GC7 was first evaluated in vitro, by using primary cultures of cortical neurons submitted to oxygen–glucose deprivation (OGD). GC7 treatment (30 µM) provided significant neuroprotection against 60-min OGD and even against a more severe 120-min OGD insult (Figure 1(a)). GC7 protection against OGD-induced necrosis was confirmed based on use of the cell death marker propidium iodide (PI) measured during 24 h following reperfusion. Reductions by factors of 4.5, 1.9 and 1.8 in the PI-positive neurons were observed immediately, 6 h and 24 h after OGD (Figure 1(b)), suggesting an inhibition of multiple steps in the neurodegenerative cascade by GC7-based inhibition of eIF5A hypusination. OGD induces energy failure, thereby disrupting transmembrane ion gradients and mitochondrial function. Specifically, extracellular glutamate elevations causes over-activation of N-methyl-D-aspartate (NMDA) glutamate receptors, promoting excessive membrane depolarization, calcium and sodium entry in the cell and calcium release from the mitochondria, and reactive oxygen toxicity. 5 We therefore investigated whether GC7 could protect primary neuronal culture against NMDA glutamate receptor- and hydrogen peroxide-induced neurotoxicity. GC7 treatment fully abolished neuronal cell death induced by a 30-min exposure to 100 µM NMDA and reduced neuronal death induced by a 30-min exposure to 300 µM H2O2 from 33% to 15%, measured 24 h after reperfusion (Figure 1(c)). We also evaluated GC7 efficiency against neuronal death induced by KCL depolarization (Figure 1(c)), which causes voltage-gated L-type Ca2+ channels to open, triggering an overwhelming influx of Ca2+ entry into neurons. GC7 treatment failed to protect against 50 µM KCl challenge. This toxic pathway differs from the mitochondrial collapse induced by OGD and NMDA receptor overactivation.21,22 Taken together, the data point to mitochondrial protection as a pivotal step in protection by GC7.
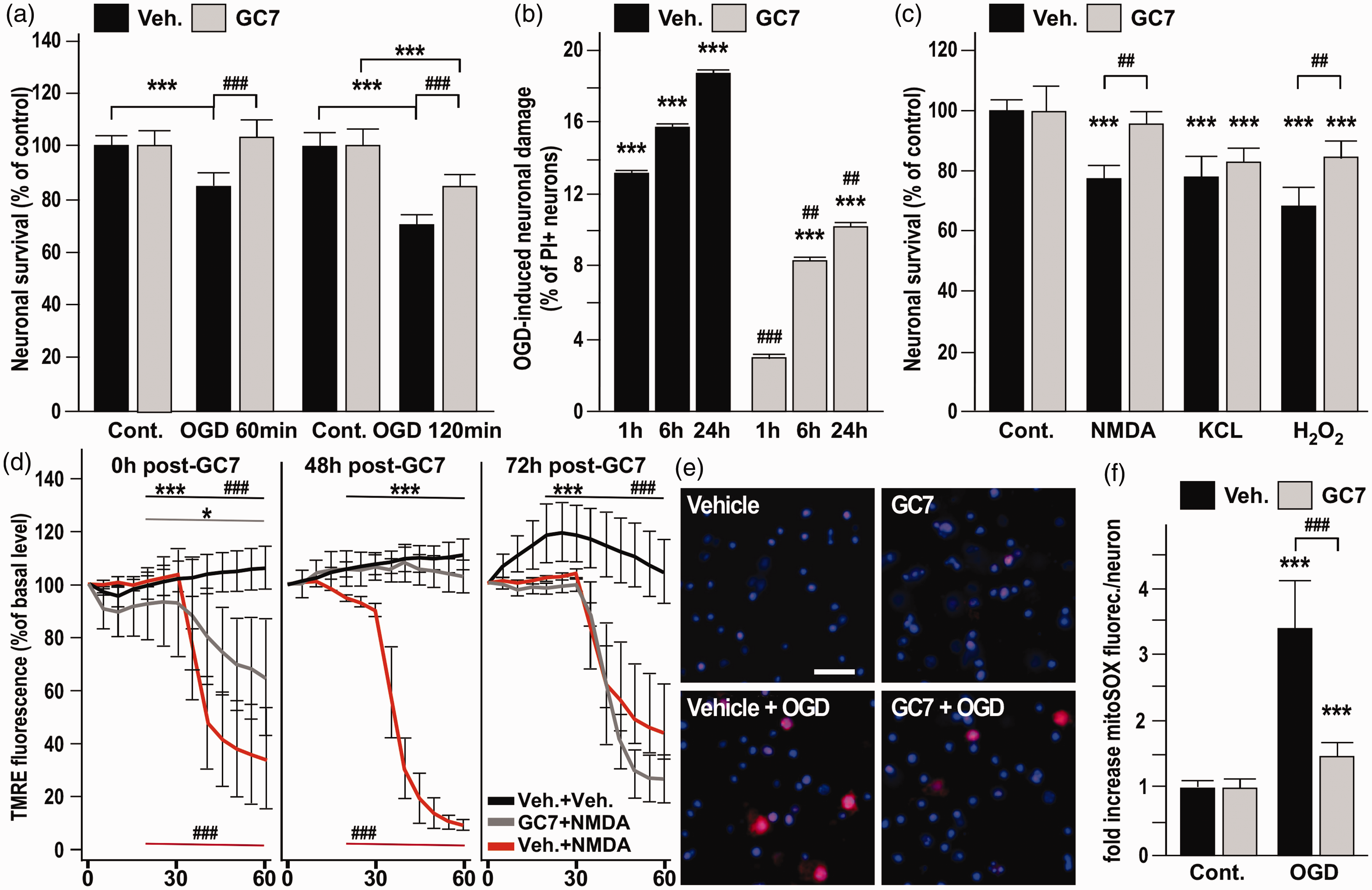
GC7 decreases neuronal death induced in vitro by ischemic-related stress by protecting mitochondria from membrane potential failure and ROS generation. (a–b) Cortical neurons exposed to GC7 were more resistant to 60 and 120 min OGD (a) and display a reduced PI-uptake measured during 24 h following 120 min OGD (b). (c) Protective effect of GC7 against 30-min exposure to 100 µM NMDA and to 300 µM H2O2, but not to 50 µM KCL. (d) TMRE fluorescence is preserved in the GC7-treated neurons indicating a protective effect in preventing the loss of ΔΨ m induced by NMDA, immediately after and 48 h, but not 72 h after the GC7 pretreatment. *p<0.05 and ***p<0.001 versus respective vehicle (Veh.)+NMDA-treated condition. ###p<0.001 versus GC7+NMDA-treated condition. (e, f) GC7 reduces OGD-induced mitochondrial superoxide generation in primary neurons stained with MitoSOX™, as shown by representative micrographs (e) and quantification of relative red fluorescence per neurons 24 h after OGD (f). ***p<0.001 versus respective control basal condition. ##p<0.01, ###p<0.001 versus vehicle-treated condition.
GC7 protects mitochondria from membrane potential failure
Since mitochondria represents an appealing target for the development of new neuroprotective pharmacological interventions,23,24 we then focus on GC7-effect on mitochondrial function, using TMRE, a voltage-sensitive marker that preferentially accumulates into the mitochondria due to the negative ΔΨ m. 25 Strikingly, the decrease in ΔΨm induced by 30 min of NMDA challenge was significantly prevented by GC7 (Figure 1(d)). To elucidate the temporal window of protection, we evaluated if GC7 preserved ΔΨ m even when the NMDA challenge was administered late after GC7 elimination from the medium (48 and 72 h). The loss of about 80% of TMRE fluorescence was still abolished 48 h after GC7 treatment, whereas 72 h after GC7 treatment no further protection of the ΔΨ m against NMDA depolarization was provided (Figure 1(d)). This identifies steady and long-lasting beneficial effects of GC7 on mitochondria integrity in GC7-treated neuronal cultures. These may not only prevent neuronal death during ischemia, but may also facilitate maintenance and recovery of neuronal function that are ATP dependent.
Preservation of mitochondria membrane potential and thereby of prevention of ROS generation are converging points for GC7 protective effect
The loss of ΔΨm is a hallmark of neuronal cell death linked to mitochondrial dysfunction causing excessive ROS production, elevated extra-mitochondrial [Ca2+], and decreased [ATP]i.26,27 The above mitochondrial results suggested that GC7 might also protect against these three prominent features occurring at the early stages of acute glutamate excitotoxicity and ischemic stroke (in both the necrotic and apoptotic components of stroke).
We therefore explored the potential of GC7 to reduce OGD-induced oxidative stress and superoxide generation in mitochondria, using the mitochondrial selective dye MitoSOX Red. 28 Remarkably, the increase in superoxide levels observed after OGD was strongly prevented in neurons treated with GC7 (Figure 1(e) and (f)), confirming a comparable mechanism of protection by inhibiting ROS production in both renal 6 and neuronal cells. In addition, mitochondria protection by GC7 prevented OGD-induced [Ca2+]i accumulation (Figure 2(a)) and [ATP]i drop (Figure 2(b)). Since the correlated rise in [Ca2+]i and drop in ATP are initiated by depolarization of the neuronal membrane in response to ischemia, our observation raise the possibility that GC7 displayed an unexpected beneficial action on membrane potential of neurons that prevents them from ischemic depolarization. Therefore, we further evaluated the effect of GC7 on the resting membrane potential. Whole-cell patch clamp recordings in cortical neurons indicated that GC7 induces membrane hyperpolarization (Figure 2(c)) that could be attributed to the opening of K+ channels, an inherent protective mechanism constituting another exciting target for neuroprotection. 29 Pharmacological inhibition of specific K+ currents showed that GC7-induced hyperpolarization was fully blocked by the specific Ca2+-activated K+ (K(Ca)) channel toxins apamin (100 nM) and charybdotoxin (100 nM), and the KATP channel blocker glibenclamide (10 µM), suggesting that GC7 did not target a specific K+ channel at the membrane, but that the enhanced neuronal K+ conductance at the membrane may occur in response to changes in intracellular concentrations of ATP or Ca2+. The KATP channels in the mitochondrial membrane are known to link the cellular energetic state to the membrane potential. 30 Their specific blockade by 5-hydroxydecanoate (5-HD, 10 µM) also fully prevented GC7-induced hyperpolarization (Figure 2(c)), pinpointing the mitochondrion as a converging point for GC7 protective effect. Indeed, the beneficial effect of GC7 strengthening the inherent protective action of increase K+ conductance is probably a by-product of GC7 effect on mitochondria related to lowered ATP production and increased [Ca2+]i in neurons cultured in Ca2+-deprived medium (Figure 2(d)).
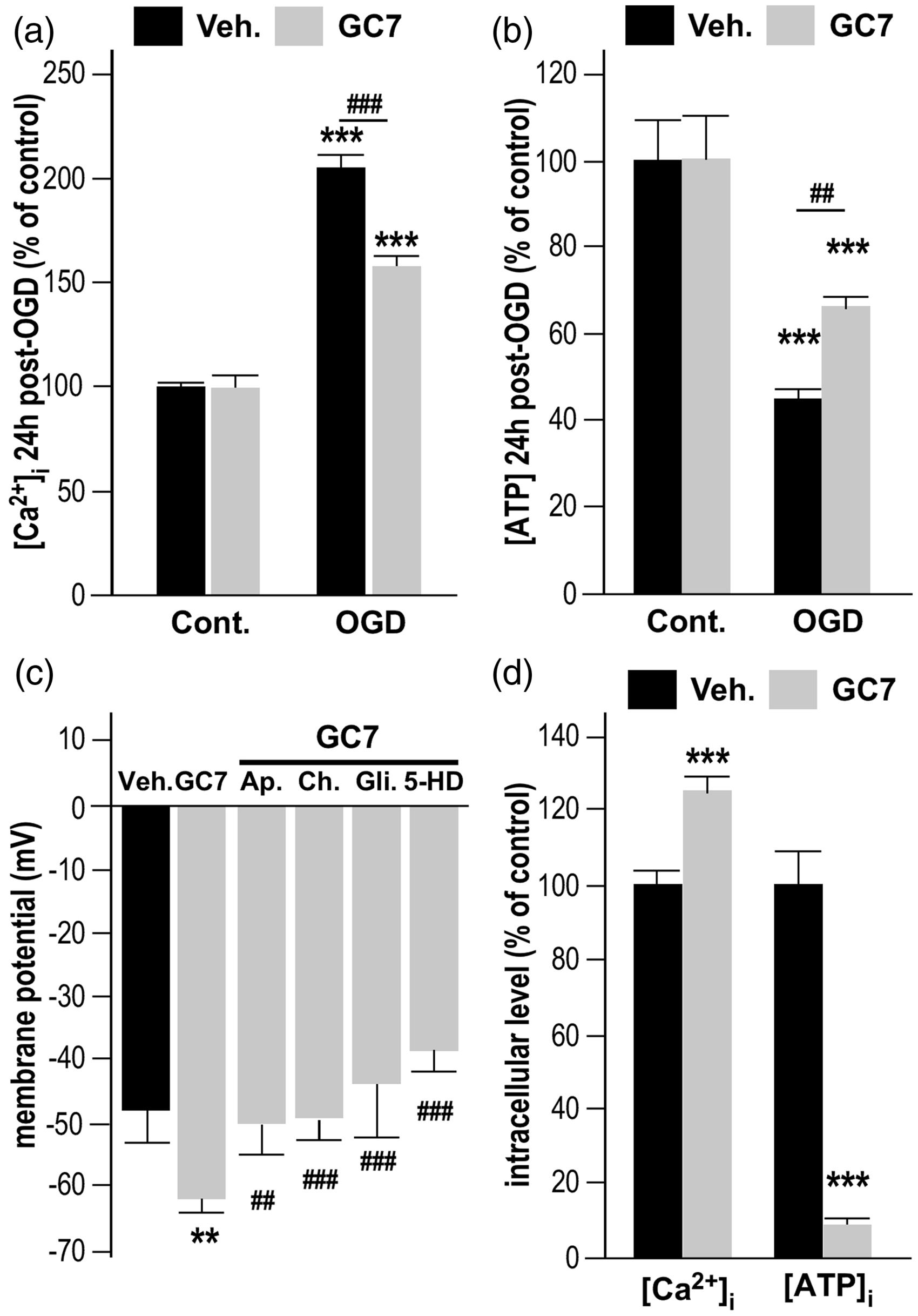
GC7 preserves the cellular energetic state and the membrane potential. (a) GC7 reduces [Ca2+]i accumulation in cortical neurons exposed to OGD. (b) GC7 also reduces OGD-induced [ATP]i drop. (c) GC7 induces a membrane hyperpolarization that was fully blocked by the specific Ca2+−activated K+ (K(Ca)) channel blocking toxins apamin (100 nM) and charybdotoxin (100 µM), the KATP channel blocker glibenclamide (10 µM), and the mitochondrial KATP channels blocker 5-hydroxydecanoate (5-HD, 10 µM). (d) When cultured in Ca2+−lacking medium, GC7-treated neurons display an elevated [Ca2+]i and lower ATP production, corroborating an adaptation of the mitochondrion to GC7 treatment. ***p<0.001 versus respective control (cont.) condition. ##p<0.01, ###p<0.001 versus vehicle-treated condition.
Both GC7 intraperitoneal pre- and post-treatment reduce brain lesion after stroke
Thus far, GC7 targeting of the mitochondria successfully achieved experimental in vitro neuronal protection through reducing mitochondrial dysfunction and ROS generation, Ca2+ accumulation, and ATP reduction. Given its in vivo beneficial effect when injected (3 mg/kg daily for three days, intraperitoneal (IP)) prior to 40 min transient unilateral renal artery ischemia, 6 we hypothesized that an IP injection of GC7 could protect the brain against acute ischemic stroke in a clinically relevant transient focal cerebral ischemia (tFCI) and reperfusion model. CG7 should share the same properties of polyamines in demonstrating some permeability across the blood–brain barrier 31 and the stroke-induced alteration of the blood–brain barrier 32 should further greatly enhance GC7 permeability. We found that not only a similar pretreatment protocol by three injections 48, 24, 2 h before tFCI (Figure 3(a)), but also a single injection 2 h before tFCI, reduced cerebral infarction size (Figure 3(b)). Moreover, a single IP injection of GC7 provided 2 h after reperfusion induced a similar brain protection (Figure 3(c)).
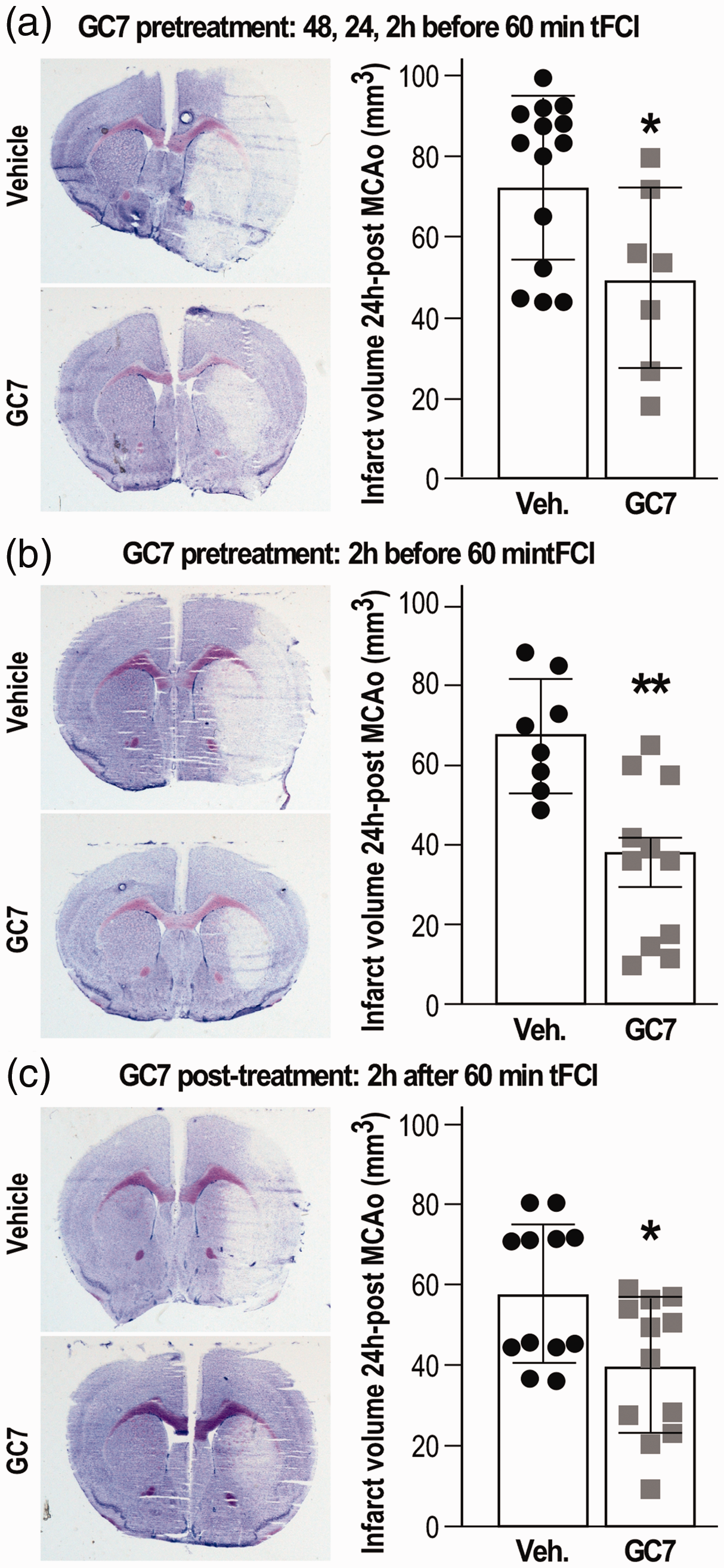
GC7 intraperitoneal injection reduces tFCI-induced infarct volume. (a) Three injections of GC7 (3 mg/kg daily for three days) or (b) a single injection 2 h before tFCI or (c) a single post-treatment 2 h after tFCI significantly reduce tFCI-induced lesion as illustrated by representative cresyl violet staining and quantitation of the infarct volume. N = 7–12/group, *p < 0.05, **p < 0.01 versus vehicle-injected mice.
GC7 post-treatment improves long-term functional recovery
The successful translation of new therapeutic opportunities will depend on their efficacy on functional stroke-induced deficits. We therefore evaluated the effect of GC7 post-treatment in the 30-min tFCI model, which is recommended for studying long-term functional outcomes in mice. 19 In this model, the temporary impairment of motor function (animals showed near baseline recovery by day 4 post-tFCI) correlates to an anatomical reorganization of undamaged brain areas and not to a decrease of infarct volume. 19 GC7 post-treatment (IP injection 2 h and two days post-tFCI) facilitated post-stroke recovery of motor functions assessed using rotarod (Figure 4(a)). As expected, the histological examination of brain sections four days after tFCI (Figure 4(b)) did not relate motor dysfunctions improvement with any reduction of the infarct. As this result indicated an overall better preservation of neuronal function by GC7, we further determined the effect of GC7 on cognitive deficits using the Morris water-maze test at two weeks after surgery. In the 30-min tFCI, the low long-term mortality that is too low to be improved by any treatment, 17 was not influenced by GC7 post-treatment (Figure 4(c)), while distinct beneficial effects could be discriminated on long-term impairment of spatial learning (Figure 4(d)) and memory (Figure 4(e)). Overall, GC7-treated mice exhibited improved searching behaviors traveling more distance in the target quadrant where the platform was previously located compared with the vehicle-treated mice (Figure 4(f)).
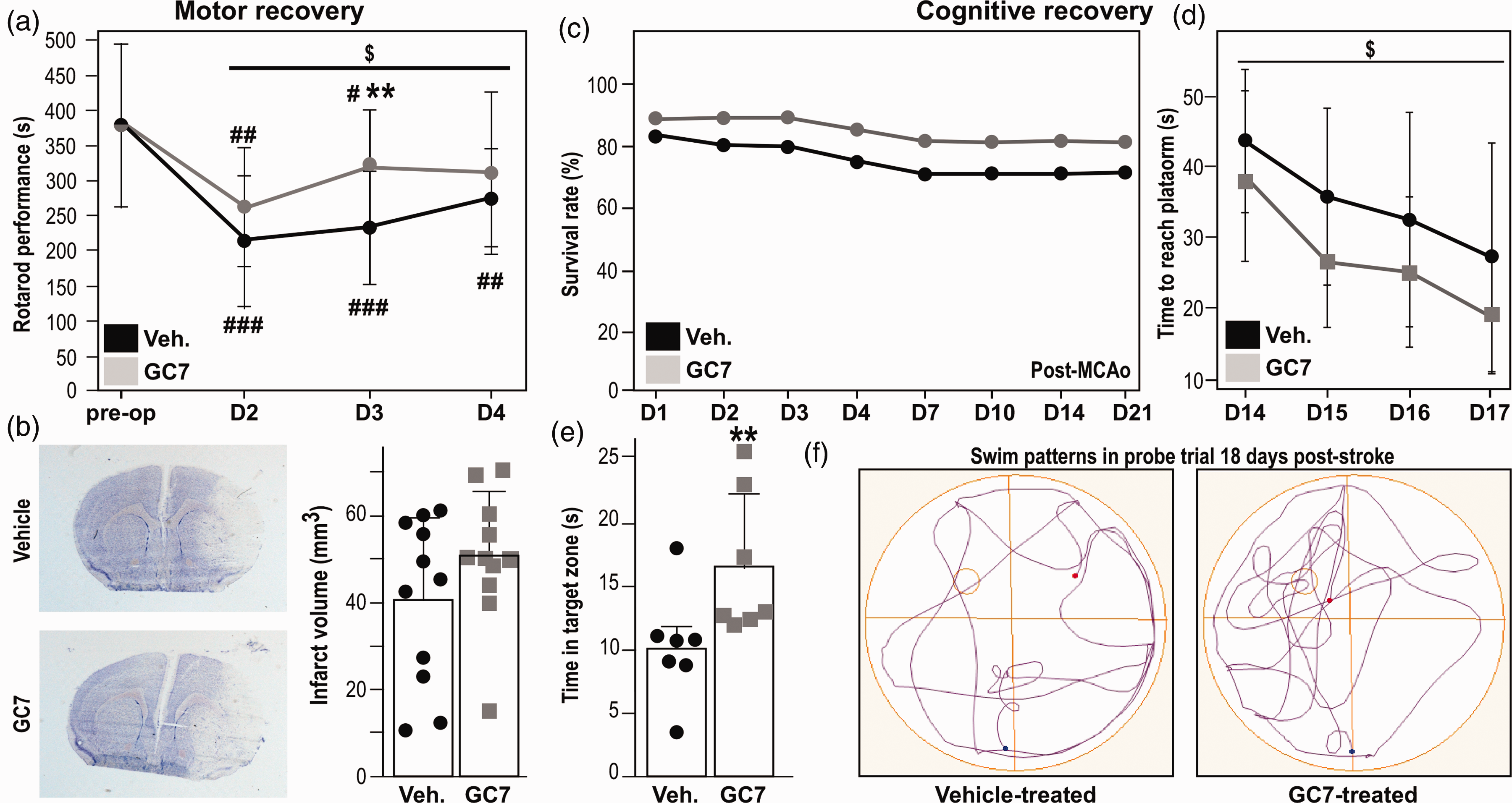
GC7 post-treatment reduces motor and cognitive impairment post-stroke. (a) The post-tFCI rotarod performance of GC7-treated mice is significantly improved during the four days of recovery (2W ANOVA: effect of group: F = 4.11, $p = 0.04594). n=11 per group at each time point) #p<0.05; ##p<0.01; ###p<0.001 versus pre-tFCI performance; **p<0.01 different from vehicle-treated control. (b) Histological examination of brain sections after 30 min tFCI did not relate motor dysfunctions improvement with any reduction of the infarct (n = 11 per group). (c) Long-term survival rate after 30 min of tFCI is similar between GC7 and vehicle-treated groups (n = 14 in each group). (d) In the Morris Water-Maze test, during the learning period, GC7 post-treatment improves latency to find the hidden platform (2W ANOVA: effect of group: F = 5.13, $p = 0.0263), and (e) during the probe trial, GC7 post-treatment increased the time spent by mice in the target quadrant (platform removed), **p<0.01, N=7 in each group. (f) Representative swim patterns 18 days post-stroke illustrate that GC7-treated mice display a better probe strategy.
Discussion
In vitro primary neuronal culture treated by GC7, the best-known inhibitor of DHS exhibited reduced cell death associated with ischemic-related stress, such as OGD, NMDA excitotoxicity and oxidative stress. The GC7 anti-ischemic effect was also found against a gold standard in vivo model of stroke. Using different tFCI durations, we demonstrated that GC7 treatment improves both neuronal survival and spontaneous functional recovery after stroke. To the best of our knowledge, it is the first proof of concept that pharmacological interfering with the polyamine-eIF5A-hypusine axis with GC7 might have novel implications for therapeutic controlling of stroke consequences. Beyond this primarily goal, our study provides new mechanistic insights identifying mitochondria as the ultimate destination in GC7 anti-ischemic effects. This echoes a recent discovery intricately linking eIF5A and cellular metabolism and respiration. 9 The fact that acute inhibition of eIF5A hypusination could dampen the Krebs cycle that highly needs oxygen for functioning while leaving glycolytic ATP synthesis operative 9 may be salutary in ischemic conditions. Consistent with these observations, by preventing the complete oxidation of glucose that produces large amount of ATP in the mitochondria compared to glycolysis, GC7 dampens the ATP production in neurons in physiological condition, while maintaining sufficient ATP to sustain transitory cell membrane integrity, functioning and survival under OGD. Moreover, it was recently suggested that, in hypoxic-ischemic brain injury, outcomes would decline as mitochondrial function becomes more dependent on oxygenation. 33 Therefore, by modifying the mitochondria dependency for oxygen supply, which is normally critical for the survival of neurons, GC7 treatment may improve the metabolism at the cellular level at a lower oxygenation level, resulting in a more favorable outcome.
In ischemic stroke where oxygen and energy supply are temporary defective, glycolysis-derived lactate, a crucial energy source used by neurons and other cells types was recently recognized for dampening neuronal excitability, triggering neuroprotection and improving neuronal plasticity. 34 Not only it is possible that GC7 also exerts a direct protective effect on astrocytes, but improving neuronal survival and function should help support the metabolic coupling between neurons and astrocytes and therefore the dynamic cooperation between these two cell types. Maintaining some neuronal activity would stimulate astrocytic lactate formation and release to support the metabolic/functional needs of adjacent neurons and oligodendrocytes. 35
Another type of positive feedback loop between neutrons and astrocytes – and even other cell types – that may also boost the tissue viability by helping with the functionality of surrounding cells is the transfer of healthy mitochondria. 36 Since the consensus that mitochondrial function reflects stroke disease states, and is readout of neurobehavioral recovery, transfer of healthy mitochondria to ischemic cells has become an appealing therapeutic approach for stroke that may account for the protective action of stem cell therapy. 37 Therefore, GC7-induced preservation of functional mitochondria fosters a therapeutic pool of healthy mitochondria available for transfer to dampen secondary cell death processes, including ROS generation and inflammation. Overall, re-orienting cells metabolism to adapt to the ischemic situation, the inhibition of eIF5A hypusination may present a crucial advantage in the less-severely ischemic “penumbral” tissue that suffer more of the limited availability of oxygen than glucose to preserve the cell and mitochondria integrity.
While our new results on mitochondrial membrane potential, ROS generation and ATP production reveal the potential of GC7 to preserve mitochondrial function after stroke, another parameter that would have been interesting to measure is the post-stroke mitochondrial oxygen consumption rate (OCR) with and without GC7 treatment in both the parenchyma and the cerebral vessels. Indeed, the response or sensitivity to ischemia/reperfusion may be different depending on the cell type, especially in vessels. Moreover following experimental stroke, OCR increases in the ischemic middle cerebral arteries (MCAs) compared with contralateral and naïve MCAs. 38 Showing that GC7 could correct such stroke-induced change in MCAs OCR would further support the concept that GC7 protection against ischemia may extend to the cerebro-vasculature by preserving mitochondrial bioenergetic function.
Interestingly, mitochondria is recognized as a common and crucial biochemical target of several FDA-approved drugs that have not been followed up clinically for stroke, while available and/or approved for other applications. 39 Therefore, our study should lead to increasing interest in developing and testing new inhibitors of eIF5A and rapidly translate to patient care, considering the well-documented safety and tolerability of the compounds targeting the polyamine biosynthetic pathway. Indeed, some of them are extensively studied in cancer,40,41 including in phase I clinical trial for neuroblastoma, the most common cancer in children, 42 non-small cell lung cancer 43 and metastatic breast cancer. 44 A striking example is DFMO, α-difluoromethylornithine a pharmacological inhibitor of the ornithine decarboxylase, which was used for the treatment of African sleeping sickness 45 and positively evaluated in phase III randomized trial against anaplastic gliomas 46 and is also under investigation as a chemopreventive agent 47 was shown to protect both cardiomyoblasts 48 and neuronal cultures against ischemia. 49 Likewise, targeting eIF5A is under investigation for treating several human cancers 50 and the safety and tolerability of intravenous infusion of chemical inhibitor of eIF5A have already been evaluated in Phase I–II in patients with relapsed or refractory B-cell malignancies. 51 Taking advantage of the clinical data on pharmacokinetics, toxicity, safety and tolerability (e.g. knowledge of the absence of dose limiting toxicities, DLTs) would serve as basis in shaping the direction of novel “drug repurposing/repositioning” research study, in which a clinically approved drug is investigated for a novel application that is for the present application of its therapeutic potential in ischemic stroke.
Interestingly, the specific inhibitory effect of GC7 on eIF5A hypusination has been investigated in different organs, in both male and female, in mice52,53 and rats 6 and these previous in vivo studies in mice showed evidence that GC7 does not induce noticeable side effects. Nevertheless, based on sex differences to stroke vulnerability 54 and, perhaps, to neuroprotective treatment as well, the effect of GC7 should be tested in both sexes for safety and tolerability. Despite GC7, the best-known inhibitor of DHS displaying an IC50 of 17 nM, 55 was not used in human clinical trials; some studies with molecules exhibiting an inhibitory effect on DHS have been published and used in clinical trials. Semapimod also called CNI-1493 or AXD455 inhibits DHS with an IC50 around 2 µM. 56 Semapimod that is well tolerated by patients 57 was tested for Crohn’s disease58,59 and for the consequence of Endoscopic Retrograde CholangioPancreatography. 60 From these studies led in humans, it is tempting to reasonably hypothesize that acute targeting of DHS with a compound like GC7 presenting an higher affinity than those use in clinical trial should not lead to significant adverse effects. Moreover, the effectiveness of stroke reperfusion therapy remains limited because the majority of stroke patients lack timely access to intravenous thrombolysis or endovascular therapy, underscoring an unmet clinical need for adjunctive neuroprotective treatments. 61 Delaying the need for oxygen, thereby “buying time” by extending the therapeutic window for reperfusion therapy, makes this an innovative and promising pharmacological agent for future clinical protocols related to ischemic challenges in combination with reperfusion therapy.
Thereby, our findings should contribute accelerating clinical trial development by proposing a new research opportunity circumventing crucial issues regarding the translation of preclinical developments to the bedside.
Finally, the novelty of eIF5a as a therapeutic target against brain ischemia and the potential of GC7 in stroke therapy advocate potential promise in screening for GC7-structurally-related eIF5A hypusination-inhibitors and targeting signaling pathways altered by GC7 as a potential therapeutic strategy in humans suffering from ischemic diseases.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by a grant DPM 20121125559 from the Fondation pour la recherche médicale (FRM), and a grant from the Société d’Accélération de Transfert de Technologie (ValorPACA/SATT Sud-Est) from France.
Acknowledgements
We are very grateful to Dr. Hamid Moha Ou Maati for his skillful technical support and advices with the electrophysiology, Dr. Edith Hamel and Dr. Joseph S. Tauskela for their critical reading of the manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
MB, EG, NM, JF and NB performed the experiments. MB, EG, MT and NB analyzed the data. MB, CH, MT and NB wrote the paper.