
Abstract
Excitotoxicity and microglia/macrophage over-activation are the important pathogenic steps in brain damage caused by ischemic stroke. Recent studies from our group suggest that the neurons in ischemic penumbra generate an anti-inflammatory cytokine, interleukin-4 (IL-4). This neuron-produced IL-4 could subsequently convert surrounding microglia/macrophages to a reparative (M2)-phenotype. The present study was designed to establish the mechanisms by which neurons under transient ischemic condition produce/secrete IL-4. We employed primary rat cortical neurons and a validated in vitro ischemic injury model involving transient oxygen–glucose deprivation (OGD). We discovered that only sublethal OGD induces IL-4 production/secretion by neurons. We then showed that excitotoxic stimulus (an integral component of OGD-mediated damage) involving N-methyl-D-aspartate (NMDA), and not kainate receptor, triggers neuronal IL-4 production/release. Of note, oxidative stress or pro-apoptotic stimuli did not induce IL-4 production by neurons. Next, using the calcineurin inhibitor FK506, we implicated this phosphatase in activation of the nuclear factor of activated T-cells (NFAT; a transcription factor activated through calcineurin-mediated dephosphorylation) and propose that this pathway is involved in transcriptional upregulation of the IL-4 synthesis in NMDA-treated neurons. Finally, using a transfer of culture medium from NMDA-conditioned neuron to microglia, we showed that the neuronal IL-4 can polarize microglia toward a restorative, phagocytic phenotype.
Introduction
Following cerebral ischemia-induced injury, microglia sense the presence of various molecules (damage-associated molecular patterns (DAMPs)) generated as a consequence of cellular damage and react by triggering pro-inflammatory responses involving secretion of various cytokines, chemokines, proteolytic enzymes, and mediators of oxidative/nitrosative damage. This phase of acute microglia response is predominantly deleterious to the neighboring brain cells and represents an important component of secondary brain damage.
Our recent studies using a transient middle cerebral artery (MCA) occlusion model suggest that in response to ischemia, the surviving neurons at borders of ischemic core produce and secrete interleukin (IL) IL-4. 1 Normally, IL-4 acts as a pleiotropic cytokine with a potent anti-inflammatory role achieved via suppression of type I (classical) inflammation. 2 Besides modulation of Th2 immune responses, 3 IL-4 was shown to polarize macrophages to their alternative phenotype, often referred to as M2 or the healing phenotype.4,5 In the brain, IL-4 is involved in microglia polarization.1,6 It is proposed that in microglia/macrophage, IL-4 activates the activator of the signal transducer and activator of transcription 6 (STAT6), which consequently promotes the transcription of peroxisome proliferator-activated receptor-γ (PPARγ), a key transcription factor regulating microglia/macrophage healing functions.1,7,8 As the pro-inflammatory phenotype of microglia/macrophages could be detrimental to the adjacent neurons, we propose that the reprogramming of microglia/macrophages from a pro-inflammatory to a “healing” phenotype mediated by IL-4 released by ischemia-sensing neurons is a self-defense response allowing neurons to avoid further injury.
In our previous work, we established that ischemia is capable of inducing IL-4 synthesis and secretion, but the mechanisms involved in IL-4 production by neurons remained elusive. Thus, the objective of this study was to identify the signaling pathway involved in IL-4 production and secretion by neurons in response to ischemia, and to further explore the role of neuron-derived IL-4 in microglia polarization. Our novel findings reveal that IL-4 transcription in neurons is regulated through a pathway involving ischemia-induced (1) glutamate release, (2) activation of N-methyl-D-aspartate (NMDA) receptor, (3) activation of calcium/calmodulin-dependent protein phosphatase, calcineurin (CN), and (4) activation of the nuclear factor of activated T-cells (NFAT; transcription factor activated through dephosphorylation by CN). Furthermore, we show that neuron-derived IL-4 is involved in modifying microglia phagocytic phenotype.
Materials and methods
Animals
All studies involving animal tissue followed the guidelines outlined in Guide for the Care and Use of Laboratory Animals from the National Institutes of Health and were approved by the Animal Welfare Committee of The University of Texas Health Science Center at Houston. While our studies are based on using primary brain cell cultures, we followed the ARRIVE guidelines as best as possible in reporting results of this work. Protocols for the isolation of the primary prenatal (neurons) and postnatal (microglia) tissue were performed as previously reported1,9,10 and are briefly outlined below.
Primary cortical neuron cultures
The cortices of E18 fetal Sprague Dawley rat embryos were dissected and dissociated by trituration as described previously.1,9 The dissociated cells were plated on poly-L-lysine-coated 6-well plates in Neurobasal medium with B27 at a density of 5 × 105 cells/ml. The cells were maintained in a CO2 incubator (5% CO2, 21% O2) at 37.0 ± 0.5℃. Half of the culture medium was changed every three days. Neurons in our cultures were ready for experiments after 11 days in culture when they demonstrated the extensive network of neurites. Using MAP2 immunohistochemical staining, we confirmed that more than 90% of living cells were MAP2+ neurons.
Primary brain glial culture and microglia isolation
The cortices of E18 fetal Sprague Dawley rat embryos were dissected and dissociated by trituration as described previously.1,10 The dissociated cells were plated in 75 cm2 tissue culture flasks in DMEM with 10% fetal bovine serum, and maintained in a CO2 incubator (5% CO2, 21% O2) at 37.0 ± 0.5℃. Half of the culture medium was changed every three days. After a total of 12–15 days in culture, the astrocytes form a confluent layer signifying that the culture was ready for microglia isolation. The loosely adherent microglia were harvested by shaking at 220 r/min for 30 min. The detached microglia were collected and re-plated onto poly-L-Lysine coated 6-well plates with or without German glass inserts at a density of 1–4 × 105 cells/ml. Using CD68 immunostaining, we confirmed that microglia cultures were more than 96% pure 24 h after re-plating.
Immunofluorescence of MAP2
MAP2 immunofluorescence was used to label neurons. The primary neuron cultures grown on German glass were fixed with 2% PFA in PBS for 15 min at room temperature and incubated in chicken anti-MAP2 (ab5392; Abcam) overnight at 4℃. Rabbit anti-chicken IgG–Alexa Fluor 488 (Invitrogen) was used to visualize MAP2-labeled neurons. DAPI was applied to counterstain the cell nuclei. The images of MAP2-labeled neurons were captured by Zeiss Confocal microscopy LSM 800.
Oxygen–glucose deprivation injury model
Eleven-day-old primary cortical neurons in culture were subjected to oxygen–glucose deprivation (OGD) injury, as we reported previously. 1 The culture media was replaced with Neurobasal medium without glucose. The cultures were placed in a gas-tight humidified chamber and flushed with 5% CO2/95%N2 for 10 min. After flushing, the cultures were kept in OGD status for the designated time in the incubator. At the end of OGD, glucose at 4.5 mg/ml was added to the cultures, and the cells were returned to their original culture condition and incubated for 6 h for reperfusion.
Administration of reagents
All reagents were purchased from Sigma-Aldrich unless otherwise stated. After 11 days of culture, primary neurons in 6-well plates were exposed to glutamate, hydrogen peroxide (Fisher Scientific), staurosporine, NMDA, or kainate in the culture medium for 5 min to induce acute stress. Culture medium was then replaced with fresh medium and the cells were returned to incubator for 6 h. MK-801, D-serine, glycine (Fisher Scientific), or FK-506 (Fujisawa Healthcare) were added 2 min before exposure to NMDA.
Cytotoxicity assay
The degree of cell injury was assessed by determining the amount of lactate dehydrogenase (LDH) released into the culture medium using CytoTox 96 Non-Radioactive Cytotoxicity Assay kit (Promega). The assays were performed following the manufacturer’s instructions. Briefly, 50 µl of medium from cultured cells was collected and mixed with 50 µl of CytoTox 96 reagent in a 96-well flat clear bottom plate. The plate was incubated for 30 min at room temperature and then recorded the absorbance at 490 nm. The result of each sample was calculated by averaging the results from triplicate wells. The cell injury index was determined by comparing the LDH release under each experimental condition to the naïve control.
In selected experiments, CytoTox 96 Non-Radioactive Cell Proliferation Assay kit (MTT assay, Promega) was used as an additional indicator of cell injury. The assays were performed according to manufacturer’s instructions. Briefly, 5 × 105 cells/ml of neurons were plated in a 96-well plate. Six hours after five min of exposure to different concentrations of glutamate, dye solution was added into each well and the cells were incubated for 2 h in a CO2 incubator (5% CO2, 21% O2) at 37.0 ± 0.5℃. After adding the stop solution, the absorbance was recorded at 570 nm. The result of each sample was calculated by averaging the results from triplicate wells. The percentage of cell viability was determined by comparing the absorbance value of each condition to the naïve control.
RNA isolation and reverse transcription-quantitative polymerase chain reaction
PCR primers.

Enzyme-linked immunosorbent assay for IL-4
The amount of IL-4 in the culture medium was measured using the Rat IL-4 Platinum ELISA kit (eBioscience) following the manufacturer’s instructions. Briefly, 50 µl of medium from cultured cells was collected and incubated with biotin-conjugate in the supplied 96-well plate for overnight at 4℃. The wells were then washed three times and then incubated with Streptavidin-HRP for 1 h at room temperature. After removing Streptavidin-HRP, TMB substrate solution was added to the wells and incubated for 10 min for color development. After adding stop solution, the absorbance of each well was read on a spectrophotometer at 450 nm. The standard curve was established with the concentration of 0–100 pg/ml. The result of each sample was measured by averaging the results from duplicate wells.
Electrophoretic mobility assay for NFAT
The activity of NFAT was assessed by using NFAT EMSA kit (Signosis) following the manufacturer’s protocol. Briefly, the nuclear extract from the cells was acquired using Nuclear Extraction kit (Signosis) and was incubated with NFAT probe. The protein/DNA complexes were then separated using a non-denaturing polyacrylamide gel. After electrophoretic transfer onto a nylon membrane, the probe was quantified using Streptavidin-HRP and chemiluminescent substrate. Optical density was determined using Kodak Analysis (EDAS) 290 system.
Ischemia model (unilateral MCA/CCAo) in rat
All animal studies followed the guidelines outlined in Guide for the Care and Use of Laboratory Animals from the NIH and ARRIVE guidelines (Animal Research: Reporting in Vivo Experiments) and were approved by the AWC of the UTHSC at Houston.
Focal ischemia was induced by left MCA and left common carotid artery (CCA) occlusion, as we described. 1 Briefly, three-months-old male Sprague Dawley rats were anesthetized with 0.35 g/kg i.p. injection of chloral hydrate. Following a small craniotomy, the left MCA rostral to the rhinal fissure, proximal to the major bifurcation of the MCA was occluded using stainless steel wire (Small Parts Inc, Miami, FL). The left CCA was occluded using atraumatic Heifetz aneurysm clips. Reperfusion was established at 1 h after occlusion. Interruption of flow through the MCA was inspected under the microscope and verified by cerebral blood flow rate (CP) measurement using a laser Doppler flowmeter (model BPM2, Vasamedics Inc., St Paul, MN). There was no mortality associated with this procedure.
CN activity assay in cultured neurons and ischemic brain from rat
The activity of CN was assessed by using a Cellular CN Activity Assay kit (Abcam) following the manufacturer’s protocol. Briefly, cultured neurons or brain tissue were rinsed with Tris buffer and homogenized in the lysis buffer, following the manufacturer’s instructions. The homogenates were centrifuged, supernatants were desalted by gel filtration, and 5 µg of total protein from each sample was used in the assay. The reactions were conducted according to the manual and were terminated after 30 min at 30℃. After 20 min of incubation for color development, the absorbance of each well was determined using a spectrophotometer at 650 nm. The standard curve was established for 0–2 nM PO4. Since the activities of phosphatases other than CN may also contribute to the results of the assay, EGTA buffer was used to chelate calcium in the reaction, thus abolishing the activity of CN. The activity of CN was then calculated by subtracting phosphatase activity with EGTA from total phosphatase activity.
Conditioned medium transfer to test microglia polarization
The medium from the cultured neurons transiently exposed to different experimental conditions (including control, NMDA, MK-801 + NMDA) was harvested and centrifuged at 400 g for 5 min. The supernatant (500 µl from neuron-conditioned medium) was combined with 500 µl of fresh DMEM, added to the microglia-containing wells, and incubated for 24 h before harvesting microglia for the gene expression analysis, as previously described. 1 For the naïve and positive controls, 500 µl of fresh Neurobasal medium with or without 1 ng/ml of rat recombinant IL-4 (ProSpec) combined with 500 µl of fresh DMEM was used. Also, 1 µg/ml of IL-4 neutralizing antibody (αIL-4Ab; BD Bioscience 555080) or IgG isotype control antibody (BD Bioscience 550878) was used to verify the causal role of IL-4.
Microglia-mediated phagocytosis of dead neurons
The assessment of microglia-mediated phagocytosis of dead neurons (DNs) was performed as we reported, 11 with minor modifications. Briefly, the cortical neurons from E16 prenatal rat pups at four days in culture were subjected to gamma irradiation (25 Gy) to induce cell death. After washing in PBS, the DNs were re-suspended in Neurobasal medium at 1–5 × 108 cells/ml, and added to the microglia cultures at a ratio of 20:1 (DNs:Microglia) for 2 h. The DNs were immunolabeled with rabbit anti-Tuj1 (PRB435P; Covance) to allow for counting of internalized DNs in microglia. The microglia were visualized using mouse anti-CD68 (MCA341; Bio-Rad) and mouse anti-HO1 (OSA-111; Enzo). The nuclei were stained with DAPI. The images were captured using a Zeiss confocal microscope LSM 800. Fifty microglia from each condition were analyzed for the presence of DN with ZEN blue edition software (Zeiss), according to intensity pixel count.
Statistical analysis
All statistical analyses, including correlation analysis between IL-4 mRNA expression and NFAT activity, were performed using the GraphPad Prism 7 and InStat programs. Two-way analysis of variance (ANOVA) was used to assess data with two grouping variables. Remaining data were analyzed using two-way ANOVA. The Tukey test was used for pairwise comparisons. Non-paired t-test was used when two groups were compared. All data are presented as mean ± SD.
Results
Sublethal OGD triggers neuronal IL-4 production and release
To study the mechanisms underlying the production and release of IL-4 by neurons in response to ischemia, we subjected rat primary cortical neuron in culture to OGD to simulate ischemic stroke-induced insult. We used 5 - and 20-min durations of exposure to OGD to achieve minor and more pronounced levels of injury, respectively (Figure 1(a)). Six hours after OGD, culture medium was collected for measurement of LDH (to gauge the level of injury) and IL-4 content in the culture media (ELISA). At the same time, cells were harvested and processed for the gene expression analysis (TaqMan real-time PCR). The results of the LDH assay (referred to as cell injury index) revealed an increased level of injury caused with 20 min of OGD as compared to 5 min of OGD (Figure 1(b)). Both the gene (Figure 1(c)) and protein (Figure 1(d)) levels of IL-4 significantly increased under exposure to shorter, less damaging (based on LDH release) OGD (5 min). These results support our earlier findings that only sublethal (less damaging) ischemic insult to neurons induces neuronal IL-4 translation, production, and release.
Brief ischemic insult (OGD model) triggers IL-4 production and release by neurons. (a) Eleven-day-old rat primary neurons in culture were grown on German-glass and exposed to OGD for 5 or 20 min. At 6 h post-exposure, neurons were fixed with 2% PFA and stained for the presence of MAP2 (green immunofluorescence) and nuclei (blue; DAPI); only 20 min of OGD resulted in histologically detectable injury. (b–d) Primary neurons in cultures were subjected to 5 or 20 min of OGD. Six hours after OGD, culture media was collected and cell injury was assessed through measuring LDH release (b) and IL-4 protein concentration using ELISA (d). At the same time point, neurons were collected for IL-4 mRNA analysis using Taqman probe-based real-time PCR system (c). The cell injury index was calculated by comparing the LDH released under each condition to the vehicle control. The data are mean ± SD (n = 3 independent experiments with triplicates at each experiment). *p < 0.05. **p < 0.01. One-way ANOVA followed by Tukey test.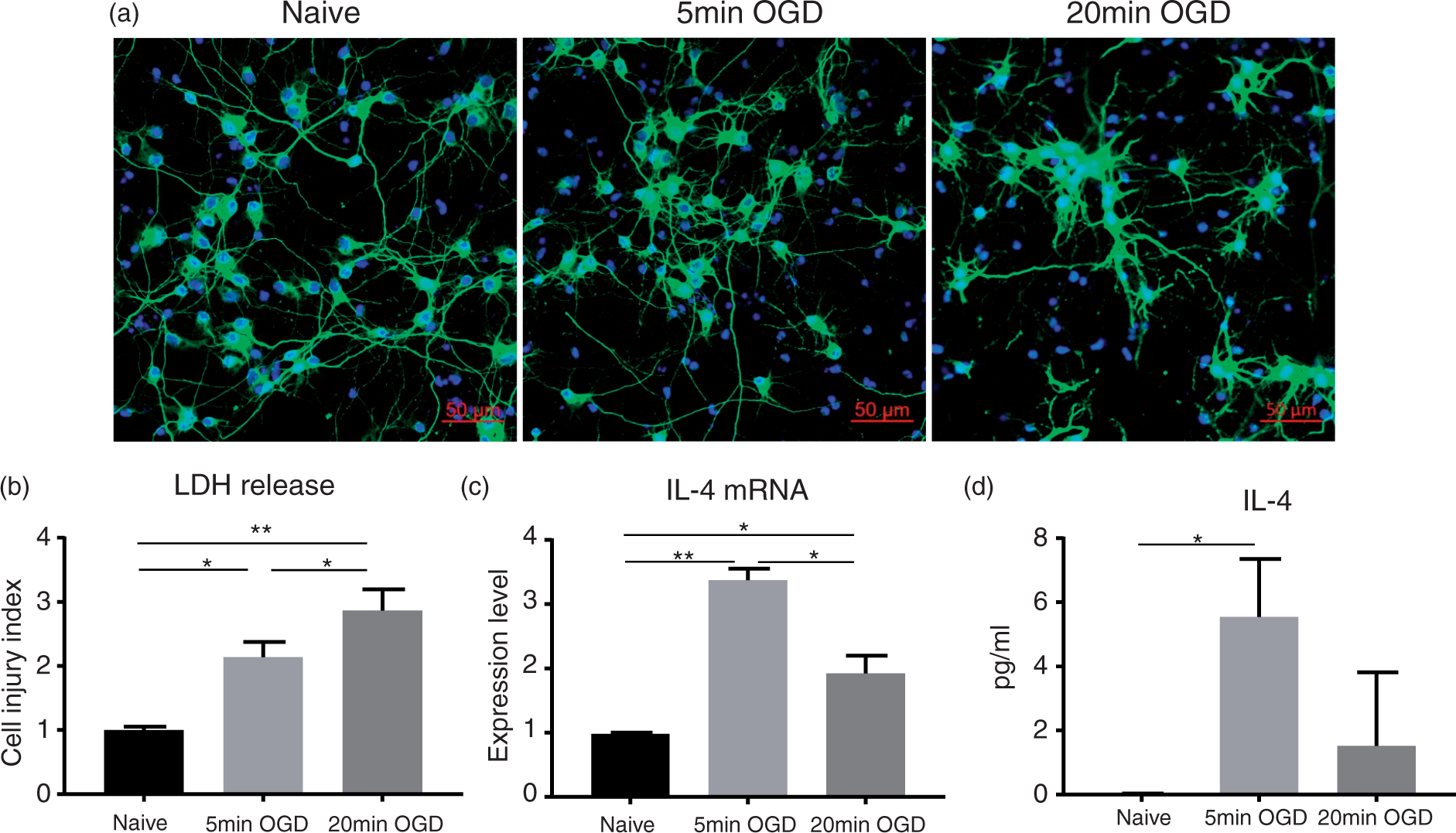
Neuronal IL-4 production is triggered by a sublethal glutamate-mediated excitotoxic insult
Cerebral ischemia-induced injury is a composite of many complex biochemical events that includes oxidative stress, apoptosis, excitotoxicity, and others.12,13 OGD-induced injury shares many of these biochemical events with the in vivo insult.14,15 Thus, to dissect which component of ischemic injury is involved in IL-4 gene induction in neurons, we transiently exposed primary neurons in culture to hydrogen peroxide (H2O2; to induce oxidative stress), staurosporine (STAU; to mediate pro-apoptotic stimulation), and glutamate (to induce excitotoxicity). First, based on the LDH release profile, we established a dose-response curve for glutamate and H2O2, allowing us to determine the neurotoxic concentrations of these agents (Figure 2(a)). An example of neuronal response to increased glutamate concentration is shown in Figure 2(d). We used 30 nM staurosporine, a dose that is well established to induce apoptosis.
16
To establish the impact of these agents on IL-4 production, we added them to the cultured neurons for 5 min followed by the replacement of medium to terminate the stimulus. After an additional 6 h of incubation, we collected the neurons for RNA analysis by RT-PCR (Figure 2(b)) and culture medium for IL-4 protein analysis by ELISA (Figure 2(c)). Of all conditions tested, only exposure to 50 µM glutamate induced neuronal IL-4 production and release (Figure 2(d) and (e)). These results suggest that IL-4 production is not a random response of neurons to harmful stimuli and that ischemia/OGD-mediated IL-4 production is mediated at least in part through activation of glutamate receptors.
Mild excitotoxicity triggers neuronal IL-4 production. Eleven-day-old primary neuron cultures were incubated with glutamate, H2O2, or staurosporine (STAU) at the indicated concentrations for 5 min, followed by culture media replacement. After 6 h of incubation, medium was collected for the assessment of LDH (a) and IL-4 protein concentration (c). The cell injury index was calculated by comparing the LDH released under each condition to the vehicle control. At the same time point, the neurons were collected for IL-4 mRNA analysis (b). Representative immunofluorescence of MAP2 (green) and staining of nuclei (blue; DAPI) (d) and MTT viability assay (e) at 6 h after transient (5 min) exposure of neurons to glutamate at the indicated concentration. The results showed low-grade injury to neurons exposed to 50 µM of glutamate. The data are mean ± SD (n = 3 independent experiments with triplicate at each experiment). **p < 0.01, compared with the naïve group. Two-way ANOVA followed by Tukey test.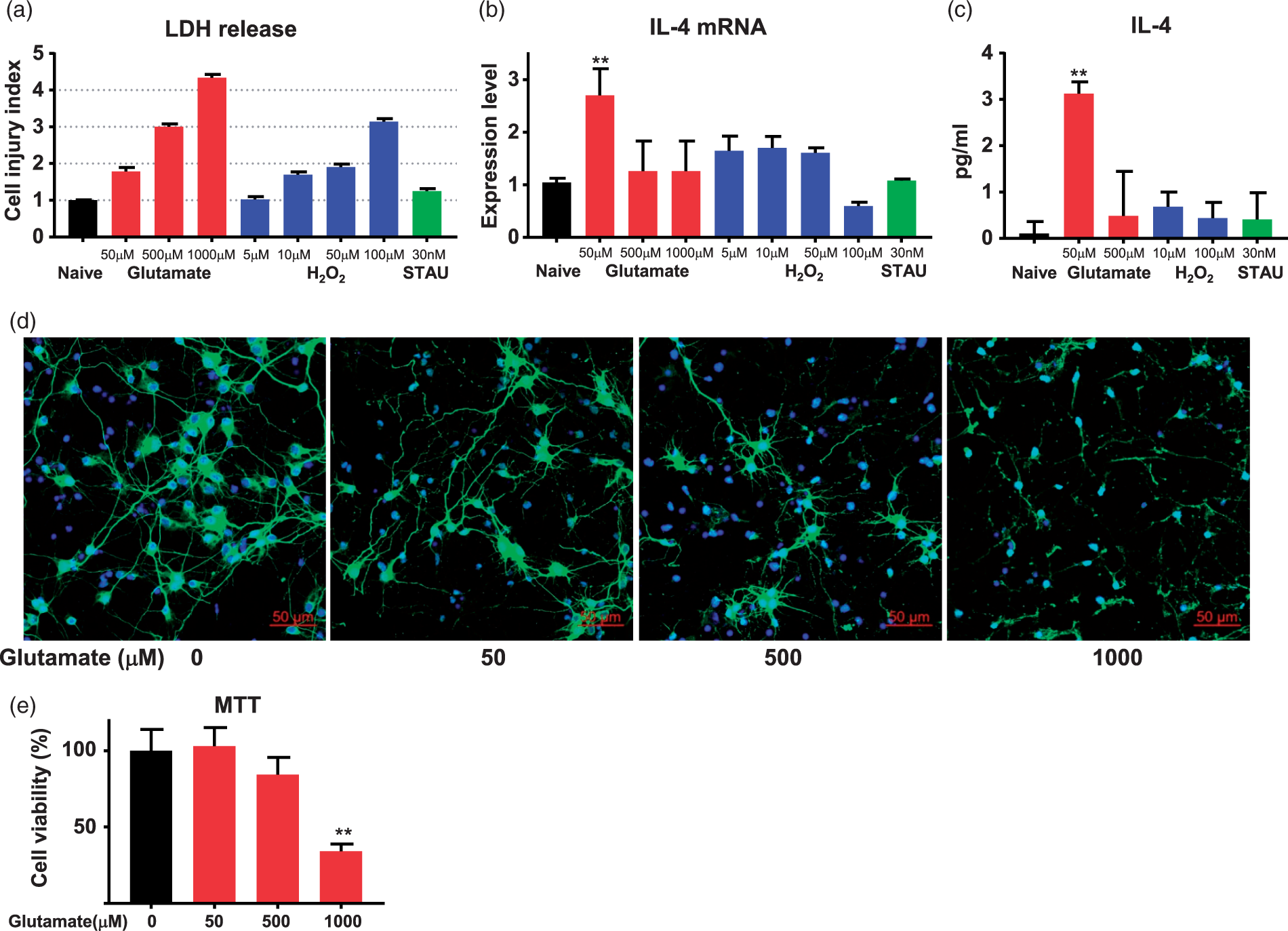
NMDA glutamate receptor mediates neuronal IL-4 production
Glutamate receptors comprise the ionotropic receptors, such as NMDA, AMPA, and kainate (KA) receptors, and metabotropic receptors. Among these receptors, the NMDA receptor (NMDAR) is regarded as an early and central contributor to glutamate-mediated ischemic injury and excitotoxicity.
17
Thus, to further understand glutamate-mediated neuronal IL-4 production, we investigated two ionotropic glutamate receptors – the NMDA and KA receptors. Again, we utilized primary neuronal cultures and transiently exposed them to high or low doses of NMDA or KA. We selected the range of concentrations of NMDA and KA that induces a mild and a more pronounced level of injury as in previous experiments (Figure 3(a)). In subsequent experiments, we exposed neurons to these predetermined concentrations of NMDA and KA. Six hours post-exposure, we harvested the neurons and culture medium for IL-4 gene expression and protein release. We found that the low concentration of 50 µM NMDA significantly increased neuronal IL-4 gene expression (Figure 3(b)) and protein release (Figure 3(c)). However, neither the high lethal dose of NMDA (500 µM), nor either dose of KA (50 µM or 500 µM) induced neuronal IL-4 production, despite the fact that the sublethal concentration of KA produced similar injury levels as the dose of NMDA that produced a mild level of injury.
NMDA but not kainate receptor mediates neuronal IL-4 production. (a–c) Eleven-day-old primary neuron cultures were transiently exposed to NMDA or kainite (KA) at the indicated concentrations for 5 min, followed by replacement to new Neurobasal medium to terminate exposure to excitotoxins. After 6 h of incubation, culture medium was collected for released LDH (a) and IL-4 protein (c), and neuron cells were harvested for IL-4 mRNA expression analysis (b). The cell injury index was calculated by comparing the LDH released at each condition to the naïve control. (d–f) Eleven-day-old primary neuron cultures were pretreated with MK-801, D-serine, or glycine at the indicated concentration for 2 min prior to the addition of 50 µM NMDA. After 5 min of incubation, the culture medium was replaced with fresh Neurobasal medium and the cells were incubated for 6 h. Culture medium was collected for analysis of released LDH (d) and IL-4 protein (f), and neurons were harvested for IL-4 mRNA determination (e). The data are mean ± SD (n = 3 independent experiments with triplicates at each experiment). *p < 0.05, **p < 0.01, compared with the naïve group unless indicated. One-way ANOVA followed by Tukey test.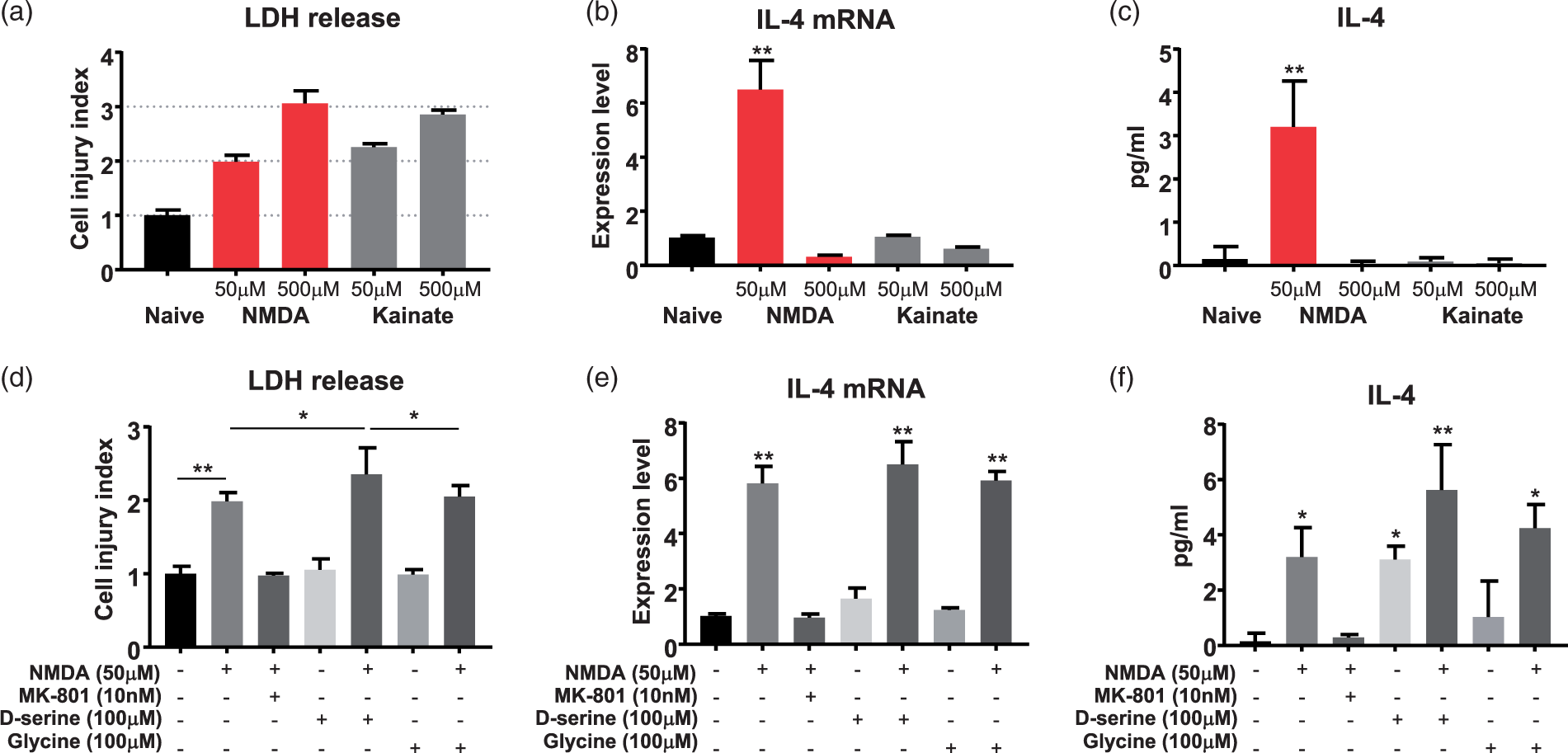
Next, to further validate the role of NMDARs in IL-4 induction by neurons, we tested if MK-801, a noncompetitive inhibitor of NMDAR, can block the effects of NMDA on neurons. We also sought to determine if the known co-activators of NMDAR, D-serine or glycine, are required in this process. The cultured neuronal cells were incubated with MK-801, D-serine, or glycine for 2 min prior to transient exposure to 50 µM NMDA (IL-4-inducing). The neurons and culture medium were harvested for gene, LDH, and IL-4 protein analysis at 6 hours after treatment with NMDA. The cell injury index (LDH release; Figure 3(d)) showed that 10 nM MK-801 fully blocked the effect of NMDA on LDH release. Conversely, D-serine or glycine alone did not cause toxicity to the neurons. However, when combined with 50 µM NMDA, D-serine augmented the LDH release (suggesting increased excitotoxic effect), while glycine showed no effect. While we have no experimental explanation for such differences, they may be due to the higher affinity of D-serine to the coactivator binding site than glycine. 18 The results of RT-qPCR (Figure 3(e)) and ELISA (Figure 3(f)) showed that MK-801 prevented the neuronal induction of both IL-4 mRNA and protein. However, NMDAR co-activators glycine and D-serine did not further induce IL-4 production. These results further support the role of NMDAR in mediating neuronal IL-4 production.
CN and NFAT regulates neuronal IL-4 production in response to NMDA
After establishing the causal role for NMDARs in neuronal IL-4 production, we next sought to explore the mechanisms downstream of the NMDAR that could regulate this process. One potential pathway leading to IL-4 transcription is through the transcription factor NFAT, as has been shown in some lymphocytes. 19 Interestingly, neurons express NFAT family members as part of activity-dependent gene transcription. 20 The activation of NFAT is primarily modulated through its dephosphorylation by the calcium-dependent phosphatase CN, an enzyme that is abundant in neurons and could be activated in response to NMDA. 21
Thus, to determine if NMDA activates CN in neurons, we measured the CN phosphatase activity in NMDA-treated cortical neurons. And to determine if this occurs in vivo, we assessed CN phosphatase activity in focal ischemia-affected brains, which are known to experience excitotoxic insult.
First, we exposed neurons in cultures to 50 µM NMDA with or without pre-treatment with MK-801, a prototypic NMDA blocker, or FK-506, a prototypic inhibitor of CN.
22
One hour after exposure to NMDA, the neurons were processed for CN activity (Figure 4(a)). As anticipated, NMDA significantly increased the CN activity in the cortical neurons and this activation was reversed by blocking NMDAR with MK-801. We also showed that FK506 predictably blocked CN activity. Second, we measured CN activity in the ischemic and contralateral cerebral cortex harvested from rats subjected to 60 min of MCA/CCA occlusion and 5 min reperfusion. The activity of CN was significantly higher in the ischemia-affected cortices as compared to contralateral cortices (Figure 4(b)). Altogether, our data suggest that CN activity is quickly increased in response to NMDA and ischemia.
FK-506, a calcineurin inhibitor, prevents NMDA-induced IL-4 production. (a) The activity of calcineurin (CN) in cultured neurons 1 h after transient exposure to NMDA, MK-801, and FK-506 was measured by CN phosphatase activity assay kit. (b) The activity of CN in ischemia-affected ipsilateral and contralateral cerebral cortices from three Sprague Dawley rats subjected to 60 min MCA/CCA occlusion and 5 min reperfusion. Eleven-day-old primary neuron cultures were pretreated with FK-506 for 2 min prior to the addition of 50 µM NMDA. After 5 min of incubation, the culture medium was replaced with fresh Neurobasal medium and the cells were incubated for 6 h. Culture medium was then collected for LDH release (c) and IL-4 protein analysis (e), and neurons were harvested for IL-4 mRNA expression analysis (d). The cell injury index was calculated by comparing the LDH released at each condition to the naïve control (f) The activity of NFAT 6 h after exposure of neurons to NMDA, MK-801, FK-506, D-serine, and OGD was measured by NFAT EMSA. IL-4 mRNA expression by neurons under the same experimental conditions as in (f) was determined using Taqman probe-based real-time PCR system (g). The expression of IL-4 mRNA under all the assessed conditions showed a strong correlation with the activity of NFAT (h). All the data are mean ± SD (n = 3 independent experiments with triplicates at each experiment). *p < 0.05, **p < 0.01, ***p < 0.001 compared with the naïve group unless indicated in the figure. One-way ANOVA followed by Tukey test. Correlations between NFAT activity and IL-4 mRNA were calculated using Pearson coefficient, R2 = 0.898, p < 0.01.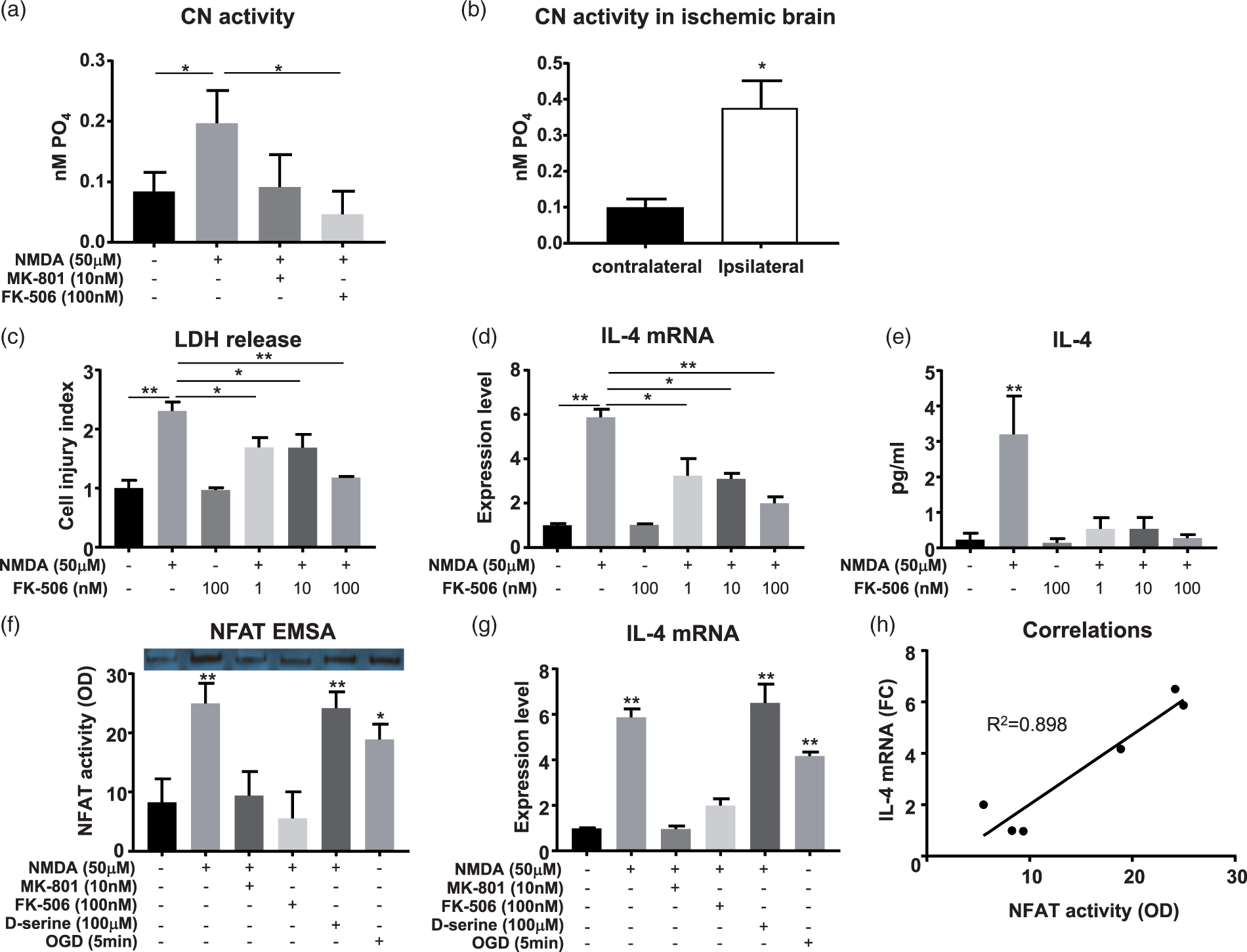
Understanding that NMDAR-mediated induction of IL-4 (Figure 3(b)) coincides with CN activation (Figure 4(a)), our next step was to determine if NFAT-dependent transcription that is normally enhanced with CN activation (and blocked with FK506), could control the synthesis of IL-4 in the neurons. We treated cortical neurons in culture with FK-506 prior to transiently exposing them to the 50 µM NMDA (to induce IL-4 production). At 6 h, we collected the neurons and culture medium for IL-4 determination and establishing cell injury index (Figure 4(a) to (e)). The injury index revealed that FK-506 alone is not toxic to neurons, even under high (100 nM) concentrations. Moreover, in a dose-dependent manner, FK-506 partially blocked excitotoxic stress (LDH release) produced by NMDA. The RT-qPCR analysis (Figure 4(d)) demonstrated that FK-506 reduced IL-4 mRNA expression induced by NMDA in a dose-dependent manner. IL-4 ELISA (Figure 4(e)) showed that FK-506 also blocked the presence of IL-4 in the culture media containing neurons treated with 50 µM NMDA.
To further investigate the relationship between the CN-NFAT pathway and neuronal IL-4 gene expression, our next approach was to directly measure the activity of NFAT in the NMDA-treated neurons. To achieve this task, we pre-treated neurons in cultures with MK-801, D-serine, or FK-506 before transiently exposing them to 50 µM NMDA or to 5 min OGD to trigger IL-4 production. The neurons were harvested 6 h after exposure to NMDA or 5 min after OGD. Neuronal nuclei proteins were extracted for electrophoresis mobility shift assay (EMSA) to measure the activity of NFAT, which also is an indirect indicator of CN-mediated dephosphorylation of NFAT (prerequisite for its activation). This experiment showed that NMDA alone, NMDA with D-serine, and OGD significantly enhanced the activity of NFAT in neurons (Figure 4(f)). Both MK-801 and FK-506 effectively abolished the NFAT activation achieved with NMDA. In addition, we found that the activity of NFAT in neurons correlated with IL-4 mRNA expression (R2 = 0.898), strongly suggesting that the CN-NFAT pathway acts as a regulator to IL-4 gene transcription in neurons (Figure 4(g) and (h)).
Neuronal IL-4 drives microglia toward a phagocytic phenotype
After investigating the mechanisms underlying the induction of IL-4 by neurons, we sought to elucidate the potential function of this neuron-secreted IL-4, especially as it relates to neuron-microglia crosstalk. It is established that IL-4 induces alternative activation of macrophages,4,5,23 suggesting that the neurons under NMDA stimulation can modify microglia to their alternative phenotype via secretion of IL-4. To test this notion, we performed conditioned medium transfer experiments (Figure 5(a)). The neuronal cultures were exposed to NMDA for 5 min to induce IL-4 production. Six hours later, this conditioned medium or medium from untreated neurons was added to primary microglia in culture. We then tested the effect on the expression of selected genes to characterize the status quo of microglia polarization. Fresh Neurobasal medium containing recombinant IL-4 (rIL-4) served as a positive control. To exclude the possibility that some other neuron-secreted factor and not IL-4 is involved in modifying microglia behavior, in additional experiments we added an IL-4 neutralizing antibody (αIL-4Ab) or IgG isotype control antibody to the neuron-conditioned medium. The relative gene expression profile is presented as log2FC compared to medium transferred from neuron cultures that was not exposed to NMDA. GAPDH (Figure 5(b)) was used as an internal control.
Microglia phenotype is modified by neuron-conditioned medium through IL-4. (a) A diagram of the conditioned medium transfer experiment. Neurons were transiently exposed to 50 µM NMDA, with or without 10 nM MK-801 (to block NMDAR activation by NMDA) for 5 min, followed by replacement of the medium to remove NMDA and MK-801. After 6 h of incubation to allow neurons to produce and release IL-4, conditioned medium was harvested from each of the experimental conditions and transferred individually to the primary microglia in culture. Some groups were exposed to conditioned medium containing α-IL-4Ab (1 µg/ml) to neutralize IL-4 secreted by neurons. IgG isotype antibody (1 µg/ml) was used as the control for anti-IL-4Ab. Fresh neuronal media with or without rIL-4 (1 ng/ml) and α-IL-4Ab was used as an additional control. After 24 h of incubation, we harvested microglia for RT-qPCR to assess indicated genes related to microglia phenotype (b–i). The data are presented as log2FC compared to medium transferred from untreated neuron cultures (n = 3 independent experiments with triplicates at each experiment). GAPDH was used as an internal control. (j) Representative micrographs illustrating rat primary microglia (green, labeled with anti-CD68 and anti-HO-1 antibodies) at 2 h after initiation of phagocytosis of apoptotic neurons (red, labeled with anti-Tuj1 antibody). Nuclei were stained with DAPI (blue). Prior to adding apoptotic neurons for the assessment of phagocytosis, the microglia were incubated for 24 h with the neuron-conditioned medium prepared as described in (a; above). After 2 h of exposure to apoptotic neurons, the microglia containing engulfed neurons were fixed and processed for immunofluorescence staining. (k) The phagocytic ability of microglia was quantified by counting internalized neurons based on pixel intensity of Tuj1 signal inside microglia. The data are expressed as mean ± SD (n = 50 microglial cells per condition). *p < 0.05, **p < 0.01, ***p < 0.001.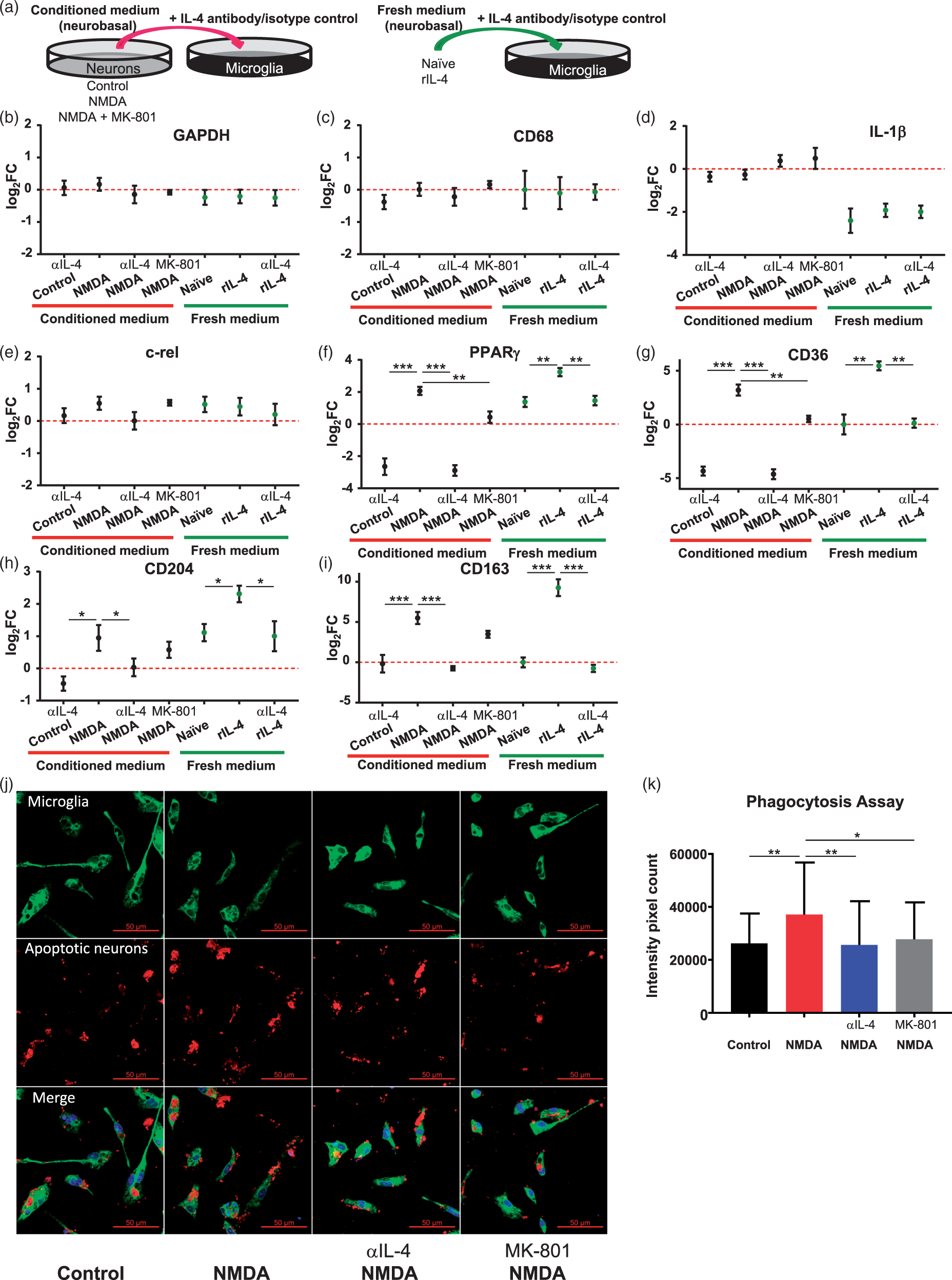
We employed CD68 as a marker for microglia, and it remained unchanged throughout all conditions (Figure 5(c)), which in conjunction with the unchanged level of the internal control GAPDH, indicates that the microglia cell number remained constant throughout the experiment. The expression of inflammatory genes, including IL-1β (a pro-inflammatory cytokine, Figure 5(d)) and c-Rel (a subunit of NF-κB, Figure 5(e)) was not affected by any of the experimental interventions. In the case of IL-1β, the expression level for this gene was lower in fresh medium (independent of our experimental interventions) than in the presence of neuron-conditioned medium, suggesting that some factors intrinsic to neuron-conditioned medium keep expression of IL-1β elevated.
As expected, the expression of PPARγ (Figure 5(f)), a downstream gene regulated by STAT6 through the IL-4 receptor, was enhanced in the positive control (rIL-4), and the enhancement was abolished by αIL-4Ab. NMDA-conditioned medium similarly enhanced the expression of PPARγ in microglia, which again was abolished by αIL-4Ab, suggesting that it was the IL-4 in the NMDA-conditioned medium that enhanced the expression of PPARγ in microglia. As we demonstrated above, pre-treatment with MK-801 suppressed the production of neuronal IL-4 induced by NMDA, which may explain why conditioned medium from neurons exposed to NMDA and MK-801 failed to induce the expression of PPARγ in microglia. CD36 (Figure 5(g)), CD204 (Figure 5(h)), and CD163 (Figure 5(i)) are scavenger receptors involved in phagocytic functions of microglia and are important markers for the M2 healing phenotype of microglia. The results of gene analysis showed that the expression of these genes followed the same pattern as PPARγ. Medium from NMDA-exposed neurons enhanced the expression of these scavenger receptors, and αIL-4Ab abolished this enhancement. Conditioned medium obtained from neurons treated with MK-801 prior to adding NMDA also failed to induce CD36, CD204, and CD163 gene expression.
To confirm that the gene expression profile in microglia (described above) indeed renders the phagocytic phenotype of microglia in response to neuron-derived IL-4, we performed phagocytosis assays by measuring the engulfment of apoptotic neurons by primary microglia exposed to neuron-conditioned medium (Figure 5(j)). Primary microglia in cultures were incubated for 24 h with conditioned medium from neurons that were exposed to NMDA, NMDA in the presence of IL-4 neutralizing Ab, or NMDA in the presence of MK-801, before exposure to apoptotic neurons. After 2 h of incubation, the neurons engulfed by microglia were labeled with anti-Tuj1 antibody and counted based on intensity pixel count. In agreement with the gene analysis results, conditioned medium from NMDA-exposed neurons (containing IL-4; Figure 3(c)) significantly enhanced the phagocytic capacity of microglia, while the presence of αIL-4Ab abolished this enhancement (Figure 5(k)). Also, medium from neurons pretreated with MK-801 before exposure to NMDA (containing no IL-4; Figure 3(f)) did not enhance microglia phagocytic ability. Our data demonstrated the function of neuron-derived IL-4 in driving microglia toward the pro-phagocytic phenotype.
Discussion
In this study, we established a new model of interactions between stressed neurons and microglia. Our data suggest that in response to sublethal ischemia, neurons secrete IL-4 as a signal to communicate with microglia to modulate their “healing” phenotype. Specifically, by employing an in vitro model of ischemia (OGD), we found that excitatory responses, involving glutamate and mediated by NMDAR, induce IL-4 transcription through the CN-NFAT pathway (Figure 6). By using neuron-conditioned medium transfer to cultured microglia in the presence or absence of αIL-4Ab (to neutralize neuron-secreted IL-4), we demonstrated that neurons produce sufficient amounts of IL-4 to induce microglia polarization. Based on gene expression profiling, it appears (as anticipated) that neuronal IL-4 induces the microglial M2 “healing” phenotype. This phenotype is associated with enhanced phagocytic activities, shown herein with the microglia phagocytosis assay (Figure 5(k)). In the context of cerebral ischemia, it could translate into improved brain repair and better post-stroke recovery.
1
An illustration of proposed mechanisms of neuronal IL-4 production. The IL-4 produced by neurons under sublethal ischemia is mediated by NMDA receptor signaling and regulated through the CN/NFAT pathway.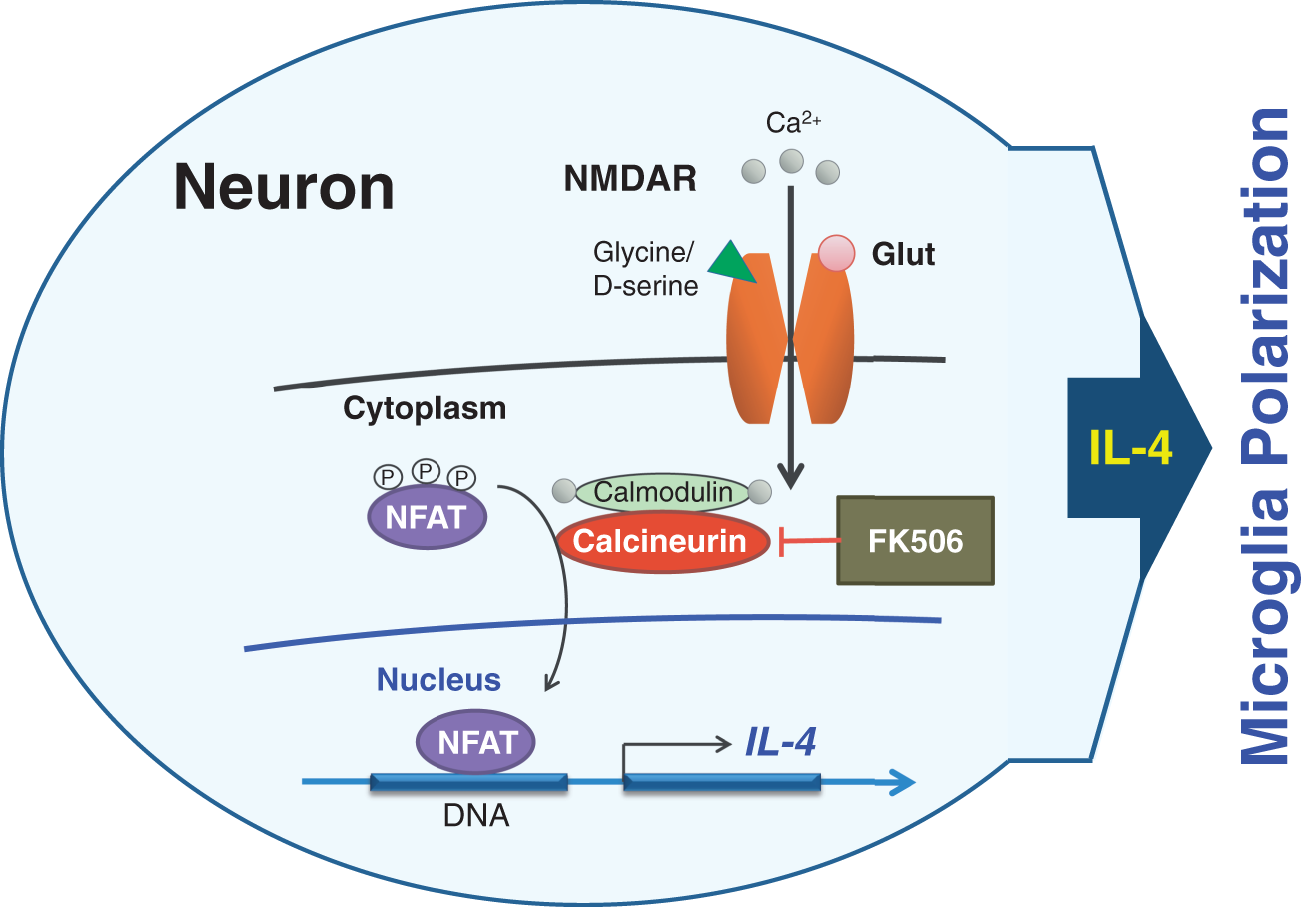
IL-4 is a potent pleiotropic regulator of inflammatory and immune pathways.24,25 Described as a product of activated T cells stimulating B cell proliferation,4,26 IL-4 was subsequently recognized as an essential effector of Th2 immune responses and M2 alternative macrophage polarization.4,27,28 Importantly, our group and others showed that rIL-4 used as a therapeutic agent is effective in reducing damage and promoting recovery after cerebral ischemia.1,6,29 Using mice expressing an IL-4 reporter construct allowing for visualization of cells expressing IL-4, we have recently reported that neurons surrounding the infarct core, and as such likely experiencing lower-grade ischemia, express IL-4. This observation was the first to report that IL-4 could also be produced by neurons. Based on the known role of IL-4 in microglia/macrophage modulation, we suggested that neuron-derived IL-4, similar to what has been suggested for lipocalin-2, 30 could support neuron-to-microglia communications, allowing neurons to directly prevent microglial overactivation that could affect the integrity of neighboring neurons.
One of the main objectives of this study was to understand how ischemia regulates IL-4 production by neurons. IL-4 is proposed to play a regulatory function in learning and memory,31,32 a process that normally involves many aspects of excitatory neurotransmission, NMDAR-mediated in particular. 33 Interestingly, some evidence also exists that the brief exposure of hippocampal tissue to ischemia through recruitment of NMDAR can induce long-term potentiation, 34 a form of synaptic rearrangement that underlies synaptic plasticity associated with learning and memory. These associations provided us with a hint that excitatory pathways could also be involved during induction of IL-4 synthesis by neurons. Indeed, in addition to brief exposure to OGD, low concentrations of glutamate or NMDA also induced IL-4 production by neurons.
Physiologically, activation of NMDAR induces Ca2+ influx into neurons, a process that controls activation of various Ca2+-dependent enzymes involved in neuronal homeostasis. Unlike during physiological responses, ischemia induces massive glutamate release (especially within the ischemic core), which results in over-activation of NMDAR, leading to excessive influx of Ca2+ into the ischemia-affected neurons. This Ca2+ overload dysregulates Ca2+-dependent processes and leads to neuronal injury, referred to as excitotoxicity,35–37 and results in necrotic or apoptotic neuronal death.38,39 While various sources of intracellular Ca2+ may result in activation of a distinct subset of Ca2+-dependent enzymes, one enzyme that is activated in neurons in response to Ca2+ entry through NMDAR activation is the Ca2+/calmodulin-dependent serine/threonine protein phosphatase, CN.40,41 CN is a pleiotropic, neuron-abundant protein that regulates many vital functions ranging from plasticity to neurodegeneration. 42 Our in vitro and in vivo studies indicate that CN activity is rapidly increased in cortical neurons upon exposure to NMDA and upon induction of cerebral ischemia. Using the selective CN inhibitor FK506, 43 we showed that FK506 could reduce NMDA-induced CN phosphatase activity and block NMDA-induced IL-4 production, indicating that CN is an upstream regulator of IL-4 synthesis in neurons.
NFAT is a phosphoprotein and a transcription factor that is normally activated by CN-mediated dephosphorylation. NFAT, through cooperation with other transcription factors, promotes expression of many target genes involved in immunoregulation. 44 Besides T-cells, where the role of NFAT is well documented, the CN-NFAT pathway has also been demonstrated to regulate synaptic plasticity,45,46 suggesting that NFAT plays an important regulatory role in neurons. Although the target genes of NFAT in neurons are largely unknown, here we established a strong correlation between the activity of NFAT and IL-4 gene expression, strongly suggesting that IL-4 expression in neurons is a product of NFAT transcriptional regulation. Specifically, we showed that NMDAR activation with a sublethal dose of NMDA leads to NFAT activation in a CN-dependent manner, as CN inhibition with FK-506 suppressed NFAT activation and IL-4 production. Our findings are consistent with the work of others showing that IL-4 transcription in some lymphocytes is under transcriptional regulation of NFAT. 19
Microglia are the brain resident macrophages and the first line responders to injuries in the central nervous system. It is generally accepted that in response to ischemic insult, microglia sense the presence of DAMP molecules generated as a result of cell damage. DAMP-activated microglia generate pro-inflammatory and oxidative responses that could be damaging to neighboring neurons that survived ischemic insult. As stated earlier, IL-4 is a potent regulator of the alternative activation of microglia/macrophages,1,5,6 a process that results in polarization of microglia/macrophages from a pro-inflammatory phenotype to anti-inflammatory phenotype; this process is normally under the control of transcription factor PPARγ. This polarization process leads to increased expression of scavenger receptors and enhanced phagocytic functions, an activity essential for effective clearance of the infarcted tissue and inflammation resolution.1,10,47–49 In support of the bioactive nature of IL-4 secreted by neurons, we demonstrated that conditioned media harvested from neurons subjected to an IL-4-inducing dose of NMDA effectively stimulated the expression of PPARγ and several key scavenger receptors (CD36, CD204, and CD163) and phagocytic ability of in microglia, responses that are associated with improved post stroke recovery.10,50–54 Importantly, IL-4 neutralizing antibody blocked the gene-induction responses and phagocytic functions, suggesting that IL-4 and no other factor co-secreted with IL-4 by neurons mediated these microglia responses.
In conclusion, we have now demonstrated that neurons under mild ischemic insult use the glutamatergic/NMDA signaling pathway, Ca2+/calmodulin-dependent phosphatase CN, and NFAT-mediated transcription to induce the expression of IL-4. This neuron-secreted IL-4 then effectively participates in the modulation of the microglial phenotype.
Footnotes
Authors’ contributions
JA, S-MT, and XuZ designed and planned experiments. S-MT, XuZ, and XiZ conducted experiments. JA, S-MT, and XuZ performed analyses and wrote the manuscript.
Funding
The author(s) disclose receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by National Institute of Neurological Diseases and Stroke (NINDS), grants R01NS084292.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.