
Abstract
Reperfusion triggers an oxidative stress. We hypothesized that mild hypoxemia in reperfusion attenuates oxidative brain injury following hypoxia-ischemia (HI). In neonatal HI-mice, the reperfusion was initiated by reoxygenation with room air (RA) followed by the exposure to 100%, 21%, 18%, 15% oxygen for 60 minutes. Systemic oxygen saturation (SaO2), cerebral blood flow (CBF), brain mitochondrial respiration and permeability transition pore (mPTP) opening, markers of oxidative injury, and cerebral infarcts were assessed. Compared with RA-littermates, HI-mice exposed to 18% oxygen exhibited significantly decreased infarct volume, oxidative injury in the brain mitochondria and tissue. This was coupled with improved mitochondrial tolerance to mPTP opening. Oxygen saturation maintained during reperfusion at 85% to 95% was associated (
Introduction
Reintroduction of O2 to the ischemic brain during reperfusion is critical for tissue survival. However, the same event triggers an oxidative stress, one of the central mechanisms of reperfusion injury. Current clinical practice is the maintaining systemic oxygenation at normoxemic (oxygen saturation (SaO2)=95% to 99%) levels in infants with hypoxia-ischemia (HI)-brain injury. However, there is a large body of experimental evidence that systemic hyperoxemia during early 30 to 60 minutes of reperfusion markedly exacerbates brain injury in newborn animals subjected to HI-insult (Koch et al, 2008; Munkeby et al, 2004; Richards et al, 2007). The primary mechanism for this hyperoxemia-driven worsening of HI-brain injury is exacerbation of an oxidative stress associated with excessive generation of reactive oxygen species (ROS).
During HI, the ability of mitochondria to generate ATP by oxidative phosphorylation is severely inhibited (Caspersen et al, 2008; Gilland et al, 1998; Ten et al, 2010). Reoxygenation/reperfusion restores mitochondrial ADP-phosphorylating capacity, normalizing ATP content in the postischemic brain (Lorek et al, 1994). However, following several hours of reperfusion, mitochondria again exhibit a profound decline in ATP-generation capacity, known as a secondary energy failure (Halestrap, 2010; Kuroda et al, 1996; Lorek et al, 1994; Puka-Sundvall et al, 2000). The molecular mechanism proposed to explain secondary energy failure is the concept of mitochondrial membrane permeabilization secondary to opening of mitochondrial permeability transition pore (mPTP). Triggered by accumulation of Ca++, an opening of mPTP results in dissipation of mitochondrial proton-motive force, which renders mitochondria incapable of energy production and ultimately leads to mitochondrial swelling and release of pro-apoptotic proteins. The formation of mPTP during reperfusion can be triggered by ROS, even in the presence of intra-mitochondrial Ca++ chelator (Kim et al, 2006). We hypothesized that mild hypoxemia during early reperfusion limits O2 supply for the reperfusion-accelerated generation of ROS. This attenuates oxidative stress to the mitochondrial matrix, and improves mitochondrial tolerance to Ca++-induced mPTP opening. Because mPTP opening is considered as a key mechanism for secondary energy failure, we propose that mild hypoxemia maintained during early reperfusion will improve post-HI-brain recovery.
Materials and methods
Induction of Unilateral Hypoxia-Ischemia and Study Groups
In accordance with the National Institute of Health Guidelines for animal research, all animal procedures for these experiments were reviewed and approved by the Institutional Animal Care and Use Committee of the Columbia University. We used the Rice-Vannucci model of HI-brain injury adapted to p10 neonatal mice (Ten et al, 2003, 2004). Briefly, following a permanent ligation of the right carotid artery, at 90 minutes of recovery pups were exposed to hypoxia (8% O2 balanced N2) for 20 minutes. The ambient temperature during hypoxia was maintained at 36.5°C to 37.5°C by placing the hypoxic chamber in a neonatal isolette (Airshield, Mooresville, NC, USA). Following 5 minutes of reoxygenation with room air (RA), all mice were randomly assigned to four study groups: RA—reoxygenation continued with RA, FiO2=0.21 (initial
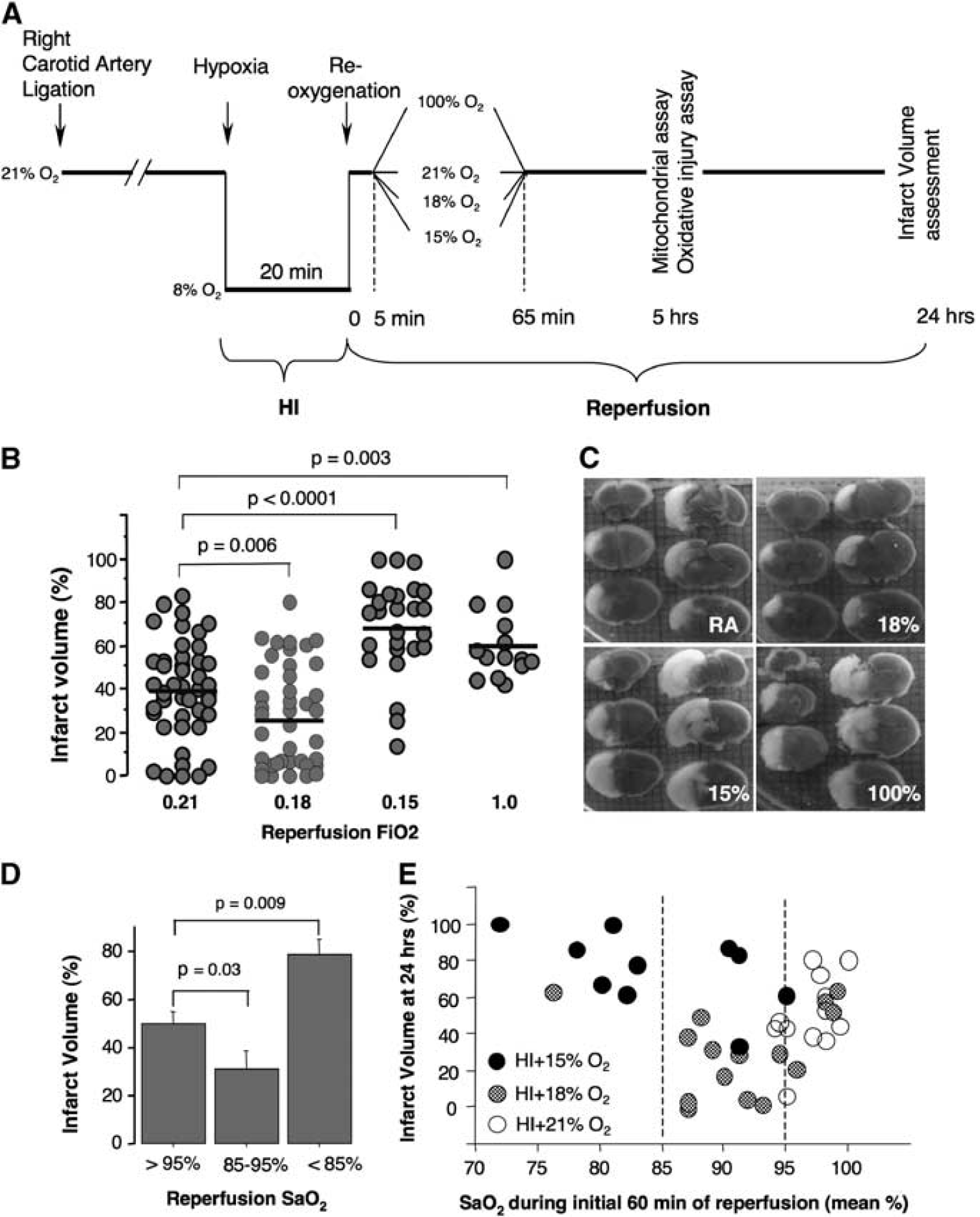
(
The exposure to different FiO2 was performed in 1 L tightly sealed glass jars at the ambient temperature 32°C. Gas flow was maintained at 2 L/min. Following 60 minutes of exposure to a different FiO2, mice were returned to their dams. To minimize a temperature-related variability in the extent of brain injury, during initial 12 hours of reperfusion mice were kept in an isolette at the ambient
Measurements of Systemic Oxygen Saturation and Cerebral Blood Flow
In randomly selected mice (
Mitochondrial Isolation
The brain nonsynaptosomal mitochondria were isolated as described (Caspersen et al, 2008) with minor modification: the tissue was homogenized manually using a dounce homogenizer (Wheaton, Millville, NJ, USA) with 0.2 mm differential (10 strokes) followed by 0.1 mm differential (10 strokes) and the final mitochondrial pellet was resuspended in 0.07 mL of albumin-free sucrose buffer (Ten et al, 2010).
Mitochondrial Ca++ Upload Capacity Assay
Mitochondrial Ca++ upload capacity (threshold for mPTP opening) was estimated using Hitachi F-7000 spectrofluorimeter equipped with magnetic stirring, thermocontrol and Calcium green 5N fluorescence probe (Ex. 488; Em. 532; Invitrogen, Carlsbad, CA, USA). In brief, isolated mitochondria (0.05 mg) were incubated in 1 mL of 10 mmol/L MOPS-Tris buffer pH 7.4 containing 120 mmol/L KCl, 1 mmol/L KH2PO4, 10 μM EGTA, 5 mmol/L succinate, 2.5 mmol/L glutamate, and 1 μM Calcium green 5N. At 100 seconds of incubation, once fluorescence reached a steady-state tracing, 10 nmol CaCl2 were added every 50 seconds. The amount of Ca++ consumed by mitochondria until mPTP opened (evidenced by the Ca++ leak-out) was recorded and expressed in nmoles of Ca++ per mg of mitochondrial protein. The incubation conditions are described in the Figure 4 legend.
Mitochondrial Respiration Assay
Mitochondrial respiration was measured using a Clark-type electrode (Oxytherm, Hansatech, UK) as described (Caspersen et al, 2008). Mitochondria (0.05 mg of protein) were added to 0.5 mL of respiration buffer composed of 200 mmol/L sucrose, 25 mmol/L KCl, 2 mmol/L KH2PO4, 5 mmol/L HEPES-KOH (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) pH 7.2, 5 mmol/L MgCl2, 0.2 mg/mL of bovine serum albumin, fatty acid free (BSA), 30 μM Ap5A (
Assessment of Oxidative Damage in the Brain Tissue and Mitochondrial Matrix
The extent of oxidative brain injury was analyzed by visual detection and semiquantification of immunopositivity for markers of lipid peroxidation (4-hydroxy-nonenal, 4-HNE) and protein peroxinitration (3-nitrotyrosine, 3-NT). In brief, at 5 hours of reperfusion, the brains were harvested from randomly selected HI-mice, fixed in 4% paraformaldehyde and soaked in 30% sucrose overnight. In all, 20-μm thick coronal sections were blocked (10% donkey serum) and incubated with rabbit polyclonal anti-4-HNE (1:500) and anti-3-NT antibodies (1:100) as described (Zhu et al, 2007). Samples were examined using Bio-Rad 2000 confocal laser-scanning device (Hercules, CA, USA) attached to a Nikon E800 microscope (San Diego, CA, USA). The 4-HNE and 3-NT immunoreactivity was analyzed by the count of the ratio of immunopositive/total cells in two nonadjacent fields (× 40) of injured cortex at three different bregma levels (−1.0, 0, +1.0 mm). Thus, six areas of cortex were analyzed for each brain sample and mean value (%) of immunopositive cells per each mouse was used for statistical analysis. Only those mice that have developed signs of brain injury (absence or residual expression of microtubule-associated protein 2 (MAP2)) were used for data analysis.
To assess the extent of oxidative injury to the mitochondrial matrix, at 5 hours of reperfusion, aconitase activity was measured in cerebral mitochondrial fraction as described (Morrison, 1954). Frozen-thawed mitochondria were mixed with the reaction buffer (50 mmol/L Tris-HCl, pH 7.4, 0.6 mmol/L MnSO4, 5 mmol/L Na citrate, 0.5 mmol/L nicotinamide adenine dinucleotide phosphate (NADP), 1 U/mL iso-citrate dehydrogenase) in a 96-well plate and the absorbance changes at 340 nm were followed for 10 minutes with a plate reader (Tecan Infinite M200, San Jose, CA, USA). The aconitase activity was expressed in mU per minute per mg of mitochondrial protein. Aconitase activity is a well-known marker for oxidative mitochondrial damage (Bulteau et al, 2003; Sadek et al, 2002).
Statistics
The analysis of variance with Fisher's
Results
The Extent of Brain Injury Depends on Oxygen Saturation During Initial Reperfusion
Mice exposed to 18% O2 exhibited a significant (
FiO2 Alters Cerebral Blood Flow Recovery During Initial Reperfusion
Analysis of CBF changes in response to reperfusion with different FiO2 revealed three patterns of CBF recovery. Mice exposed to FiO2=1.0 demonstrated a dramatic (up to 300% of the pre-HI value) elevation of the CBF in the postischemic hemisphere (Figures 2A and 2B). In contrast, those mice that were breathing with FiO2=0.15, after brief normalization of the CBF in response to initial reoxygenation with RA, demonstrated significant reduction in the CBF, consistent with the values observed during HI-insult (Figures 2A and 2B). Mice reoxygenated with RA or 18% O2 exhibited similar patterns of CBF recovery (Figures 2A and 2B). An approximation of O2 delivery to the postischemic hemisphere during the 60 minutes of reperfusion (mean CBF in percentage of preischemic value × mean SaO2) showed that reoxygenation with 15% O2 substantially limited O2 delivery (≈4,200 a.u.) compared with mice reoxygenated with RA (≈10,670 a.u.) or 18% O2 (≈9,100 a.u.).
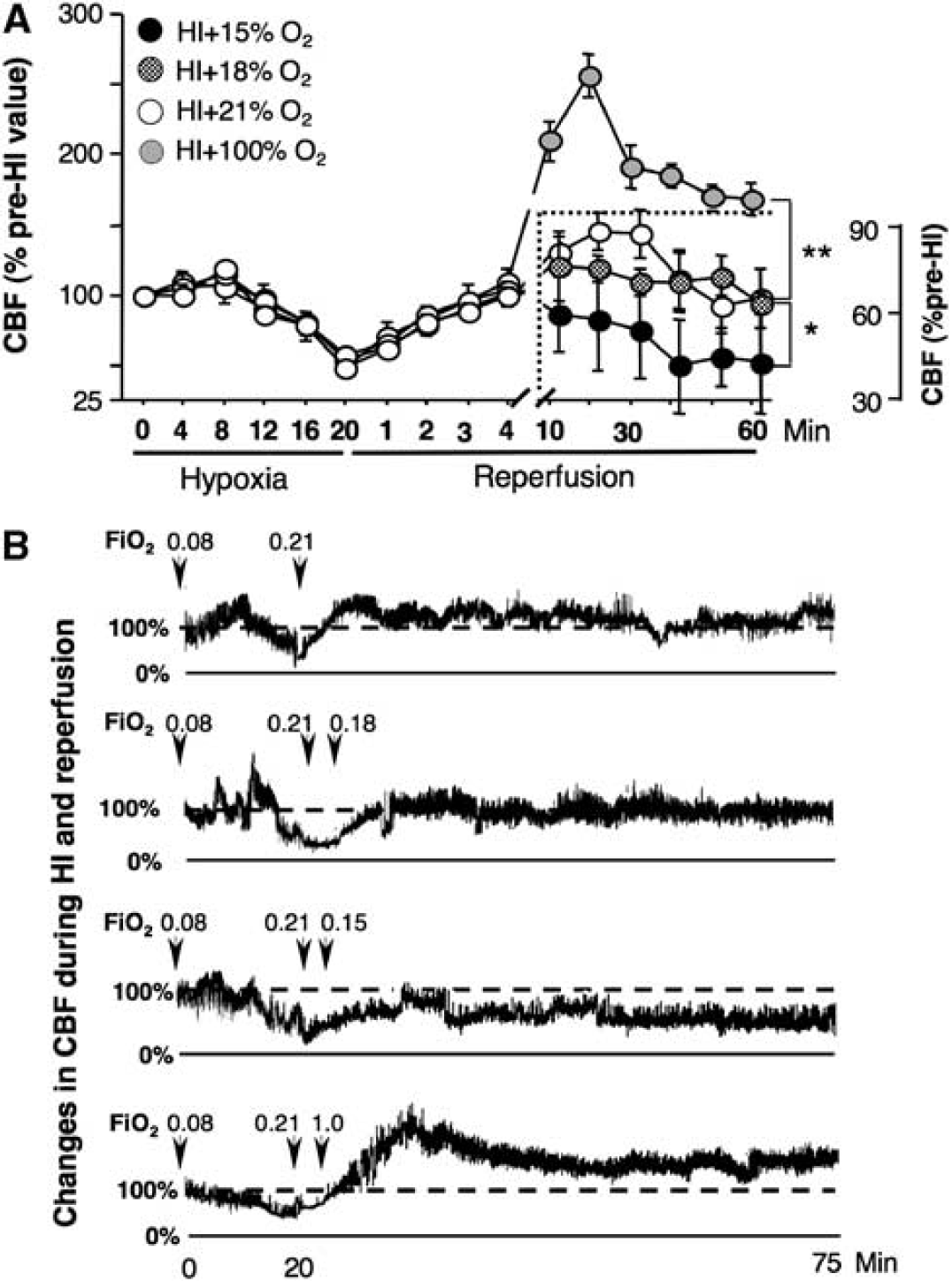
(
FiO2 During Initial Reperfusion Alters the Extent of Oxidative Damage to Brain Mitochondria and Tissue
Semiquantitative analysis of immunoexpression of 4-HNE and 3-NT revealed that at 5 hours of reperfusion HI-mice exposed to 100% O2 on reperfusion developed the most extensive oxidative injury of lipids and proteins in their brains. In contrast, mice exposed to 18% O2 exhibited significantly decreased proportion of 3-NT-positive cells compared with their RA-reoxygenated littermates (Figures 3A and 3B). The reduction in the proportion of 4-HNE-positive cells in these mice did not reach statistical significance (
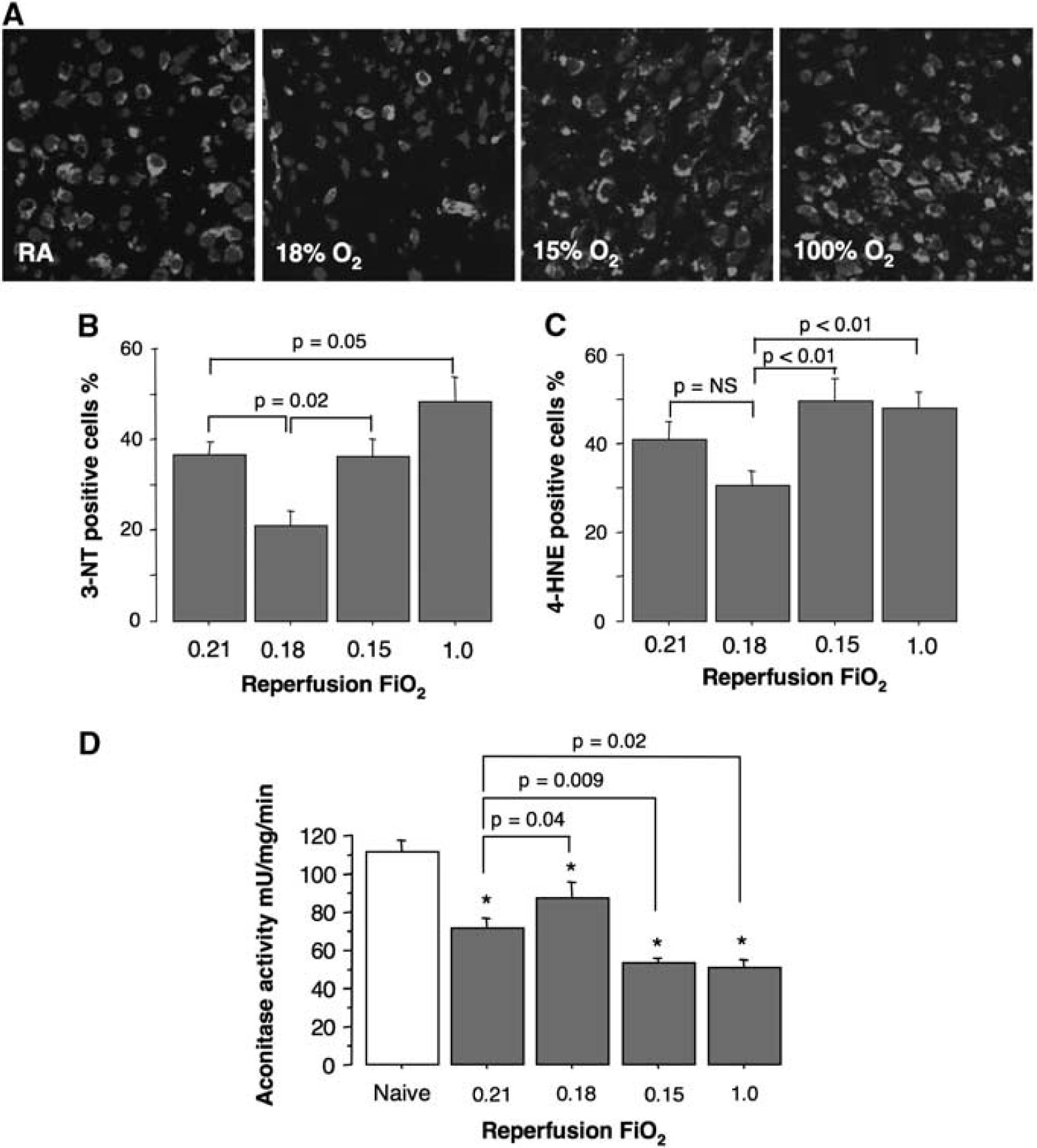
(
Mild Hypoxemia Limits Mitochondrial Permeability Transition Pore Opening and Secondary Inhibition of Oxidative Phosphorylation During Reperfusion
Compared with naives, the HI-insult significantly reduced Ca++ threshold for mPTP opening in all groups of mice (Figures 4A and 4B). However, the brain mitochondria isolated from the mice reperfused with 18% O2 demonstrated significantly better ability to buffer a Ca++ load compared with all other HI-mice (Figures 4A and 4B). In contrast, mice exposed to 15% or 100% O2 exhibited significantly poorer Ca++-buffering capacity compared not only with naive mice, but also compared with RA-reoxygenated littermates (Figures 4A and 4B). Figure 4C demonstrates that Ca++-buffering capacity assay used in this study tests cyclosporine-A-sensitive mitochondrial membrane permeabilization, which results in cytochrome-C release.
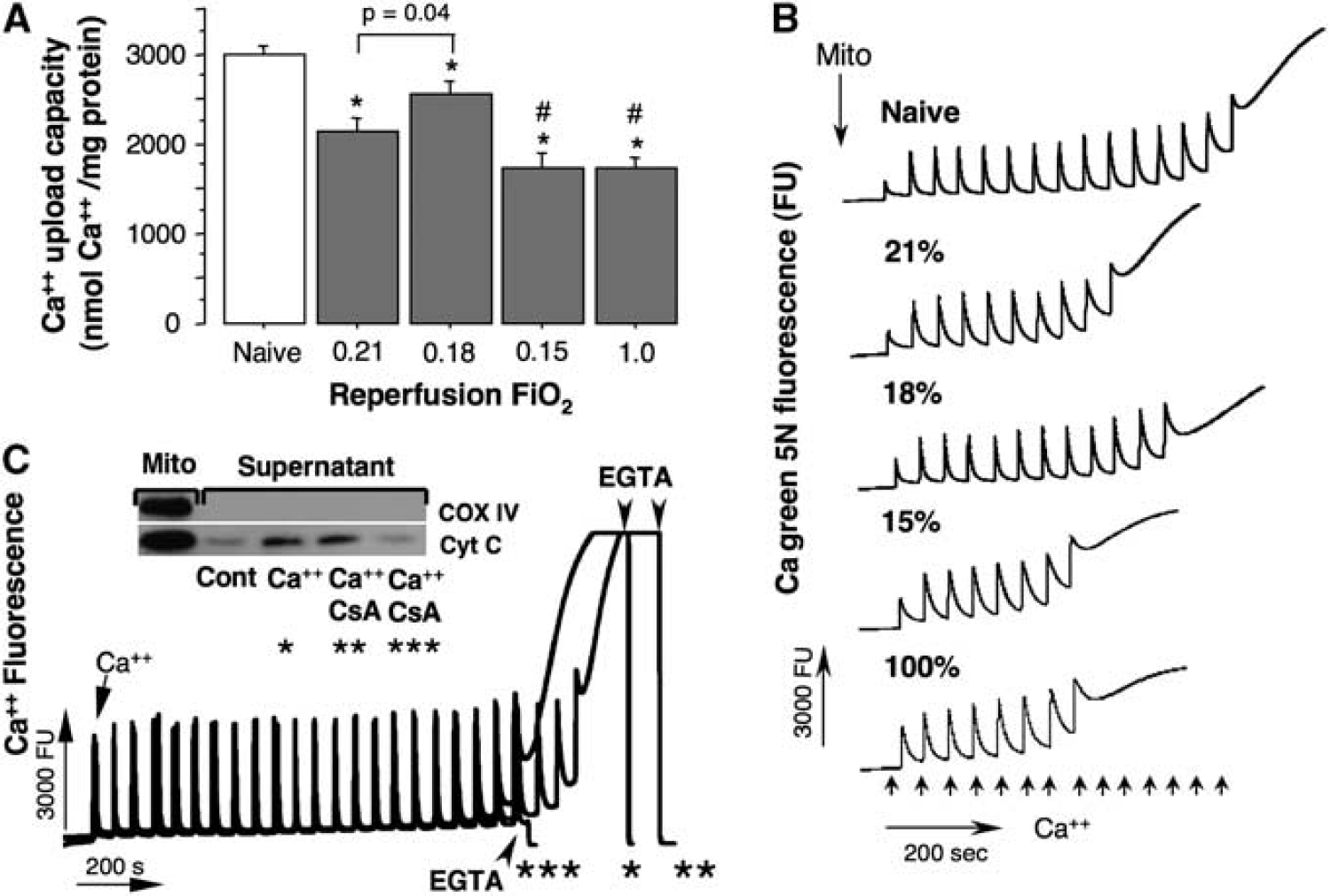
(
At the same time point of reperfusion (5 hours following HI-insult), mitochondria isolated from all groups of HI-mice demonstrated a significantly decreased state 3, ADP-phosphorylating, respiration rate compared with naive littermates (Figure 5A). However, compared with all other groups of HI-mice, mice reoxygenated with 18% O2 exhibited significantly better-preserved ADP-phosphorylating respiration rate (Figure 5A). The RCR was also significantly decreased in HI-mice (except those animals that were reoxygenated with 18% O2) compared with the naives (Figure 5B). No significant difference was detected in the state 4 (resting respiration rate) among all HI-mice (data not shown). Importantly, at this time point of reperfusion, mitochondrial ADP-phosphorylating respiration rate strongly (
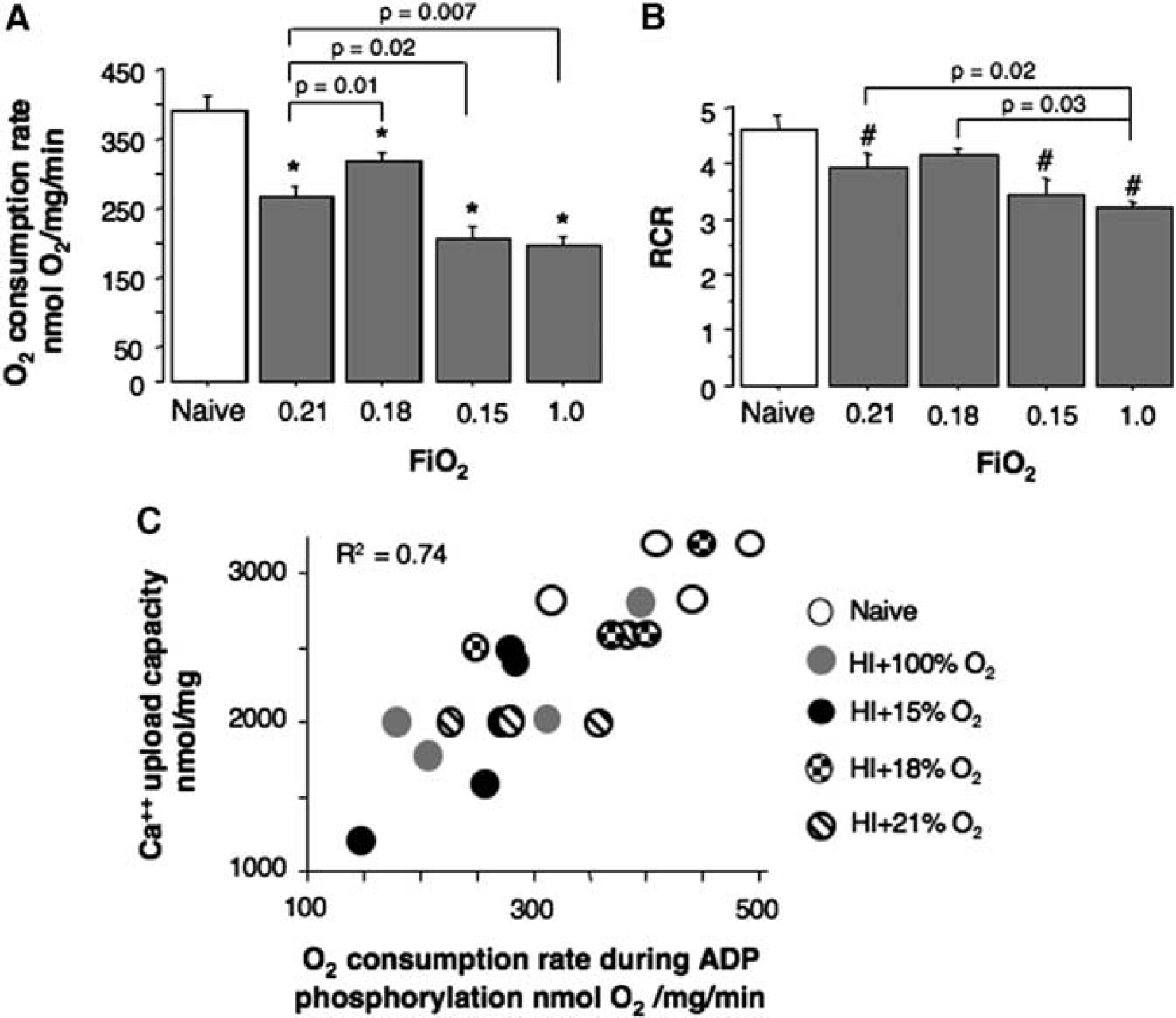
(
Discussion
This study demonstrates that changes in systemic oxygenation during the initial 60 minutes of reperfusion significantly alter the extent of HI-brain injury in neonatal mice. While, the reoxygenation with FiO2 1.0% or 0.15% markedly exacerbated brain damage, mild hypoxemia by the exposure to 18% oxygen was associated with a significant reduction in cerebral infarct volume compared with the RA-reperfused littermates. This suggests that different levels of systemic oxygenation at the initial stage of reperfusion contribute to the mechanisms involved in either propagation of HI-brain injury or cerebral recovery. There are numerous reports that hyperoxemia initiated during resuscitation and maintained for the initial 30 to 60 minutes of reperfusion is detrimental for brain recovery following HI-insult (Koch et al, 2008; Munkeby et al, 2004; Richards et al, 2007). However, it has never been shown that therapeutic maintenance of subnormal oxygenation during early reperfusion protects neonatal brain against HI-injury. It is important to note that in this study, the FiO2 was changed following 5 minutes of resuscitation initiated with RA. Earlier, we have shown that 5 minutes of reoxygenation with RA was required to achieve a full recovery of the cerebral circulation, the end point of resuscitation in this model (Presti et al, 2006). Thus, the neuroprotection exerted by the mild hypoxemia in our study is not applicable to the resuscitation paradigm, in which resuscitation with 18% oxygen was not beneficial for biochemical cerebral recovery in asphyxiated piglets (Jantzie et al, 2008).
Reintroduction of oxygen to ischemic tissue at the onset of reperfusion is the major initiating event for the reperfusion-driven oxidative stress. One of the sources for oxidative radicals in postischemic cells is mitochondrion. The ROS of mitochondrial origin can damage cells in the brain and heart during ischemia—reperfusion (Battaglia et al, 2010; Kim et al, 2006; Loor et al, 2010). We propose that mild hypoxemia applied shortly after HI limits O2 availability for the reperfusion-accelerated ROS formation in the mitochondria, and this attenuates an oxidative stress. Although, it is still debatable whether
It is important to note that data on pathogenic role of Ca++-triggered mPTP in the developing HI-brain are not straightforward. Wang et al (2009) has reported that neonatal mice (as opposite to adult mice) deficient in cyclophilin-D (the only known functional component of mPTP) exhibited exacerbation of HI-brain injury. These data challenge cyclophilin-D-dependent mPTP opening as a pathogenic mechanism for secondary energy failure in the developing HI-brain. Similarly, posttreatment with cyclosporine-A, antagonist of cyclophilin-D, did not protect neonatal rats against HI-brain injury (Puka-Sundvall et al, 2001). Recently, however, Hwang et al (2010) demonstrated that, cyclosporin-A, injected immediately after HI-insult significantly protected the developing brain, attenuating both necrotic and apoptotic cell death in neonatal rats. Similar results were obtained in neonatal rats subjected to a mild focal cerebral ischemia—reperfusion (Leger et al, 2010). In our study, the attenuation of secondary energy failure, defined by the preservation of mitochondrial ADP-phosphorylating activity was strongly (
When HI-mice were exposed to a greater degree of hypoxia (15% O2), this was associated with robust exacerbation of HI-brain injury. Compared with the littermates reperfused in normoxia and mild (18% O2) hypoxia, these mice exhibited a secondary decline in CBF, although initially, their CBF fully recovered in response to reoxygenation with RA. The degree of this reduction in CBF was comparable to that observed during HI, suggesting that the exposure to 15% O2 during reperfusion caused a secondary HI-insult, which may account for exacerbation of cerebral injury. While mild hypoxemia was associated with markedly attenuated oxidative stress, a further decrease in oxygenation resulted in an opposite effect, exacerbation of oxidative damage. This suggests that, if the brain experiences a secondary ischemic event, the oxidative injury propagates even in an environment with a low O2 content. For example, during oxygen—glucose deprivation, neuronal mitochondria markedly accelerated ROS generation in spite of the minimal O2 availability; when mitochondrial membrane potential collapsed the ROS generation decreased (Abramov et al, 2007). Similarly, cardiomyocytes exhibited enhanced mitochondrial superoxide production during simulated ischemia, leading to an opening of mPTP (Loor et al, 2010). Thus, an ischemic stimulation of mitochondrial ROS release may account for exacerbation of oxidative injury in HI-mice subjected to 15% O2 during reperfusion. Indeed, similarly to 100% O2 reoxygenated littermates, the brain mitochondria isolated from these mice exhibited a significant decrease in aconitase activity and Ca++-buffering capacity, suggesting that the severity of oxidative stress to mitochondria was comparable to that in HI-mice exposed to hyperoxia. Secondary ischemia may cause an additional intra-mitochondrial Ca++ flux (Kristian, 2004), further contributing to the loss of Ca++-buffering capacity.
To dissect-out the contribution of an oxidative stress driven by reperfusion, we exposed HI-mice to extreme hyperoxia (FiO2=1.0). Our data are in full agreement with previous reports, demonstrating exacerbation of oxidative neurologic damage in hyperoxic HI-mice compared with normoxemic littermates (Koch et al, 2008). The novel findings are that hyperoxic reperfusion was associated with dramatic overcirculation in the post-HI-brain. Given that exposure to 100% O2 during reperfusion substantially increases paO2 (Richards et al, 2007), the post-HI overcirculation is expected to result in excessive O2 delivery, potentiating generation of ROS and oxidative injury. In animals subjected to a focal brain ischemia—reperfusion injury, reactive/malignant hyperemia during reperfusion was strongly associated with exacerbation of oxidative stress, edema, intra-cranial hypertension, and the extent of injury (Heiss et al, 1997; Lee et al, 2004; Perez-Asensio et al, 2010). This part of our study offers an additional mechanistic explanation for deleterious effect of hyperoxic reperfusion on the outcome of HI-brain injury.
In conclusion, to our knowledge this is the first report, demonstrating that mild hypoxemia (SaO2=85% to 95%) maintained during early reperfusion protects immature brain against HI-insult. One of the mechanisms for this neuroprotection is attenuation of mitochondrial oxidative stress and improved mitochondrial tolerance to Ca++-triggered mPTP opening. The therapeutic effect of hypoxemic reperfusion, however, becomes detrimental if hypoxemia is severe enough to cause a secondary decline in cerebral circulation. This work offers the experimental background for the development of a novel therapeutic strategy in which the maintenance of subnormal SaO2 without deterioration of CBF during early reperfusion limits HI-injury to the immature brain.
Footnotes
The authors declare no conflict of interest.