
Abstract
SLC5A8 is a sodium-coupled monocarboxylate and ketone transporter expressed in various epithelial cells. A putative role of SLC5A8 in neuroenergetics has been also hypothesized. To clarify this issue, we studied the cerebral phenotype of SLC5A8-deficient mice during aging. Elderly SLC5A8-deficient mice presented diffuse leukoencephalopathy characterized by intramyelinic oedema without demyelination suggesting chronic energetic crisis. Hypo-metabolism in the white matter of elderly SLC5A8-deficient mice was found using 99mTc-hexamethylpropyleneamine oxime (HMPAO) single-photon emission CT (SPECT). Since the SLC5A8 protein could not be detected in the mouse brain, it was hypothesized that the leukoencephalopathy of aging SLC5A8-deficient mice was caused by the absence of slc5a8 expression in a peripheral organ, i.e. the kidney, where SLC5A8 is strongly expressed. A hyper-excretion of the ketone β-hydroxybutyrate (BHB) in the urine of SLC5A8-deficient mice was observed and showed that SLC5A8-deficient mice suffered a cerebral BHB insufficiency. Elderly SLC5A8-deficient mice also presented altered glucose metabolism. We propose that the continuous renal loss of BHB leads to a chronic energetic deficiency in the brain of elderly SLC5A8-deficient mice who are unable to counterbalance their glucose deficit. This study highlights the importance of alternative energetic substrates in neuroenergetics especially under conditions of restricted glucose availability.
Introduction
SLC5A8 was first identified by our group as a protein that was putatively involved in iodide metabolism. 1 In parallel, the human Slc5a8 gene was associated, by Li et al., 2 with tumor suppressor function in colon cancer. In 2004, two groups reported sodium-dependent inward currents induced by various monocarboxylates under voltage-clamp conditions with SLC5A8-expressing Xenopus laevis oocytes.3–5 Based on this property, it was proposed that the main physiological roles of SLC5A8 are sodium-coupled lactate absorption in the kidney and butyrate absorption in the colon.4,6,7 SLC5A8 expression was found in the apical membranes of various epithelial cell types (kidney, colon, thyroid and salivary glands). Frank et al. 7 showed that SLC5A8-deficient mice are viable, fertile and that they lack a morphological phenotype. The same group demonstrated the role of SLC5A8 in lactate reabsorption in the kidney, salivary glands and colon. Despite its observed tumor suppressor function, SLC5A8-deficient mice did not show a higher incidence of colon tumor formation than control animals. 7
In the literature, evidence for the expression of SLC5A8 in brain remains unclear. Frank et al. 7 did not detect SLC5A8 in mouse brain tissue, whereas Martin et al. 8 reported neuron-specific expression.
Recent advances in neuroenergetics demonstrated metabolic compartmentalization where astrocytes display more glycolytic activity, while neurons mostly rely on oxidative pathways. Consequently, the utilization of glucose, the major energy source of the brain, 9 differs between the two cell types. According to the astrocyte neuron lactate shuttle hypothesis, glucose is prominently utilized by astrocytes that feed neurons through the release of L-lactate, which is formed from pyruvate, the end product of glycolysis. Under normal physiological conditions, lactate is thought to be the preferred energy substrate of neurons. 10 Ketones are also capable of fulfilling the energy requirements of the brain and can become the primary energetic fuel when the entry of glucose is impaired11–16 as is the case in diabetes, during starvation and aging. Similar to the interaction between the astrocytes and neurons in L-lactate production and utilization, some researchers have proposed, based on empirical data, that ketone bodies are also shuttled between astrocytes and neurons. 17 In the neurovascular unit, which is mainly composed of neurons, astrocytes and endothelial cells of the blood–brain barrier, the transport of monocarboxylates and ketone bodies is mediated by monocarboxylate transporters (MCTs). The expression of different members of the MCT family in each cell type of the neurovascular unit allows the flux of energetic substrates, not only through the blood–brain barrier, but also among brain parenchymal cells.18,19 Since previous reports demonstrated that SLC5A8 is a Na+-dependent transporter of monocarboxylates and ketone bodies3–5,8 and given that neuronal expression of the protein has been observed, Martin et al. 8 proposed a putative neuroenergetic role for SLC5A8 in the maintenance of energy status and neuron function.
Here, we used SLC5A8-deficient mice to understand the physiological role of the protein in the brain, particularly during aging.
Materials and methods
Generation of SLC5A8-deficient mice
A neomycin-resistance cassette flanked by loxP sites and a loxP site were inserted upstream of the sixth exon and downstream of the seventh exon, respectively, at the Slc5a8 locus. Embryonic stem (ES) cells (129 Sv) were electroporated under standard conditions. Clones were selected for G418 resistance. We identified homologously recombined clones and mutant mice by PCR and confirmed by Southern blot (using 3′ and 5′ probes). Four independent ES clones harboring the recombined allele were injected into C57BL/6 blastocysts. Chimeric males derived from two independent ES clones were bred with C57BL/6 females, and their progeny was analyzed by Southern blot. Mice homozygous for the modified Slc5a8 locus were obtained. No obvious difference was detected, when compared to the parental mice. Females carrying the floxed-SLC5A8 allele in a mixed C57BL6/129 Sv background were subsequently bred with a heterozygous Cre-transgenic male (mixed 129 Sv/FVB), leading to the deletion of the sixth and seventh exons. The progeny were genotyped and mice carrying both the Cre transgene and the slc5a8 mutant allele were selected. First, males and females were mated to obtain mice carrying the slc5a8 allele but lacking the Cre transgene. These males and females were then mated to obtain homozygous SLC5A8-deficient mice. The generated SLC5A8-deficient animals were back-crossed with commercial C57BL6/J (Janvier laboratories) for seven generations. Then heterogenous males and females were mated to obtain homozygous control (wild-type) and homozygous SLC5A8-deficient mice. The homozygous strains were breeded for six generations to produce the animals used for our studies. All study protocols followed the ARRIVE guidelines (Animal Research: Reporting of In vivo Experiments) and were approved by the Ministry of education and research (MESR, reference APAFIS#4205-2016021912155771 v4) that adheres to the politics and guidelines of the National Institute of Health principles of animal laboratory care (NIH publication 86-23, revised 1995). Animals were fed a standard diet. Mice with the same mixed background were used as controls and referred to as wild-type (WT). The animals were treated in accordance with the French Agriculture Ministry guidelines, and the experiments were approved by the University of Nice Sophia Antipolis Animal Care User and Ethics Committee (Ciepal, reference NCE/2016–308).
Histology
Mice were sacrificed by cervical dislocation. Brain and kidneys were rapidly removed and fixed in 3.7% (v/v) paraformaldehyde for 24 h prior to paraffin embedding; 4 µm slices were deparaffinized, rehydrated and pre-treated using automated PT link (Dako SA, Trappes, France). The slices were stained with HES (Hematoxylin 3 min, Eosin 30 s and Safran 15 min). Image acquisitions were performed by light microscopy as described for the immunohistological experiments below. Quantification of vacuole areas in the cerebellar white matter was performed using ImageJ in a blinded assessment, that is, without revealing the identity of the samples to the analyst prior to quantification (n = 6).
Whole brains were rapidly removed from sacrificed animals and frozen in liquid nitrogen; 10 µm sections were prepared from frozen brains with a cryostat. Slices were placed on glass slides and stored at −20℃ until required for LC-MS sample preparation. Specific parts of the slices were removed from the slide by scraping. The remaining tissue was then detached from the slides with 70% methanol and incubated overnight at −20℃. Samples were then centrifuged at 13,000 g for 15 min at 4℃. Supernatants were removed, dried, resuspended in 80 µL of a 20:80 acetonitrile-H2O mixture (HPLC grade, Merck Millipore) and stored at −20℃ until use for LC-MS analysis.
Immunohistochemistry
Mouse brain sections and kidneys were fixed in 3.7% formalin for 24 h, embedded in paraffin, and sectioned at 4 µm. Sections were deparaffinized, rehydrated and antigens retrieved (pH 6.0, 10 mM Na-citrate buffer) using a PT link (Dako SA, France). Immunostaining was performed with the Dako Autostainer (Dako SA) following the manufacturer’s standardized protocol for all steps.
Monoclonal anti-SLC5A8 antibodies (see Supplementary Figure 1) were obtained by immunizing mice with purified peptides corresponding to the C-terminal domain of the mouse SLC5A8 and fusion of their splenocytes with a mouse myeloma cell line (ATCC®CRL-1580) according to standard protocols. 20 Anti-SLC5A8 antibody was used at 23.2 mg/L. Brain cell-specific antibodies included NeuN (1:50; Abcam), Olig2 (1:100; Abcam), GFAP (1:4000; Abcam) and Iba1 (1:500; RayBioteck). The secondary antibodies were EnViosion reagents (Dako SA), and were used following the manufacturer’s instructions. All sections were counterstained with hematoxyline, rinsed in water, dehydrated and mounted with cover slips. Image acquisition was performed using a Nikon microscope (Nikon France Instruments, Champigny sur Marne, France) equipped with a Nikon Camera. Quantification of brain cells was performed in a blinded assessment using ImageJ. The density of brain cells is expressed as the ratio of the brain cell-specific stained area to the area of the global ROI in the cerebellar white matter (n = 6).
Western blot analyses
Western blot analyses were performed on total tissue extracts using Tris-Glycine gels from Invitrogen (Life Technologies). Transfer to PVDF membrane was performed with the iBlot transfer system (Invitrogen), and phosphate buffer (PBS) was used as a blocking reagent. Anti-MBP antibodies (1:1000; Sigma-Aldrich) or our monoclonal anti-SLC5A8 antibodies (1:100) were used. Secondary anti-mouse antibodies (1:5000; Thermo Scientific) coupled to horse radish peroxidase were used for signal detection based on chemiluminescence (Amersham ECL plusprime). Anti-actin antibodies (1:5000; Sigma-Aldrich) were used to reveal actin as an endogenous control for protein loading in all lanes.
In vivo SPECT 99mTc-dl-HMPAO (hexamethylpropyleneamine oxime) imaging
SPECT (single photon emission computed tomography) was used for in vivo imaging of mouse brains with the 99mTc-dl-HMPAO (hexamethylpropyleneamine oxime) perfusion tracer (CERESTAB™, GE Healthcare SAS, France). The 99mTc-HMPAO injection solutions were prepared using a commercially available kit and following the manufacturer’s guidelines. Technetium pertechnetate eluate (99mTcO4−) was produced by a 99mTc-generator. HMPAO solution was prepared within 2 h prior to injection and intravenous administration was required to obtain optimal cerebral capture; 100 to 150 MBq in 0.25 mL 0.9% NaCl was injected in the animal’s caudal vein. Thirty minutes after injection, mice were anesthetized with 1.5% Isoflurane* under respiratory monitoring before starting the imaging procedure. Imaging was performed with a gamma camera coupled to a CT scanner (eXplore speCZT CT120, General Electric). The imaging procedure started with the tracking sequence (SCOUT) followed by the SPECT acquisition, coupled to the CT scanner for 30 min. Image analysis was performed using the Amide software (A Medical Image Data Examiner version 1.0.4). 21 Native values of SPECT imaging were expressed as a percentage of injected activity per volume of tissue. For this, the activity present in the animal at the time of imaging was calculated from the injected activity corrected for the imaging timeframe and the decay formula radioactivity of 99mTc (6.005 hours). CT scanner imaging provides only bone information for the mouse cranial box, and the acquired images have been fused with the anatomical atlas digital brain of the adult mouse C56Bl/6 to perform measurement of radioactivity in different brain anatomical regions. 22 All values obtained have been normalized and are given as a percentage of the average cerebral HMPAO uptake.
Urine collection and processing
Male mouse littermates (three per cage) were housed collectively in a mouse metabolic cage (Techniplast, Germany) with free access to water and standard mouse chow. After adaptation to the cage for a maximum of 48 h or until no weight loss was detected, urine was collected over a period of 24 h and stored at −20℃ until use. Urine samples were mixed with three volumes of methanol and incubated overnight at −20℃ for protein precipitation. Samples were then centrifuged at 13,000 g for 15 min at 4℃. Supernatants were removed, dried, resuspended in 80 µL of a 20:80 acetonitrile-H2O mixture (HPLC grade, Merck Millipore) and stored at −20℃ until use for LC-MS analysis.
LC-MS analyses
Chromatographic analysis was performed with the DIONEX Ultimate 3000 HPLC system coupled to a chromatographic column (Phenomenex Synergi 4 u Hydro-RP 80A 250 × 3.0 mm) set at 40℃ and a flow rate of 0.9 mL/min. Gradients of mobile phases (mobile phase A: 0.1% formic acid in water and mobile phase B: 0.1% formic acid in acetonitrile) were performed over a total of 25 min.
MS analysis was carried out on a Thermo Scientific Exactive Plus Benchtop Orbitrap mass spectrometer. The heated electrospray ionization source (HESI II) was used in positive and negative ion modes. The instrument was operated in full scan mode from m/z 67 to m/z 1000. High-resolution accurate mass (HRAM) full-scan MS and top five MS/MS spectra were collected in a data-dependent fashion at a resolving power of 70,000 and 35,000 at FWHM m/z 200, respectively. Raw data files were converted to mzXML files using MSconvert (version 2.1, ProteoWizard). 23 The post-treatment of data was performed using the MZmine2 version 2.31 (Boston, United States). Metabolites were identified using the Human Metabolome Database version 4.0.
Microarray experiments and statistical analyses
RNA was isolated with an RNeasy kit (Qiagen) from freshly removed mouse brain tissue according to the manufacturer’s protocol. RNA samples were labeled with Cy3 dye using the Low RNA Input QuickAmp Kit (Agilent), as recommended by the supplier. Labelled cRNA probes (400 ng) were hybridized on 8 × 60 K high-density SurePrint G3 gene mouse GE 8 × 60 K Agilent microarrays. Four biological replicates were performed for each experimental condition. Normalization of microarray data was performed using the Limma package (http://www.bioconductor.org). Interslide normalization was performed using the quantile methods. Means of ratios from SLC5A8-deficient versus control tissues were calculated and B-test analysis was performed. Differentially expressed genes were selected based on an adjusted p value of 0.05. Data from expression microarrays were analyzed for enrichment in biological themes (Gene Ontology molecular function and canonical pathways) and biological networks were built using Ingenuity Pathway Analysis software (http://www.ingenuity.com/).
Intra-peritoneal glucose and insulin tolerance tests
For glucose tolerance tests (GTT), mice were injected intraperitoneally with 2 g glucose (Sigma) per kg of body weight in 0.9% NaCl after an overnight fast (16 h). For insulin tolerance tests (ITT), mice were starved for 6 h and injected with 0.75 IU/kg body weight of soluble insulin (Humulin Regular; Eli Lilly & Co., Indianapolis, Indiana, USA). Blood glucose levels were measured before (t = 0) and after glucose/insulin injection at 15, 30, 60 and 120 min. Glycemia was measured on venous blood samples from the tail vein using a Glucometer OneTouch Verio (LifeScan Inc., Canada).
Statistical analyses
Statistical analyses were performed using Microsoft Excel software for all experiments. Continuous variables are presented as mean with standard deviation. Student’s t-tests were used to compare different conditions. A p-value < 0.05 was considered to be significant.
No animals have been excluded from any of our experiments. Mice were tail-marked before being transferred from the housing to the research facilities by our technical staff in charge of animal breeding. The investigators responsible for the above-mentioned experiments had no knowledge of the genotype (WT or SLC5A8-deficient) of the animals prior to carrying out the experimental procedures and data collecting.
Results
Immunohistology of SLC5A8-deficient and wild-type mouse brains
The histological phenotype of 3-, 12- and 20-month-old SLC5A8-deficient mice (KO) was examined and compared to wild-type mice (WT). Hematoxylin and eosin (H&E) staining of sagittal brain slices revealed large vacuoles with spongiotic appearance throughout the white matter of the cerebellar arbor vitae of all 20-month-old SLC5A8-deficient mice examined (12.3 ± 3.5% of the cerebellar arbor vitae area vs. 0.8 ± 0.7% of the same area of wild-type mice at the same age). They were not observed either in brains of 12-month-old mice and 3-month-old mice, SLC5A8-deficient nor controls (Figure 1(a)). Using Western Blot analyses of 3-month and 20-month-old WT and SLC5A8-deficient mice (n = 3), no myelin loss was found in the cerebellum of all SLC5A8-deficient mice compared to wild-type mice indicating normal myelination and myelin maintenance (Figure 1(b)). Because empty vacuoles, at high magnification, were crossed by MBP-stained tissue strands, we concluded that these vacuoles may be located within myelinic regions suggesting myelin sheath swelling (Figure 1(c)). Segmental intramyelinic oedema without myelin loss was detected in all other brain regions containing white matter (corpus callosum, internal capsule, fiber bundles of caudoputamen and brainstem). According to the intramyelinic location of vacuoles, no vacuole was observed in the gray matter of these animals (Figure 1(d)). In addition, no cytoplasmic vacuole was revealed in oligodendrocytes and neurons by immuno with anti-Olig2 antibodies and anti-NeuN antibodies, respectively (Figure 1(d) and (e)). Microglia was studied by immunostaining using anti-Iba1 antibodies. In 20-month-old SLC5A8-deficient mice, Iba1-positive staining revealed the activation of microglial cells characterized by amoeboid morphology and increased cell volume. In wild-type mice and in young null mice, Iba1 staining revealed the “resting” phenotype of microglial cells with a relatively small cellular body and long branching extensions (Figure 1(f)).
Elderly SCL5A8-deficient mice (KO) present diffuse leucoencephalopathy characterized by intramyelinic oedema without myelin loss but associated with microglial activation. The diffuse leucoencephalopathy is illustrated in the white matter of cerebellum (Arbor vitae). (a) Hematoxylin and Eosin (H&E) coloration of paraffin-embedded sections of cerebellum from control and SLC5A8-deficient mice (KO) (optical microscopy ×10) and quantification of vacuolated area in the cerebellar white matter (n = 6). (b) Myelin binding protein (MBP) staining of cerebellum from control and SLC5A8-deficient mice (KO) (optical microscopy ×10) and quantification of MBP in cerebellum using Western blot analyses (n = 3). (c) Cerebellar white matter vacuoles at high magnification (×100) using MBP staining in control (WT) and SLC5A8-deficient mice (KO). (d) NeuN (d), Olig2 (e) and Iba1 (f) immunohistochemcal staining in elderly control (WT) and SLC5A8-deficient mice (KO) cerebellum (×10) associated with a quantification of the expression of each cerebral cell type (n = 6).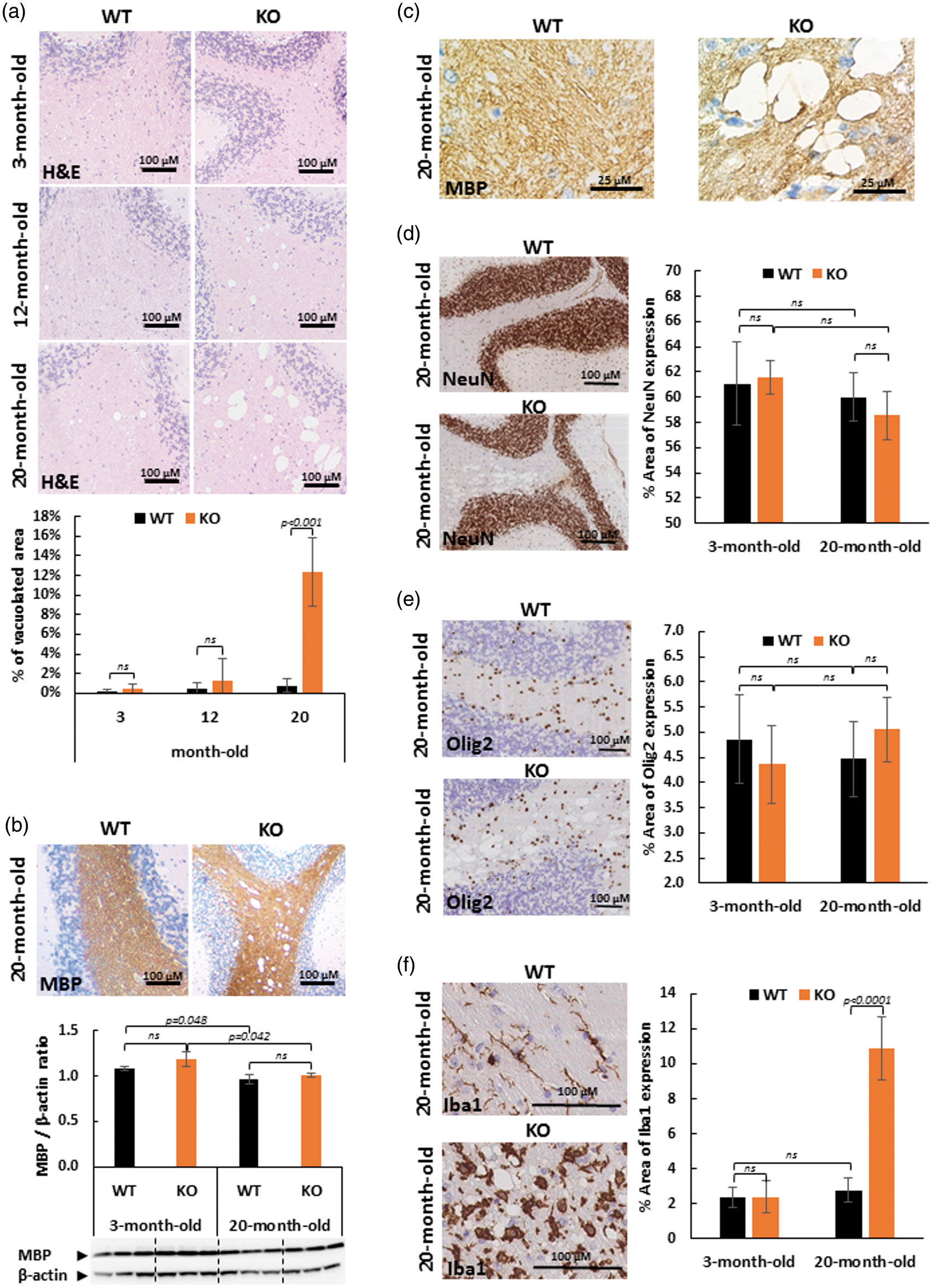
Therefore, we concluded that elderly SLC5A8-deficient mice suffer diffuse leucoencephalopathy characterized by segmental intramyelinic oedema without myelin loss and associated with new microglial activation.
In vivo SPECT imaging with 99mTc-dl-HMPAO (hexamethylpropyleneamine oxime) of SLC5A8-deficient and wild-type mouse brains
Because blood flow in the brain is tightly coupled to local brain metabolism and energy consumption, 99mTc-dl-HMPAO (hexamethylpropyleneamine oxime) perfusion tracer was used to assess regional brain metabolism. It has to be noted that brain 18F-FDG PET imaging could have been a possible alternative approach but it was not used since it can only assess cerebral glucose metabolism. Cerebral 99mTc-dl-HMPAO (CERESTAB*) uptake was measured by SPECT imaging in SLC5A8-deficient and wild-type mice aged 4 and 12 months, respectively. 99mTc-dl-HMPAO uptake was measured in the neocortex, caudo-putamen, thalamus as well as the cerebellum and normalized to the mean brain uptake (Figure 2). No difference in gray matter HMPAO uptake, represented by the neocortex and thalamus regions, was detected between SLC5A8-deficient and wild-type mice at both ages. In contrast, mixed brain structures containing gray and white matter like caudo-putamen and cerebellum, presented significant hypoperfusion in 12-month-old SLC5A8-deficient mice when compared to wild-type mice at the same age (Figure 2). Since hypoperfusion was only found in mixed brain structures, we concluded that the alterations to cerebral energy metabolism in 12-month-old SLC5A8-deficient mice were limited to the brain white matter.
Elderly SLC5A8-deficient mice presenting regional metabolism disturbances in the cerebellum and caudoputamen. 99mTc-HMPAO regional distribution according to phenotype (slc5a8-deficient mice (KO) and control (WT)) and age (4 and 12 months). Uptake of 99mTc-HMPAO was normalized to mean brain uptake (n = 4). NC: neocortex; CP: caudoputamen; Th: thalamus; BS: brainstem; Cer: cerebellum.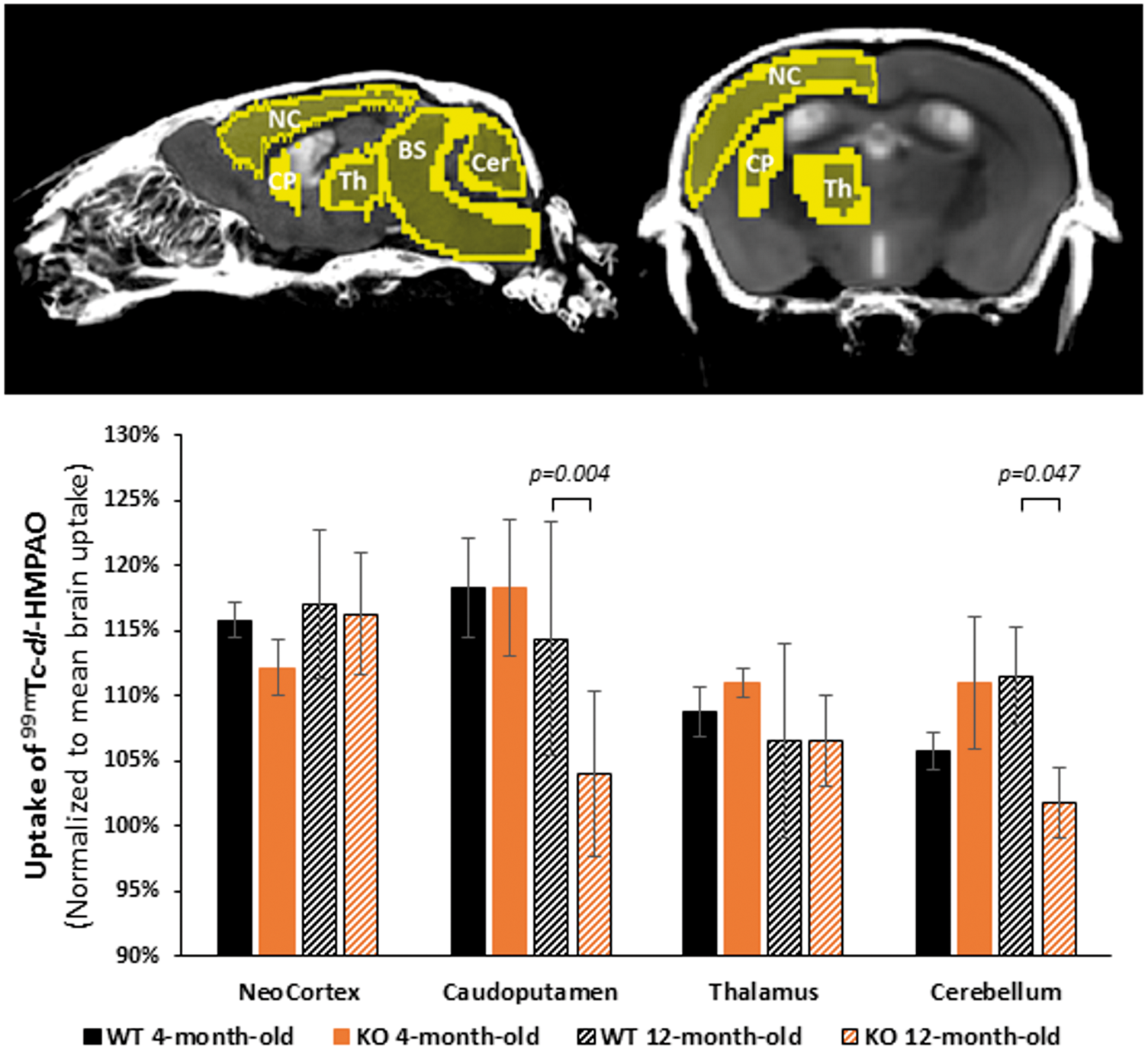
Expression of SLC5A8 in wild-type mice
To further analyze SLC5A8 expression in different brain regions of wild-type mice, immunohistochemistry was performed with a novel anti-SLC5A8 antibody that we generated and characterized (Supplementary Figure 1). No SLC5A8-specific staining was detected in the brains of 3-month and 20-month-old control mice nor in SLC5A8-deficient mouse brains at the same ages (Figure 3(a)). In contrast, strong SLC5A8-specific staining was revealed with the same anti-SLC5A8 antibody in the kidney of three-month-old wild-type mice while it was not detected in the kidney of three-month-old SLC5A8-deficient mice, as expected (Figure 3(b)). In accordance with the literature, expression of the SLC5A8 protein was typically localized in the brush border of proximal convoluted tubules in the kidney of wild-type mice. Using Western Blot analyses, no significant difference in SLC5A8-expression was observed between kidneys from young and elderly WT mice (Figure 3(c)).
Immunohistochemistry using an anti SLC5A8-specific antibody (a) SLC5A8 expression is not detected in WT and SLC5A8-deficient mouse cerebellum, (b) SLC5A8-specific staining is revealed in renal proximal tubules of WT mice but not in tubules from SLC5A8-deficient mice ((g) = renal glomeruli, n = 6). Scale bar: 100 µM. (c) Quantification of SLC5A8 protein using Western Blot analyses of 3-month and 20-month-old WT and SLC5A8-deficient mice (n = 3).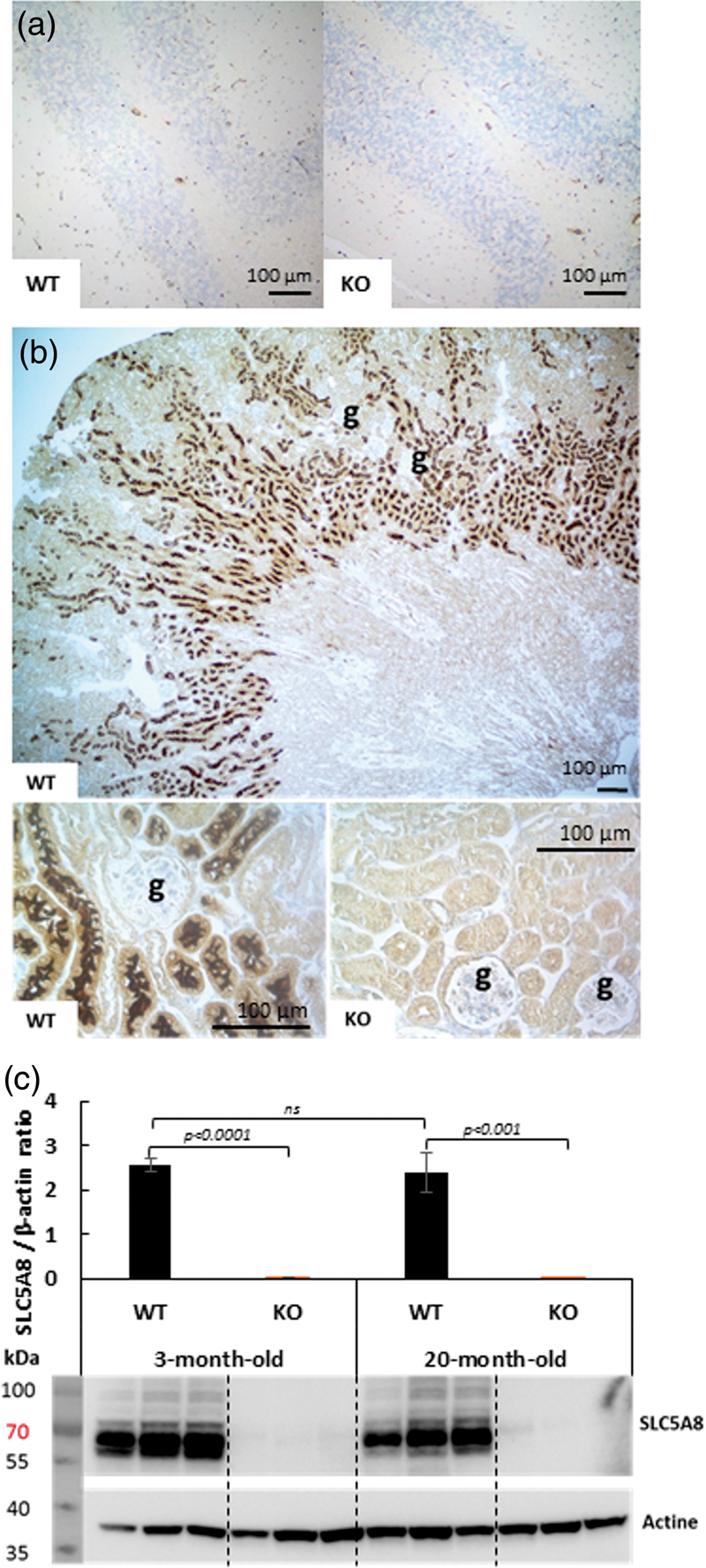
LC-MS analyses of 24-h urine from SLC5A8-deficient and wild-type mice
Since the SLC5A8 protein was not detected in the brain of wild-type mice, it was hypothesized that the observed cerebral phenotype of SLC5A8-deficient mice could be associated with a systemic effect due to the loss of energetic substrates during renal filtration. Frank et al. 7 reported lactaturia in SLC5A8-deficient mice but the authors found no alteration in plasma L-lactate levels in these mice compared to control animals. Similar to L-lactate, ketones are considered to be the brain’s main alternative energy source to glucose 13 but no information about their circulating and/or tissue levels as well as their excreted amounts have been reported in SLC5A8-deficient mice.
The physiological role of the SLC5A8 protein in renal reabsorption was studied through a comparison of the urinary metabolome of wild-type and SLC5A8-deficient mice. Twenty-four-hour urine collections from null mice and control animals were analyzed by LC-MS. β-hydroxybutyrate (see Supplementary Figure 2 for its identification) was found to be significantly increased in the 24-h urine of SLC5A8-deficient mice compared to wild-type mice (Figure 4(a)). KO/WT ratios of this ketone body were higher for 18-month-old mice (approximately 12-fold) than for 3-month-old mice (approximately 7-fold). No difference was noted for the urinary creatinine levels between all animals, which allowed us to make the above comparison without normalization. As described by Frank et al.,
7
we found lactaturia in SLC5A8-deficient mice (3.5-fold) and also an increased level of pyruvate in the urine of SLC5A8-deficient mice compared to control animals (5.7-fold) (Supplementary Figure 3).
SLC5A8-deficient mice present β-hydroxybutyrate deficiency due to a renal reabsorption defect in proximal tubular tubule. Level of hydroxybutyrate [M-H] m/z: 103.0387 (see Extended data Figure 2) (a) and its acyl carnitine [M + H] m/z: 248.1492 (see Extended data Figure 4) (b) in 24-h urine of wild-type and SLC5A8-deficient mice assessed by LC-MS (n = 4).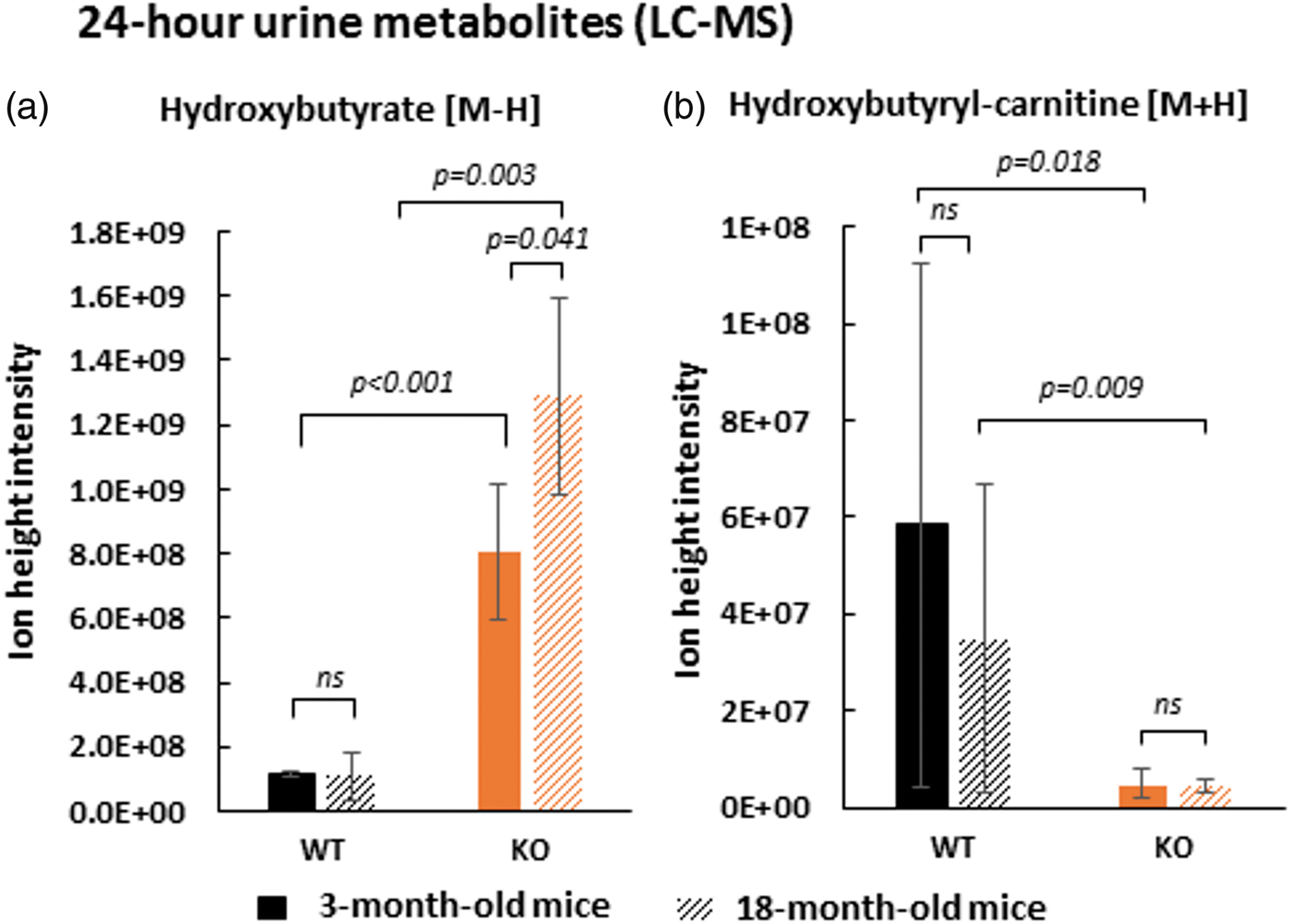
Hydroxybutyryl-carnitine, the ester of hydroxybutyryl and carnitine, is the activated form of β-hydroxybutyrate. Because the level of this acyl carnitine was known to be correlated with the systemic level of its derived short chain fatty acid, the systemic consequence of β-hydroxybutyrate loss in SLC5A8-deficient mouse urine was determined by analysis of β-hydroxybutyryl-carnitine. 24 The level of the acyl-carnitine form of β-hydroxybutyrate (see Supplementary Figure 4 for its identification) was found to be significantly decreased in SLC5A8-deficient mice versus control mice in 24-h urine samples suggesting a systemic impact due to ketone body loss in the urine of SLC5A8-deficient mice (Figure 4(b)). No significant difference was observed for the KO/WT ratio of hydroxybutyryl-carnitine between 3-month-old mice (≈12-fold) and 18-month-old mice (≈8-fold). The level of two additional ketones, acetoacetate and acetone, could not be detected by LC-MS. Acetoacetate was assessed by a semi-quantitative urinalysis reagent test (Legall method). This colorimetric test gave positive staining for high levels of acetoacetate in the urine of SLC5A8-deficient mice, while the test was negative (no staining) for the urine of wild-type mice. In conclusion, SLC5A8-deficient mice showed a defect in renal reabsorption of both major ketone bodies.
LC-MS analyses of SLC5A8-deficient and wild-type mouse brains
Since SLC5A8-deficient mice presented strong ketonuria, cerebral β-hydroxybutyrate levels in SLC5A8-deficient mice were compared to those of wild-type mice using an LC-MS approach. First, β-hydroxybutyrate levels were measured in homogenized whole brains of 24-month-old mice. SLC5A8-deficient mouse brains contained 28% less β-hydroxybutyrate than brains of control animals (Figure 5(a)). Second, the levels of β-hydroxybutyrate were assessed in different cerebral areas. To exclude the possibility that the previously described results are due to aging, three-month-old mice were used for these experiments. Rostral-mesencephalon brain sections were chosen including the occipital neo-cortex (grey matter) and the mesencephalon (white matter) (Figure 5(b)). The level of β-hydroxybutyrate was 27% lower in coronal mesencephalon sections of SLC5A8-deficient mice than in the same brain sections of control animals. It can be concluded that SLC5A8-deficient mice suffer from low cerebral β-hydroxybutyrate levels, particularly in the white matter, most probably due to the loss of ketone bodies via the urine. Figure 5(c) shows the ATP/AMP ratio in the whole cerebellum of the different animal groups (see Supplementary Figures 5 and 6 for the identifications by LC-MS). We found that elderly SLC5A8-deficient mice had a significant lower level of this indicator of cellular energy status when compared to control mice.
Depletion of β-hydroxybutyrate ([M-H] m/z: 103.0387) levels in brain tissue of SLC5A8-deficient mice assessed by LCMS. Measurements were performed on crushed fresh brain tissue of older mice (24 months) (n = 5) (a) and from rostral-mesencephalon frozen sections of young mice (3 months) (n = 4) (b). Frozen sections were analyzed whole and after micro-dissection of the neocortex (grey matter) and the mesencephalon (white matter) as specified on the cutting plane below. (c) ATP/AMP ratio in the whole cerebellum of 3-month and 20-month old WT and SLC5A8-deficient mice (n = 6).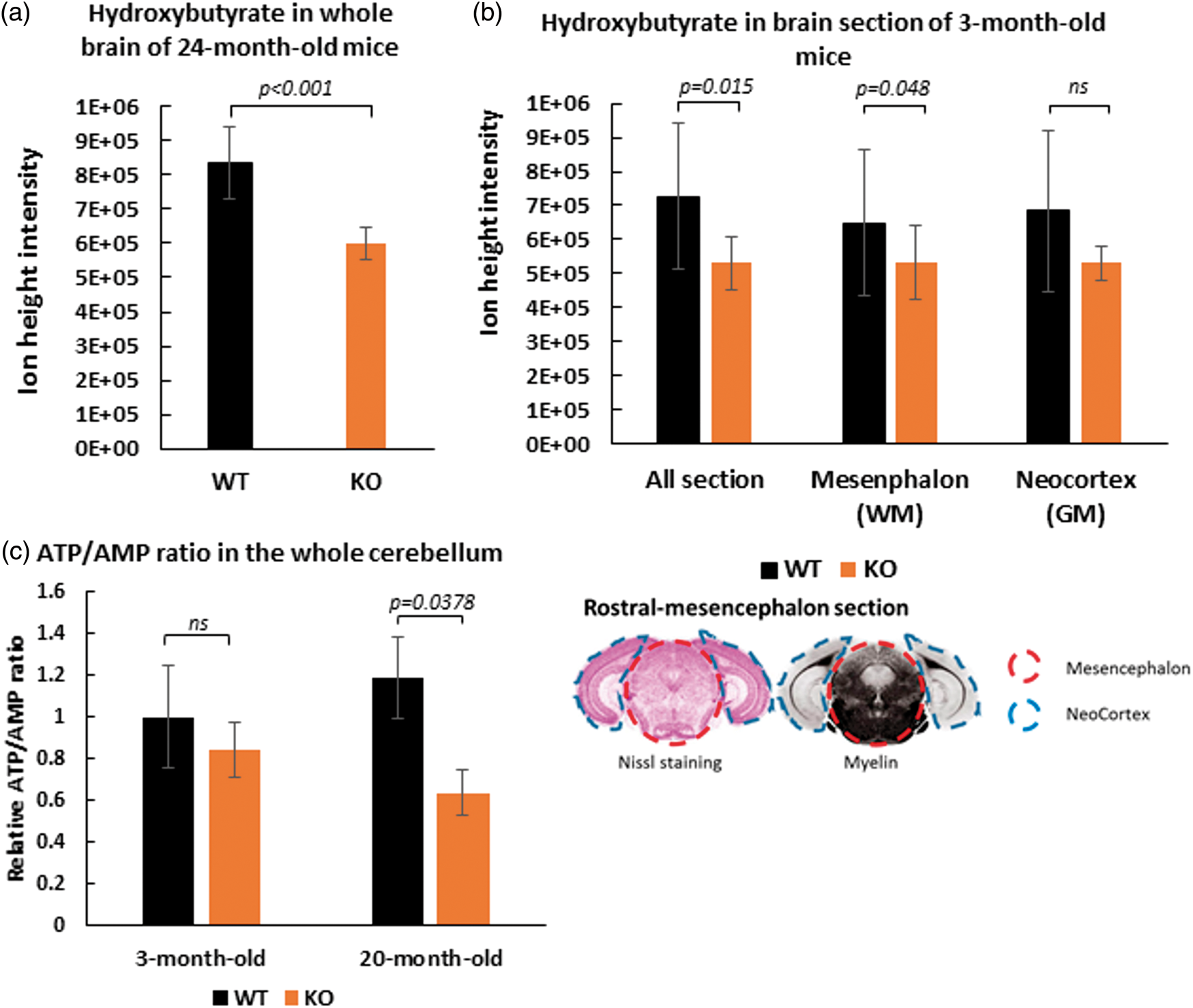
Transcriptomic analyses on SLC5A8-deficient and wild-type mouse brains
In the brain, ketones are produced by ketogenic amino acid and fatty acid breakdown in astrocytes. Transcriptomic analyses (NCBI GEO platform Accession Number GSE117705) on whole brain homogenates were performed from SLC5A8-deficient and wild-type mice at ages of 4 and 18 months to study the expression of genes encoding proteins that are involved in ketogenic amino acid and lipid catabolism.25–28
Significant up-regulation of BCAT2, Aass and Tdo2 was found in slc5a8-null mice at 4 and 18 months of age. These genes encode proteins involved in the degradation of the ketogenic amino acids leucine, lysine and tryptophan. BCAT2 encodes the mitochondrial enzyme “branched chain amino acid transaminase 2,” which catalyzes the first step of the leucine degradation pathway leading to the production of acetyl-CoA, acetoacetate and β-hydroxybutyrate. Aass stands for aminoadipate-semialdehyde synthase, a bifunctional enzyme responsible for the first two steps of the lysine degradation pathway. Tdo2, or tryptophan 2,3-dioxygenase, catalyzes the first and rate-limiting step of tryptophan catabolism. The Pts gene product, 6-pyruvoyltetrahydropterin synthase, is involved in the transformation of the ketogenic amino acids phenylalanine, tyrosine and tryptophan into non-energetic degradation products. Pts was found to be down-regulated in SLC5A8-deficient mice suggesting metabolic reorientation that may allow for the production of ketones from these amino acids instead of non-energetic molecules. In addition, an up-regulation of LAT4 (SLC43A2) expression was found in SLC5A8-deficient mice but this finding was limited to 18-month-old animals. Interestingly, the LAT4 gene encodes a member of the amino acid transporter-3 family (SLC43), which mediates the transport of branched-chain amino-acids including leucine. Taken together, these results show enhanced transcription of genes encoding proteins that are involved in ketogenic amino-acid catabolism and transport in the brain of SLC5A8-deficient mice. This finding suggests that increased ketogenic amino acid catabolism and ketone production in the brain of these animals may serve to compensate for urinary ketone loss.
Phospholipase A2 (Pla2g4e) gene transcripts were observed to be up-regulated in the brain of SLC5A8-deficient mice of both ages when compared to brains of corresponding control mice. Phospholipase A2 found in brain tissue has been associated with the cPLA2-sphingomyelinase pathway in which brain lipids are utilized to generate fatty acids. In astrocytes, fatty acids can be converted into ketone bodies, which might fulfil the energy needs of neurons. 28 In 18-month-old SLC5A8-deficient mice, the level of transcripts from two additional genes encoding lipolytic enzymes, phospholipase C (Plcd4) and phospholipase D2 (Pld2) was also up-regulated when compared to control mice.
It is concluded that, in SLC5A8-deficient mice, the ketogenic amino-acid and lipid catabolism pathways are activated as an adaptive compensatory response to β-hydroxybutyrate deficiency in the brain. As would be expected, this compensatory mechanism intensified in the aging brain.
In addition, enhanced levels of Fxyd2 gene transcripts coding for an auxiliary subunit of the Na+-K+-ATPase were detected in SLC5A8-deficient elderly mice. FXYD2 proteins regulate Na+-K+-ATPase activity by increasing the apparent affinity of the transporter for ATP, while decreasing its Na+ affinity. Increased expression of the Fxyd2 gene occurs in order to maintain the Na+-K+-gradient across the plasma membrane when ATP production is low. 29 This finding is in accordance with our hypothesis that SLC5A8-deficient mice struggle to meet the energy demands of the brain, particularly during aging.
Glucose and insulin tolerance tests
We previously showed that SLC5A8-deficient mice exhibit white matter degeneration in the aging brain that is most probably linked to ketone body deficiency. The principal reason for ketone bodies to become the primary energy source in the brain is because of reduced availability of glucose, as occurs during prolonged starvation or diabetes, for example. Our animals had free access to drinking water and food, and did not suffer starvation. Therefore, glucose metabolism was assessed by performing glucose tolerance tests (GTT) on 3-month and 18-month-old SLC5A8-deficient and control mice. These tests did not reveal any glucose metabolism disorder in young SLC5A8-deficient mice versus control mice (Figure 6(a) and (b)). In contrast, aged SLC5A8-deficient mice displayed significantly higher (170.3 ± 27.1 mg/dL) overnight fasting capillary glycaemia compared to control mice (139.6 ±13.6 mg/dL). GTT in aged SLC5A8-deficient mice revealed that blood glucose levels following an intra-peritoneal glucose injection were significantly higher than in controls, suggesting glucose intolerance (Figure 6(b)). ITT was performed on the same SLC5A8-deficient mice that presented glucose intolerance. ITT is an established method for quantifying insulin resistance that may be at the origin of the observed glucose intolerance in SLC5A8-deficient mice. Our results showed that the decay rate of capillary glycemia was not significantly lower in elderly SLC5A8-deficient mice leading us to conclude that these mice presented glucose intolerance due to islet function but not insulin action (Figure 6(c)). In accordance with this conclusion, elderly SLC5A8-deficient mice had a significantly smaller body weight than control mice (Figure 6(d)).
Elderly SLC5A8-deficient mice present glucose intolerance due to islet function but not insulin action. (a) Fasting glycemia with 3-month-old and 18-month-old, control and SLC5A8-deficient mice (n = 5). (b) Glucose Tolerance Test (GTT) with 3-month-old and 18-month-old, control and SLC5A8-deficient mice (n = 5). (c) Insulin Tolerance Test (ITT) with 18-month-old SLC5A8-deficient mice (n = 6). (d) Mice body weight according to age in WT and KO mice (0–12 months, n = 45; 13–24 months, n = 14; >24 months, n = 7).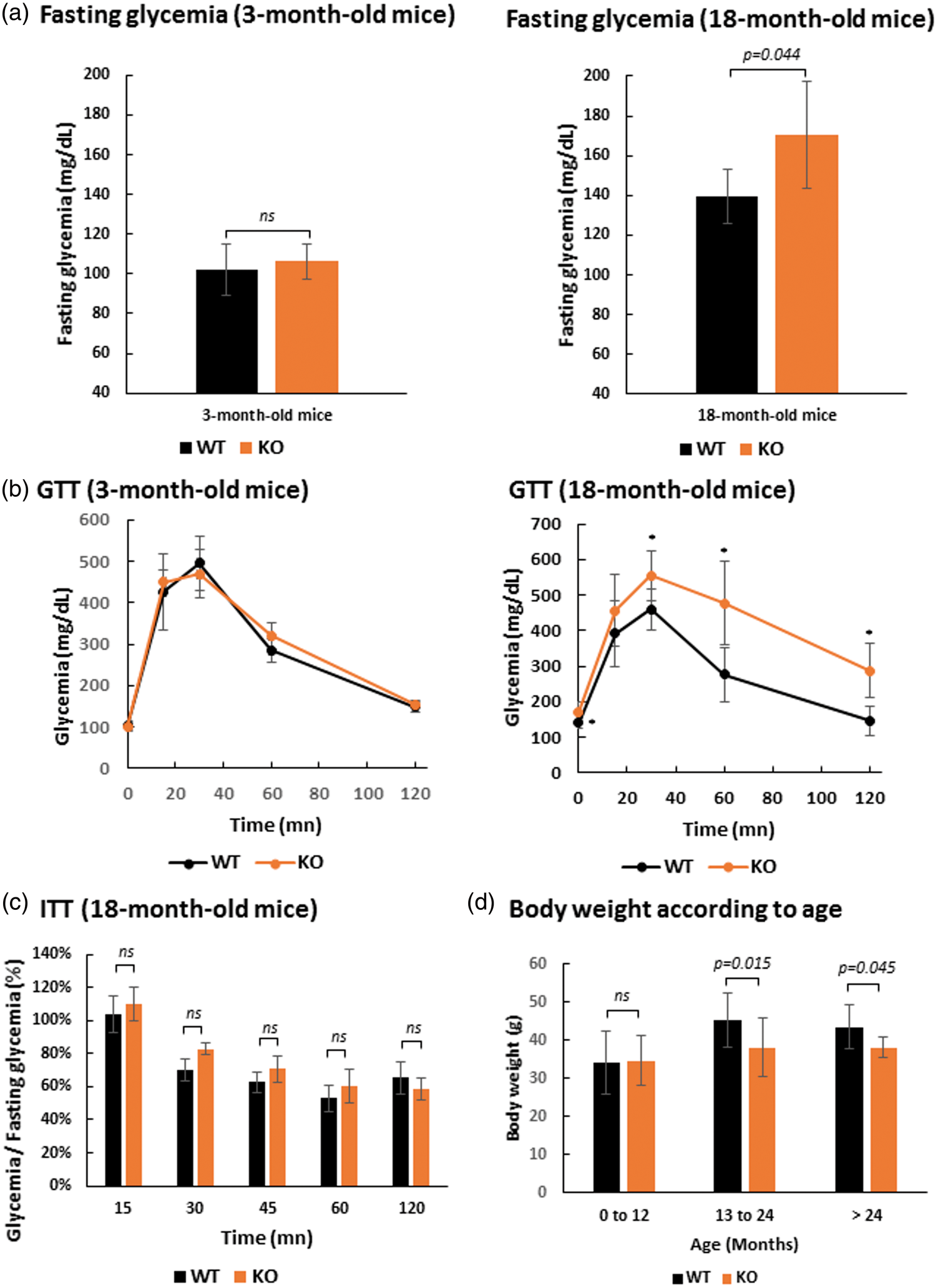
Discussion
Here we report, for the first time, that aged SLC5A8-deficient mice present a cerebral ketone body deficiency due to a defect in renal ketone reabsorption. Ketone bodies are an alternative energy source for the brain, particularly when the glucose supply is impaired. Our studies showed that aging SLC5A8-deficient mice exhibit brain white matter lesions. This cerebral phenotype is most probably due to a chronic energy deficit caused by ketone deficiency and glucose intolerance.
Aged SLC5A8-deficient mice show diffuse leucoencephalopathy characterized by intramyelinic oedema without myelin loss associated with microglial activation. According to Van Der Knaap’s classification of myelin disorders, intramyelinic oedema without myelin loss is associated with metabolic disorders. 30 Intramyelinic oedema is commonly referred to as a water/ion homeostasis disorder in peri-axonal regions or between lamellae, the so-called “panglial syncytium.” Strong evidence has suggested that alterations in the “panglial syncytium” could lead to myelin ultra-structural lesions with intramyelinic oedema. 31 Studies of transgenic knockout mouse models for connexins 43/30, Kir 4.1, ClC2 or GlialCAM (a ClC2 chaperone) have reported an intramyelinic oedema phenotype in the brain.32–36 In humans, loss-of-function mutations in ClC2 lead to megalencephalic leukoencephalopathy with subcortical cysts, a disease characterized by chronic brain white matter vacuolar oedema.37,38 Interestingly, chronic brain hypoperfusion models also lead to segmental intramyelinic oedema in the white matter. 34 In TgPAC-Notch3R169C mice, a pre-clinical mouse model of small vessel disease named CADASIL (Cerebral Autosomal Dominant Arteriopathy with Subcortical Infarctus and Leukoencephalopathy), early changes in white matter are linked to intramyelinic oedema with microglial activation. 39 We observed similar changes in the white matter of our aging SLC5A8-deficient mice as those described by Wakita et al. 40 with a chronic hypoperfusion mouse model obtained by the permanent bilateral occlusion of common carotid arteries. Since brain hypoperfusion is associated with energy deficit, this finding suggests that the intramyelinic oedema and glial activation are associated with chronic energy deficiency. In a mouse model for maple syrup urinary disease, an inherited disorder of branched-chain amino acid metabolism, vacuolization and disruption of white matter was reported and attributed by the authors, in part, to long-term cerebral energy deprivation. 41 For several groups, these histological lesions might be the consequences of a water/ion homeostasis defect in the “panglial syncytium” due to the failure of key ATP-dependent ion pumps.31,39,41 Our SPECT HMPAO study of brain regional metabolism also supported the hypothesis that chronic energetic failure leads to intramyelinic oedema with glial activation.
Martin et al. 8 remain the only group that reported widespread expression of the SMCT1 mRNA in the mouse brain (mainly in cortex and hippocampus) by in situ hybridization analyses (ISH). They also performed immunofluorescence analyses and proposed that the SMCT1 protein is expressed exclusively in neurons. Results shown in this publication are in contrast with results presented here and all other analyses performed up to now on SLC5A8 expression in the mouse brain. First, Frank et al. 7 did not observe SLC5A8 expression in the mouse brain by ISH analysis and Western blot. The same conclusion was supported by data from the Allen mouse brain atlas (http://mouse.brain-map.org) obtained by ISH. 42 Furthermore, in a RNA-sequencing transcriptome database (http://www.brainrnaseq.org) of glia, neurons, and vascular cells of the mouse or human brain, transcripts of slc5a8 were not found.43,44 Other RNA-sequencing databases like the expression atlas (http://www.ebi.ac.uk/gxa) gave the same result. 45 In a proteomic study of membrane proteins in different areas of the rat brain, no peptide corresponding to the SLC5A8 protein was found despite the identification of a large number of membrane proteins. 46 Therefore, we hypothesized that the white matter phenotype of aging SLC5A8-deficient mice underlies a metabolic disorder which is caused by the lack of SLC5A8 expression in a peripheral organ (and not by a lack of cerebral expression). We assume that chronic brain energetic failure, demonstrated by the prototypic histological lesions and energetic metabolism disturbances assessed by SPECT HMPAO in the white matter, in addition to the detection of lower ATP/AMP ratio levels in brains of elderly SLC5A8-deficient mice, was in fact the result of the loss of energetic substrates in urine. In accordance with the literature, we showed that SLC5A8 is strongly expressed in the kidney proximal tubule, which is the major resorptive segment of the nephron.4,7 In addition, SLC5A8 is known to mediate sodium-dependent transport of monocarboxylates and ketone bodies leading us to assume that the renal reabsorption defect of these metabolites gives rise to an energy deficiency in the brain of aging SLC5A8-deficient mice.3–5,8,47 In vivo, LC-MS analyses of 24-h urine confirmed lactaturia in SLC5A8-deficient mice, as described by Frank et al., 7 and it was further supported by increased urinary levels of other monocarboxylates. But most interestingly, these studies showed strong hyper-excretion of the ketone body β-hydroxybutyrate in the urine of SLC5A8-deficient mice. We then turned to consider the systemic effects of this strong β-hydroxybutyrate loss, and the consequences for the brain. Interestingly, the serum levels of ketone bodies were not affected by the renal loss. However, the acyl-carnitine form of β-hydroxybutyrate (hydroxybutyryl-carnitine) was found to be significantly decreased in 24-h urine of SLC5A8-deficient mice. Because the level of this acyl carnitine is known to be correlated with the systemic level of its derived short chain fatty acid, 24 we conclude that, despite the constant serum level, the loss of β-hydroxybutyrate through urine in SLC5A8-deficient mice has significant effects on its availability for the energy metabolism of these animals.
Our data clearly show that SLC5A8-deficient mice suffer β-hydroxybutyrate insufficiency in the white matter, while normal levels were obtained for the grey matter. This may be due to more efficient β-hydroxybutyrate uptake across the blood–brain barrier in cortical regions where monocarboxylate transporters are more abundant.27,48,49 It has been shown in a human brain ischemic model that white matter is more vulnerable to energy crisis than grey matter. 50 Whole-brain transcriptomic data showed that SLC5A8-deficient mice activate degradation of ketogenic amino-acids, leucine in particular, and that lipids most probably compensate for the β-hydroxybutyrate deficiency caused by urinary ketone loss. In accordance with our results, Klosinski et al. 28 recently proposed that lipolysis of myelin lipids in the brain is an adaptive response that generates ketone bodies to fulfil the energy needs of neurons. 28 The authors presented evidence that the loss of myelin sheath integrity, observed only by electron microscopy, is induced by activation of the cPLA2-sphingomyelinase pathway. 28 Consistently, our SLC5A8-deficient mice presented enhanced expression of phospholipase A2 in the brain, which is most likely an indication of increased lipolysis.
Despite the permanent loss of ketone bodies, starting from an early age, only elderly SLC5A8-deficient mice showed cerebral metabolism disturbances and detectable histological lesions in the white matter. In addition to published hypotheses on the main pleiotropic properties of ketone bodies,27,28,51 we here report further findings describing the deleterious effect of the loss of ketone bodies in elderly SLC5A8-deficient mice. Our data reveal a cerebral energy crisis in the elderly SLC5A8-deficient mice as has been demonstrated by prototypic histological lesions, disturbances of the energetic metabolism in the white matter, and a significant decrease of the ATP/AMP ratio in brain cells. In general, ketone bodies are an accessory cerebral energetic fuel but they can become the main energy source in the case of impaired glucose entry in the context of aging. In addition to aging, we demonstrated that glucose metabolism was altered in the elderly SLC5A8-deficient mice when compared to control animals, most probably due to a defect of glucose entry into brain cells. A possible explanation for this phenomenon is that the glucose intolerance in the elderly SLC5A8-deficient mice, in addition to permanent ketone body insufficiency, leads to a chronic energy crisis with a decrease of the ATP/AMP ratio in the brain of aging SLC5A8-deficient mice. The elucidation of the molecular mechanisms underlying glucose intolerance in elderly SLC5A8-deficient mice needs further investigation but we hypothesize that it was the consequence of chronic ketogenesis induced by ketone body urinary loss, as has previously been suggested. 52 The loss of this neuroprotective agent may contribute to the brain damage observed in elderly SLC5A8-deficient mice during aging.
In summary, we here describe, for the first time, a mouse model for ketone body insufficiency. The presented results, in particular the alterations to the brain white matter of these animals, highlight the major roles of ketones as alternative energy substrates when glucose metabolism is impaired due to insulin resistance and aging. At present, studies of the physiological roles of ketones are limited to animal injury models or to human cases of traumatic, ischemic or neurodegenerative diseases with the application of various ketone administration protocols or ketogenic diets. 27 We provide here a new mouse model with permanent ketone deficiency that should be very useful in the development of novel neuroprotective agents in the case of brain injury or aging.
Supplemental Material
Supplemental material for Urinary ketone body loss leads to degeneration of brain white matter in elderly SLC5A8-deficient mice
Supplemental Material for Urinary ketone body loss leads to degeneration of brain white matter in elderly SLC5A8-deficient mice by Laurent Suissa, Virginie Flachon, Jean-Marie Guigonis, Charles-Vivien Olivieri, Fanny Burel-Vandenbos, Julien Guglielmi, Damien Ambrosetti, Matthieu Gérard, Philippe Franken, Jacques Darcourt, Luc Pellerin, Thierry Pourcher and Sabine Lindenthal in Journal of Cerebral Blood Flow & Metabolism
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded by grants of the “Commissariat à l’Energie Atomique” (“Programme de Toxicologie nucléaire”), the “Département des Alpes-Maritimes” (“Appel d’offre Santé”) and the “Commissariat Général à l’Investissement” (“Recherche en matière de Sûreté Nucléaire et Radioprotection” program operated by the ‘‘Agence Nationale de la Recherche’’ (ANR), France). Luc Pellerin received financial support from the program IdEx Bordeaux ANR-10-IDEX-03-02.
Acknowledgements
We thank the IRCAN Animal Core Facility and the CHU Histology core facility for providing access to their equipment. The IRCAN Animal core facility is supported by the University Nice Sophia Antipolis. We thank the radio-pharmacy of the medical center for oncology Antoine Lacassagne for the 99mTc-HMPAO. Metabolomics analyses have been funded in by Cancéropole PACA and the Département des Alpes-Maritimes. We also thank Lyse Domenech, Marianne Goracci, Sandrine Destrée and Arnaud Borderie for excellent technical assistance. The authors acknowledge the excellent support of the Nice-Sophia Antipolis Functional Genomics Platform in which the microarray experiments were carried out. The authors wish to thank Abby Cuttriss for critical reading of the manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
Laurent Suissa – Acquisition of data, analysis and interpretation of data, literature search, and drafting of manuscript. Virginie Flachon – Engineering of deficient mice, drafting of manuscript. Jean-Marie Guigonis – Acquisition of mass spectrometry data, analysis and interpretation of data. Vivien Olivieri – Acquisition of data, analysis and interpretation of data. Fanny Burel-Vandenbos – Acquisition of histochemical data, analysis and interpretation of data. Julien Guglielmi – Acquisition of SPECT imaging data, analysis and interpretation of data. Damien Ambrosetti – Acquisition of histological data, analysis and interpretation of data. Matthieu Gérard – Engineering of deficient mice, drafting of manuscript. Philippe Franken – Acquisition of data, analysis and interpretation of data. Jacques Darcourt – Study conception and design, drafting of manuscript. Luc Pellerin – Study conception and design, critical revision. Thierry Pourcher – Study conception and design, analysis and interpretation of data, critical revision. Sabine Lindenthal – Study conception and design, acquisition of data, analysis and interpretation of data, critical revision. All the authors discussed data, edited, commented on the manuscript, and approved the final version of the manuscript.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.