
Abstract
Triggering receptor expressed on myeloid cells-2 (TREM2) is an innate immune receptor that promotes phagocytosis by myeloid cells such as microglia and macrophages. We previously showed that TREM2 deficiency worsened outcomes from experimental stroke and impeded phagocytosis. However, myeloid cells participating in stroke pathology include both brain resident microglia and circulating macrophages. We now clarify whether TREM2 on brain microglia or circulating macrophages contribute to its beneficial role in ischemic stroke by generating bone marrow (BM) chimeric mice. BM chimera mice from TREM2 knockout (KO) or wild type (Wt) mice were used as donor and recipient mice. Mice were subjected to experimental stroke, and neurological function and infarct volume were assessed. Mice with intact TREM2 in brain microglia showed better neurological recovery and reduced infarct volumes, compared with mice lacking microglial TREM2. Myeloid cell activation and numbers of phagocytes were decreased in mice lacking brain TREM2, compared with mice with intact brain TREM2. These results suggest that TREM2 expression is important for post-stroke recovery, and that TREM2 expression on brain resident microglia is more essential to this recovery, than that of circulating macrophages. These findings might suggest a new therapeutic target for cerebrovascular diseases.
Introduction
Innate immune responses accompanying ischemic stroke involve various immune cells and various pathways which interact with one other, and contribute significantly to stroke pathophysiology.1,2 Microglia are the brain’s resident immune cell and recent work has shown that these cells play a crucial role in the brain’s immune response, not only in brain ischemia, but in other brain diseases. Microglia are the brain’s resident myeloid-lineage cells involved in constant immune surveillance. 3 Like other myeloid cells, they are capable of transforming into phagocytes, and have thus been referred to as ‘brain macrophages’.1,2 To date, numerous studies4,5 have been performed to clarify the role of microglia in stroke pathophysiology, including its functions in chemotaxis, secretion of pro-inflammatory and anti-inflammatory cytokines, growth factors, chemokines, proteases, and neurotrophins. Currently, regulation and modulation of this multifunctional cell have been investigated as a novel therapeutic target in stroke.
Among these many microglial functions, its phagocytic properties have been intensely studied in several brain disease models. Several phagocytic pathways have been identified in the past, 6 and the efficient clearance of tissue debris is thought to be critical in the reconstruction and reorganization of neuronal networks. Recent work in the area of stroke has demonstrated that microglia possess both damaging and beneficial properties depending on the timing and specific phenotype. 7 We previously reported a beneficial role of microglial phagocytosis, which was promoted by the innate immune molecule, triggering receptor expressed on myeloid cells-2 (TREM2) in experimental stroke models.8,9 We showed that in a paradigm of neuroprotection by therapeutic hypothermia, TREM2 was upregulated to a greater extent in myeloid cells within the ischemic brain. By studying mice deficient in TREM2, we found that these mice had worsened neurological outcomes compared to wildtype. 8 In line with our previous reports, recent studies from other groups similarly reported a beneficial role of TREM2 in ischemic brain.10,11
TREM2 was originally described on circulating macrophages, where it bound anionic moieties on bacteria and yeast and led to their phagocytosis. 12 TREM2 is uniquely expressed on myeloid cells,13,14 as it is not observed on granulocytes and lymphoid cells such as neutrophils and lymphocytes. 12 In the brain, it is only expressed on microglia,15,16 although not all microglia express TREM2. 17 Both macrophages recruited from circulation and microglia derived from the brain itself, have the ability to phagocyte ischemia-damaged tissues. 18 Therefore, TREM2 may be acting through microglia and/or circulating macrophages to carry out its phagocytic functions in stroke. However, it is unknown which pool of TREM2 positive cells is responsible for its phagocytic functions.
To explore which pool of myeloid cells contributes to TREM2’s effect in ischemic stroke, we conducted the present study by employing a bone marrow (BM) chimeric mouse model. BM chimeric mice are created when BM from a donor strain is transplanted into recipient mice, whose marrow had been ablated. This model allows for the study of marrow related contributions to a particular pathology.18–21 This experimental approach has enabled us to distinguish resident microglia versus infiltrated circulating macrophages in vivo.3,18
Materials and methods
Animals
All experimental procedures were approved by the San Francisco Veterans Affairs Medical Center Institutional Animal Care and Use Committee, and were in accordance with National Institutes of Health and ARRIVE (Animal Research: Reporting In Vivo Experiments) guidelines.
Male TREM2 knock-out (KO) mice and wild-type (Wt) littermates, which were originally generated by Colonna and colleagues (Washington University, St.Louis, MO), 22 were used as previously described. 8 These mice were backcrossed for over five generations. Green fluorescent protein (GFP) transgenic mice were also used as donors, in pilot experiments to develop and validate the BM chimera model.
Generation of BM chimeric mice
BM from donor mice (male, two to three months of age, KO or Wt mice) was harvested from the femurs. Cells were suspended in 2% bovine calf serum (BCS). Prior to BM transplantation, recipient mice (male, two to three months of age, KO or Wt mice) were treated with busulfan (25 mg/kg/d) for five days to ablate endogenous BM. Recipient mice were given 2 × 107 donor BM cells in 200 µl PBS via a tail vein. 20 The resulting chimeric mice were housed in autoclaved cages with sterile water containing antibiotics to allow complete reconstitution of BM before use in further experiments.
In pilot studies to validate this approach in our lab, GFP transgenic mice (C57BL/6-Tg (CAG-EGFP)1Osb/J, Jackson Labs) were used as donors, while Wt mice were used a recipients. After receiving donor marrow, mice were survived 80 days, after which brains were harvested for histology and flow cytometry. Some of these mice also underwent stroke surgeries (see below).
Chimeric mice derived from the TREM2 KO strain were divided into four groups: Wt to Wt group (Wt BM transplanted into Wt mice), KO to Wt (KO BM transplanted into Wt mice), Wt to KO (Wt BM transplanted into KO mice), KO to KO (KO BM transplanted into KO mice) (Figure 1(a)). These mice also underwent stroke surgeries 80 days post transplantation.
Scheme of the experimental design and validation of the bone marrow (BM) chimera model in mice. (a) Wildtype (Wt) and TREM2 knockout (KO) mice were used as BM donors (Bone marrow) or hosts (Brain). Four permutations representing four experimental groups are studied. (b) Following endogenous BM ablation with busulfan, BM from green fluorescent protein transgenic (GFP Tg) mice was transplanted into Wt mice (n = 3). Blood samples were collected and subjected to flow cytometry. No GFP positive cells (y-axis) were present in circulating leukocytes from non-transplanted control mice (control, top graph). By 40 days post BM transplantation, ∼74% of cells in from the circulation were GFP positive (middle graph). By 80 days post transplantation (bottom graph), ∼95% of circulating cells were GFP positive, and suggests near complete turnover of the endogenous BM by the GFP transgenic BM. (c) Mice were transplanted with GFP Tg BM, then subjected to dMCAO 80 days later. Immunofluorescence of brains from a GFP BM chimeric mouse is shown one day post dMCAO. In the ipsilateral peri-infarct region, several GFP positive cells are observed (arrows) and are presumed to derive from the circulation. In the contralateral non ischemic hemisphere, no GFP positive cells could be seen, nor were any GFP positive cells observed in sham control chimeras (not shown). Thus, transplanted BM did not turnover any endogenous brain cells at the time of the stroke surgeries.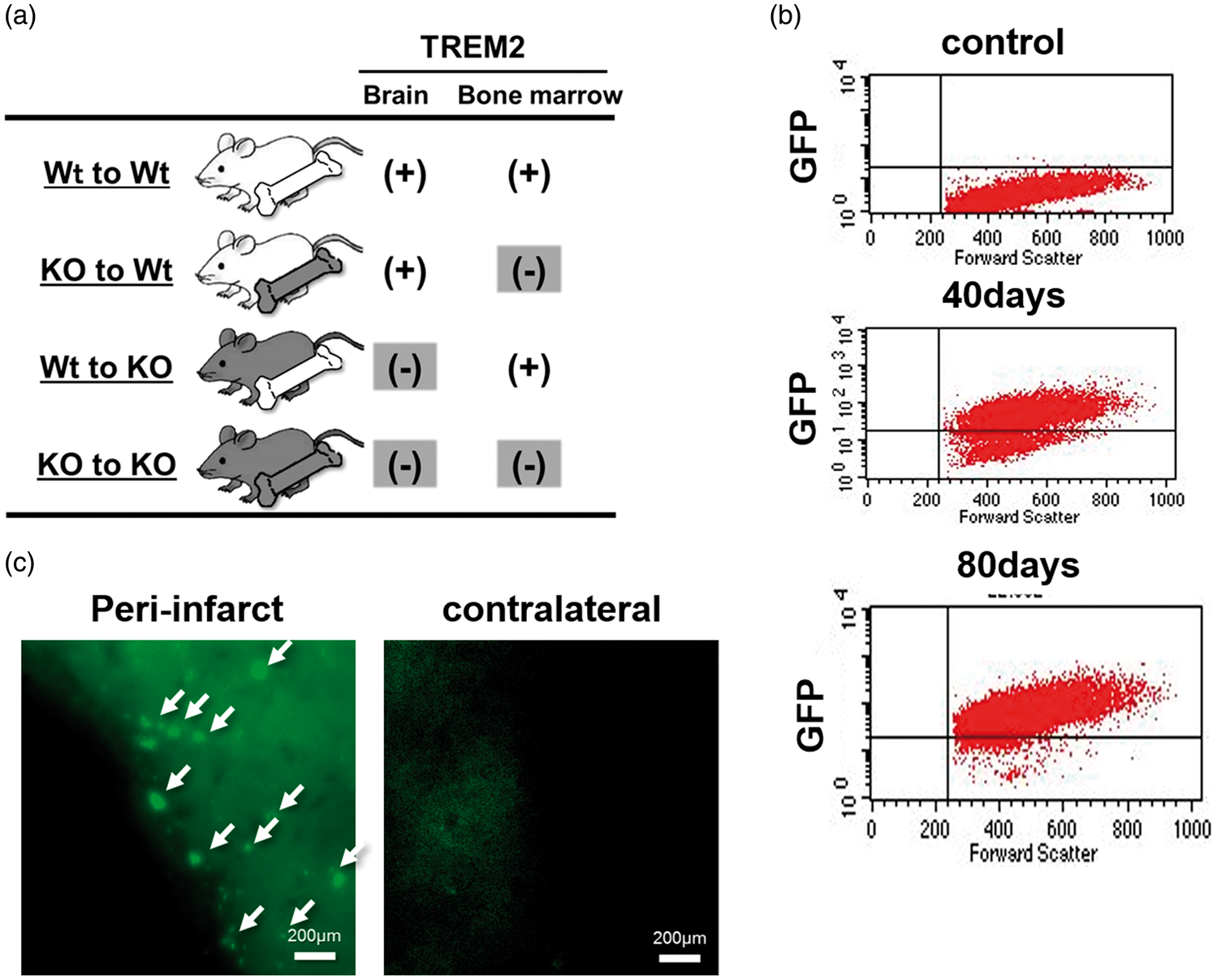
Flow cytometry
After collection of whole blood samples in EDTA at the time of euthanasia, cells were suspended in cell lysis buffer to remove red blood cells. 23 Cell suspensions were transferred to a 40 µm pore size filter, fixed in BD Cytofix (BD Biosciences, Cat No: 554714) and resuspended in 400 ul of ice cold FBS (BD Pharmingen, Cat No:554656). Samples were analyzed on a FACSCalibur flow cytometer (Becton Dickinson Immunocytometry Systems, BDIS) at the San Francisco VAMC Flow Cytometry Core facility using FL-1, FSC and side scatter detectors. Green eGFP fluorescence was measured in the FL1 channel. On each sample, 10,000 events were acquired within the single gate. The analysis of data was performed with CELLQuest software (BDIS).
Stroke model
All surgical techniques were performed under aseptic conditions. After approximately two months following BM transplantation, stroke surgery was performed under gas anesthesia. Anesthesia was induced by the inhalation of 3.0% isoflurane in medical air: O2 (80%:20%). Surgical planes of anesthesia were assessed by the absence of hindleg withdrawal in response to a pinch. A 1 cm skin incision was created between the left margin of the orbit and the tragus, and the temporalis muscle was incised. A focal cerebral infarct was induced by permanent occlusion of the distal left middle cerebral artery (dMCAO) as previously described.8,24 Briefly, a small craniotomy was made above the proximal segment of middle cerebral artery (MCA) and the MCA was exposed after the dura was opened and retracted. The MCA was occluded by coagulation at the MCA segment just proximal to the olfactory branch. Rectal temperature was maintained between 36.5℃ and 37.5℃ during the procedures by using a thermometer connected to a heating pad. At the end of surgery, incision sites were closed and animals were allowed to recover. Mice were returned to their cages and allowed free access to food and water, and were housed in a climate-controlled environment (25℃). Mice were included in the study if they showed neurological deficits as defined by a Bederson score of at least 3 (circling behavior) at 24 h post dMCAO. 25
Behavior assays
Neurological assessments were performed before surgery and 2, 7, and 14 days after stroke as previously described. 26 All studies were recorded using a video camera, and scoring was performed by two different investigators blinded to experimental conditions.
Elevated body swing test
To measure motor deficits, mice were suspended vertically by the tail with their heads elevated three inches above the test bench. A lateral swing was counted each time the animal moved its head >10° away from the vertical axis. Counts were carried out for four 15-s sessions, and the frequency of right-biased swing was calculated.26,27
Adhesive removal test
Circular adhesives (4 mm diameter) were attached to the palm of each forepaw, and mice were observed for 2 min and scored for identifying the time it took for the mouse to detect the adhesive (contact time), and the time it took to remove it (removal time).
Histology
Fourteen days post stroke, animals were euthanized with an anesthesia overdose, perfused transcardially with ice-cold normal saline. Brains were harvested, sunk in 30% sucrose overnight and frozen at −80℃. Frozen sections (20 µm thickness) were cryosectioned in the coronal plane.
Measurement of infarct volume
Brain sections were stained with cresyl violet to measure infarct size as previously described.8,28 Coronal sections from six brain regions between 1.5 mm anterior and 1.5 mm posterior to the bregma) were collected at 500 µm intervals throughout the ischemic lesion and stained with cresyl violet. We identified areas of damage depending on microscopic evidence of cell death (loss of normal brain architecture, presence of gliosis, loss of tissue, damaged cell morphology). The infarct areas in each section were measured using an image analysis system (ImageJ) and calculated by subtracting the normal ipsilateral area from that of the contralateral hemisphere to correct for brain edema.
Immunohistochemistry and immunofluorescence
Tissue sections were stained by peroxidase-labeled Griffonia simplicifolia isolectin-B4 (IB4) (10 g/ml; L5391, Sigma) followed by diaminobenzidine (Vector Laboratories) to identify microglia and other myeloid lineage cells. To visualize the lipid-rich foamy macrophages (phagocytes), Oil red O staining (O0625, Sigma) was used according to the instructions of the manufacturer followed by hematoxylin counterstaining. In order to assess TREM2 expression on these immune cells, sections were colabeled for CD11b, to identify microglial/macrophages/monocytes, and TREM2. Sections were reacted with primary antibodies against CD11b (rabbit polyclonal, 1:200; ab75476, Abcam) and TREM2 (rat monoclonal, 1:50; MAB17291, R&D Systems) at 4℃ overnight. Sections were then reacted with Alexa Fluor 488-conjugated goat anti-rabbit IgG (1:400, Life Technologies) and Alexa Fluor-594 conjugated goat anti-rat IgG (1:200, Life Technologies) at room temperature for 1 h. To assess for TREM2 expression on phagocytes, sections were colabeled with the macrophage marker CD68 plus TREM2. Sections were incubated with primary antibodies against CD68 (rabbit polyclonal, 1:1000; MBS175328, My Bio Source) and TREM2 (rat monoclonal, 1:200; MAB17291, R&D Systems) at 4℃ overnight. Sections were then reacted with Alexa Fluor 488-conjugated goat anti-rabbit IgG (1:400, Life Technologies) and Alexa Fluor 594-conjugated goat anti-rat IgG (1:200, Life Technologies) at room temperature for 1 h.
Positive cells were counted under optical microscopy (BX-53 microscope, Olympus) or laser confocal microscopy (LSM510, Zeiss), as previously described.8,21 Briefly, a section 1 mm anterior to the bregma was used to quantify the cell numbers, and positive cells were counted from five random nonoverlapping high-power fields (HPFs) (×400 for optical microscopy, and ×630 for laser confocal microscopy) within the cortex adjacent to the outer boundary of the infarct, as delineated by the hematoxylin stain.
Statistical analysis, RIGOR criteria
All experimental groups were studied in a randomized fashion, and rating and lesion analyses were performed by investigators blinded to experimental conditions. Sample size calculations were performed based on our prior studies with this stroke 8 and BM chimeric 21 models. All statistical analyses were performed using Sigma Stat version 3.1 (Systat software). All data were presented as the mean ± SD. The differences between the two groups were compared using an unpaired t-test, and multiple comparisons were performed using one-way ANOVA followed by Bonferroni’s post hoc test where data were normally distributed. For behavior test analysis, two-way ANOVA was applied. Non-parametric tests of Mann–Whitney or Wilcoxon Rank Sum were applied where data were not normally distributed. P < 0.05 was considered significant.
Results
All animals survived the stroke and sham surgeries. There were no mortalities and no exclusions.
Validation of the BM chimera mouse model
Our prior work using BM chimeras involved the ablation of endogenous marrow by irradiation. 21 In this approach, animals required sedation and head shielding prior to irradiation which led to near complete turnover of endogenous marrow without detection of transplanted marrow cells into the brain or evidence of brain injury that could have been caused by the irradiation. In our current approach, we elected to ablate BM pharmacologically with busulfan, which was shown in prior reports to have fewer complications such as stimulation of the endogenous immune system, and ease of implementation. 20 To validate this approach in our hands, BM from GFP transgenic donor mice was transplanted into Wt host mice (n = 3). Blood samples were collected and subjected to flow cytometry before, 40 days and 80 days after BM transplantation (Figure 1(b)). As expected, flow cytometry showed no GFP positive cells (presented by y-axis) in circulating leukocytes from non-transplanted control mice. Among chimeric mice, 74% of leukocytes were GFP positive by 40 days and ∼95% by 80 days.
These GFP chimeric mice were also subjected to stroke (dMCAO), and brain were collected one day later. Figure 1(c) shows several GFP positive cells within the peri-infarct areas of the brain (arrows), and is consistent with infiltrating leukocytes into areas of infarction. On the contralateral non-ischemic side (contralateral), no GFP positive cells were observed. Further, in GFP chimeric mice that underwent sham surgery, no GFP positive cells could be seen in the intact brain (not shown). These data indicate that our model not only led to near complete turnover of the host’s endogenous BM, but that there was no turnover of any brain cells from the transplanted marrow after 80 days. Thus, this model could be used to differentiate contributions from brain-derived versus marrow-derived immune cells.
Mice with intact TREM2 in brain microglia had more favorable neurological recovery
Sensorimotor tests were carried out prior to stroke and 2, 7 and 14 days later. Mice with intact TREM2 in the brain (Wt to Wt and KO to Wt groups; n = 6/group) performed better on the elevated body swing test (Figure 2(a)) on days 7 and 14, compared with those that lacked TREM2 in the brain (Wt to KO and KO to KO groups). Similarly, these same groups performed better on two different measures from the adhesive removal test (Figure 2(b)). There were no significant differences in neurological recovery when Wt mice were transplanted with TREM2 KO BM (KO to Wt vs. Wt to Wt). TREM2 KO mice did not have better neurological recovery when transplanted with Wt BM (Wt to KO vs. KO to KO). These results indicate that amongst mice with resident brain cells containing intact TREM2 fared better, regardless of the status of TREM2 in their BM, compared to those that lacked TREM2 in the brain. Since we and others have previously shown that TREM2 is expressed exclusively in myeloid cells,8,13,14,16,17 it is reasonable to conclude that the effects of TREM2 occur within the brain through microglia.
Neurological assessments showed worsened deficits in mice that lacked TREM2 in the brain. (a) Elevated body swing test showed significantly worse performance in Wt to KO and KO to KO groups, compared to Wt to Wt and KO to Wt groups 7 and 14 days post stroke as evidenced by greater right sided bias. (b) Adhesive removal test showed worsened sensorimotor function 7 and 14 days in Wt to KO and KO to KO groups compared to Wt to Wt and KO to Wt groups as evidenced by both longer contact and adhesive removal times. (*P < 0.05, **P < 0.01; n = 6/group).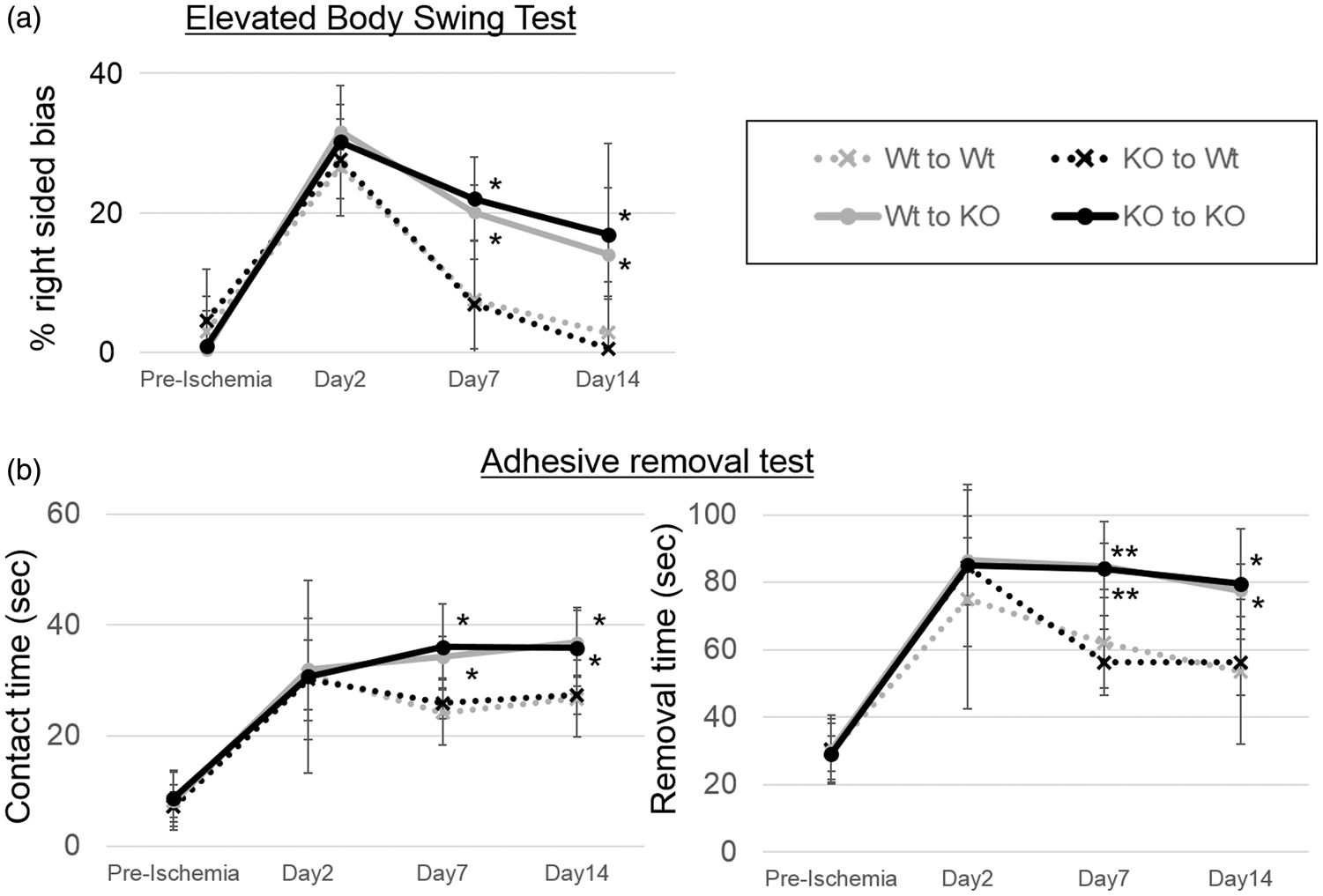
Mice with intact TREM2 in brain microglia had smaller infarcts
Infarct size 14 days after dMCAO from all four BM chimera groups were compared (Figure 3). In line with neurological assessments, groups with intact TREM2 in brain microglia (KO to Wt (12.9 ± 5.8%; n = 6) and Wt to Wt (11.6 ± 1.2%) groups; n = 6) had significantly smaller infarct volumes compared to groups lacking brain TREM2 (KO to KO (24.1 ± 9.3%; n = 6) and Wt to KO (18.1 ± 5.2%; n = 6) groups). Further, replacement of intact TREM2 (Wt to KO) in TREM2 deficient mice (KO to KO) showed a trend towards reduced infarct volume. Also of note, amongst brains of both groups of KO mice, we failed to observe tissue resorption by 14 days, while brain tissue loss was seen in Wt mice (Figure 3(a)). Areas of reactive gliosis were observed within all ischemic brain regions, but this was not significantly affected by the absence of TREM2.
Ischemic lesion size in BM chimera mice. (a) Representative cresyl violet-stained sections 14 days post-dMCAO of each experimental group are shown. Of note, brains from the KO to KO and Wt to KO mice showed areas of pallor within the ischemic territory and lack of tissue resorption. In contrast, brains from the Wt to Wt and KO to Wt brains showed more brain tissue loss compared to KO. Reactive gliosis (darker staining within the ischemic lesions) was observed in all brains, but not significantly affected by TREM2 deficiency. (b) Quantification of infarct sizes are shown. Groups with intact brain TREM2 (KO to Wt (12.9 ± 5.8%) and Wt to Wt (11.6 ± 1.2%) groups) had significantly smaller lesions compared to groups that lacking brain TREM2 (KO to KO (24.1 ± 9.3%) and Wt to KO (18.1 ± 5.2%) groups). (*P < 0.05, **P < 0.01; n = 6/group).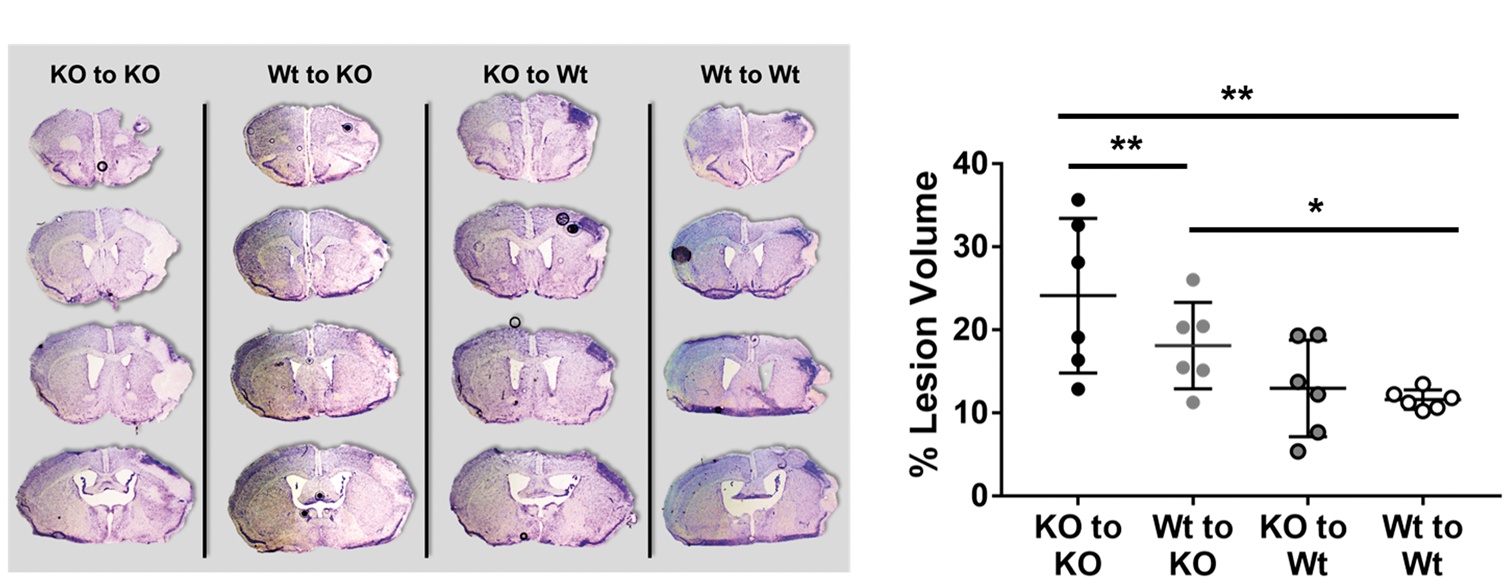
Fewer activated microglia/macrophages in TREM2 KO mice, graded response with BM transplantation
Activated microglia and macrophages were identified by IB4 staining, and were counted at the infarct border 14 days after stroke (Figure 4). The number of activated microglia and macrophages was significantly decreased in descending order: Wt to Wt, KO to Wt, Wt to KO, KO to KO (n = 6/group). In addition, we investigated whether there may be a strong correlation between infarct volume and the number of IB4 positive cells by regression analysis. As shown in Supplemental Figure 1, our model showed a negative correlation between number of activated myeloid cells and infarct volume (r = −0.63, P < 0.001).
Reduced microglia/macrophages in mice lacking brain TREM2. Microglia/macrophages were identified by isolectin-B4 (IB4) staining. Deeper staining with thickened or absent processes and enlarged cell bodies were used to identify activated microglia/microglia. (a) Representative images of IB4 staining of each group are shown. (b) Counts of IB4 positive activated microglia/macrophages were highest amongst Wt to Wt mice (133.3 ± 67.0 cells/HPF) and the lowest in the KO to KO group (17.0 ± 12.4 cells/HPF). The other two experimental groups had intermediate counts, but there were more positive cells in the KO to Wt group (104.0 ± 45.1 cells/HPF) compared to Wt to KO (36.4 ± 17.8 cells/HPF). (*P < 0.05, **P < 0.01; n = 6/group).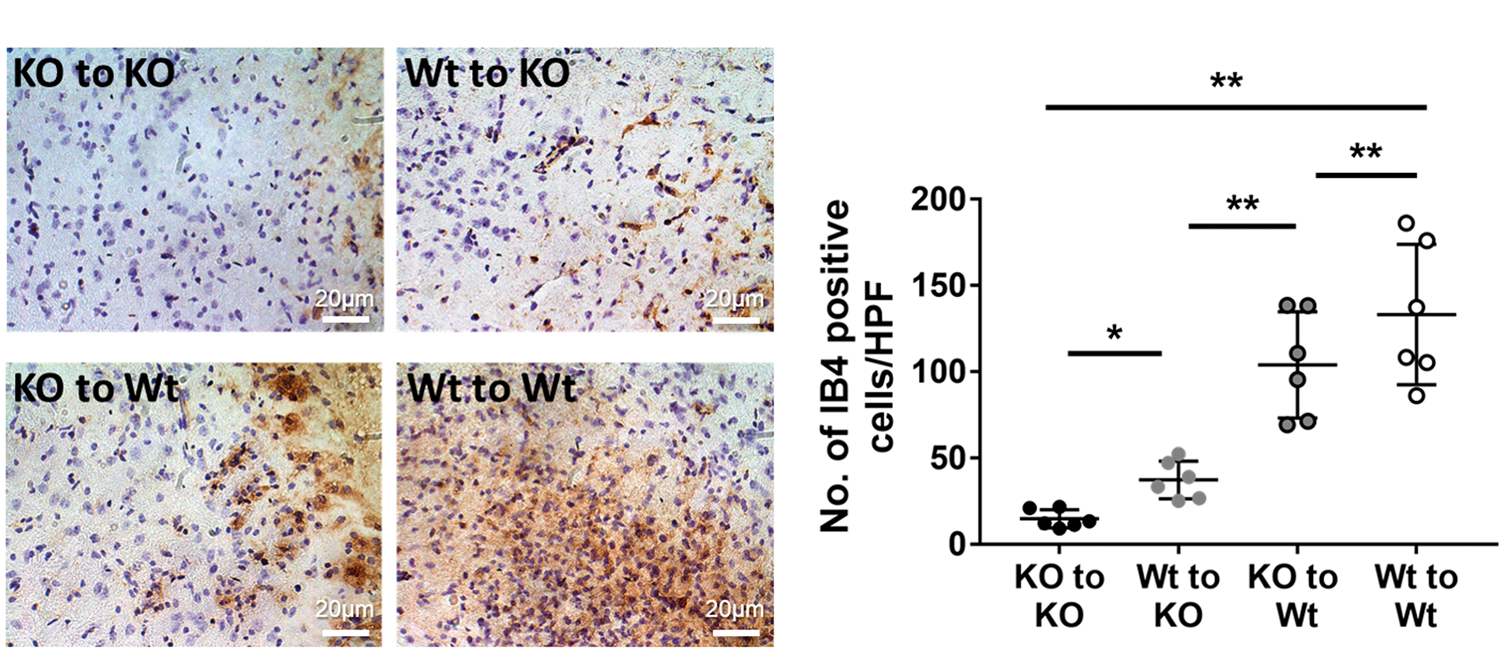
Colabeling with CD11b and TREM2 was performed to identify the expression of TREM2 on microglia/macrophages (Figure 5). TREM2 (red) colocalized exclusively to CD11b positive (green) cells (Figure 5(a)). Total numbers of colabeled cells (Figure 5(b)) followed the same pattern as that seen with IB4 staining. The numbers of TREM2/CD11b colabeled cells were significantly decreased in descending order: Wt to Wt, KO to Wt, Wt to KO, KO to KO (n = 6/ group). These results suggest that intact TREM2 on brain microglia, rather than on circulating microphages, contributed more to the accumulation of these cells to the infarct borderzone. It should also be pointed out that the few TREM2 positive cells observed among KO to KO brains appeared as faint round spots (Figure 5(a), KO to KO row), of which a few appeared to overlay some CD11b positive cells (Merge). While these likely represent artifact from the secondary antibody, they could have alternatively represented very low levels of TREM2. The latter seems unlikely, as our prior work validating the TREM2 KO model indicated total lack of TREM2 protein expression.
8
TREM2 expression amongst microglia/macrophages in BM chimera mice. (a) Brain sections were colabeled with TREM2 (red) and the microglia/macrophage marker CD11b (green). DAPI stain was used to identify nuclei (blue). Immunofluorescence stains show that TREM2 colocalized exclusively to CD11b positive cells, but not all CD11b positive cells were TREM2 positive. (b) Counts of coloabeled cells were significantly decreased in order: Wt to Wt (81.0 ± 31.0 cells/HPF), KO to Wt (66.3 ± 32.2 cells/HPF), Wt to KO (30.3 ± 9.1 cells/HPF), KO to KO (16.8 ± 10.5 cells/HPF). (*P < 0.05, **P < 0.01; n = 6/group).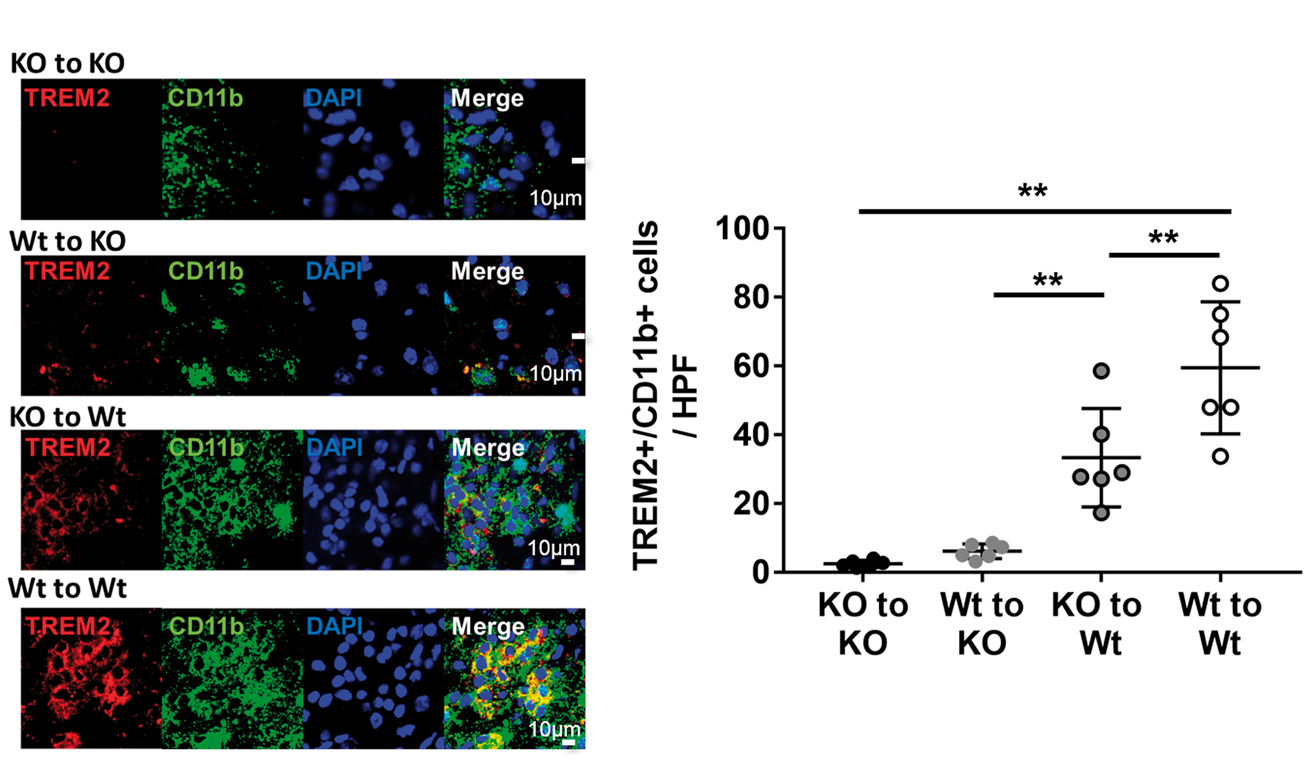
More phagocytosis observed in mice with intact TREM2 in the brain
Oil red O staining was used to detect intracellular lipids as a functional indicator of myeloid cells capable of phagocytosis (Figure 6). Experimental groups with intact TREM2 expression in the brain (KO to Wt and Wt to Wt groups; n = 6/group) demonstrated more oil red positive cells (foamy macrophages phagocytes), compared to mice lacking TREM2 in the brain (Wt to KO and KO to KO groups) (Figure 6(a) and (b)) Because TREM2 deficiency in microglia and/or macrophage is affecting on the total number of myeloid cell accumulation in ischemic brain (Figure 4), the number of oil-red O positive cells in each mice was divided by the number of IB4 positive cells. This average percentage estimates phagocytic capacity (Figure 6(c)). This result further confirmed that phagocytic capacity is significantly higher in mice with intact TREM2 in the brain, compared to mice lacking TREM2 in brain.
TREM2 deficiency in the brain leads to near complete absence of phagocytosis. Oil red staining was used to identify functional phagocytosis in brain sections. (a) Representative images of oil red staining of each group are shown. Near complete absence of oil red positive cells was seen in brains of TREM2 KO mice, compared to Wt, regardless of their marrow status. (b) Counts of oil red foamy macrophages were lowest where the host animal was TREM2 deficient (Wt to KO: 3.2 ± 6.2 cells/HPF); KO to KO:1.0 ± 1.0 cells/HPF) compared to Wt host animals (KO to Wt: 50.5 ± 20.9 cells/HPF; Wt to Wt:93.4 ± 49.4 cells/HPF). (c) To reflect the phagocytic capacity in each mouse, the number of oil red foamy macrophages was divided by the average number of IB4-stained cells. The average percentage was higher in Wt host animals (KO to Wt: 48.7 ± 15.9%; Wt to Wt: 66.2 ± 15.8%), compared to TREM2 deficient (Wt to KO: 6.9 ± 13.0%); KO to KO:5.7 ± 5.5%) (*P < 0.05, **P < 0.01; n = 6/group).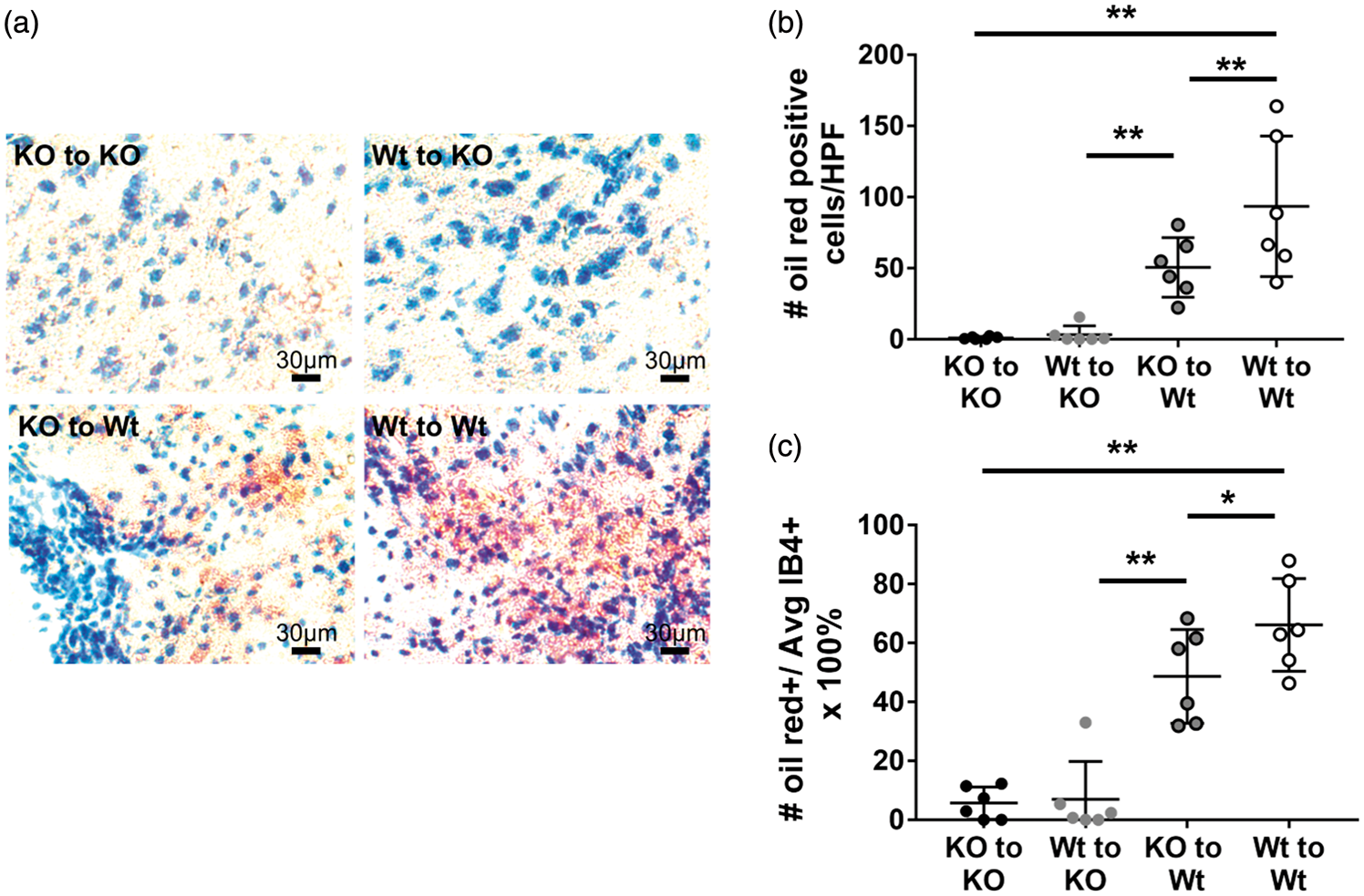
Colabeling with CD68 and TREM2 was performed to identify the expression of TREM2 on phagocytes (Figure 7). In the present study, CD68 was used as marker of phagocytic cells. First, we investigated the percentage of phagocytic CD11b positive cells (CD68+CD11b+) in each mouse (Supplemental Figure. 2). In line with oil red O staining, the result demonstrated that the percentage of phagocytes was significantly decreased in descending order: Wt to Wt, KO to Wt, Wt to KO, KO to KO.
Proportions of TREM2 positive macrophages in BM chimeric mice. Brain sections were stained with TREM2 (red) and the macrophage marker CD68 (green). (a) Representative immunofluorescence images show that all TREM2 positive cells colocalize exclusively to a subset of CD68 positive cells (Merge, yellow). Fewer CD68 positive and fewer colabeled cells were observed where the host animal was TREM2 deficient. Counts of double-labeled cells showed that both the total number of double labeled (b) and proportion of TREM2 positive cells among CD68 positive cells (c) were significantly decreased, in order of Wt to Wt (number:64.3 ± 20.0 cells/HPF; proportion 87.7 ± 10.9%), KO to Wt (number:36.7 ± 18.4 cells/HPF; proportion 56.8 ± 18.8%), Wt to KO (number:12.5 ± 6.3 cells/HPF; proportion 47.7 ± 21.6%), KO to KO group (number:0.7 ± 0.9 cells/HPF; proportion 4.4 ± 5.8%). (*P < 0.05, **P < 0.01; n = 6/group).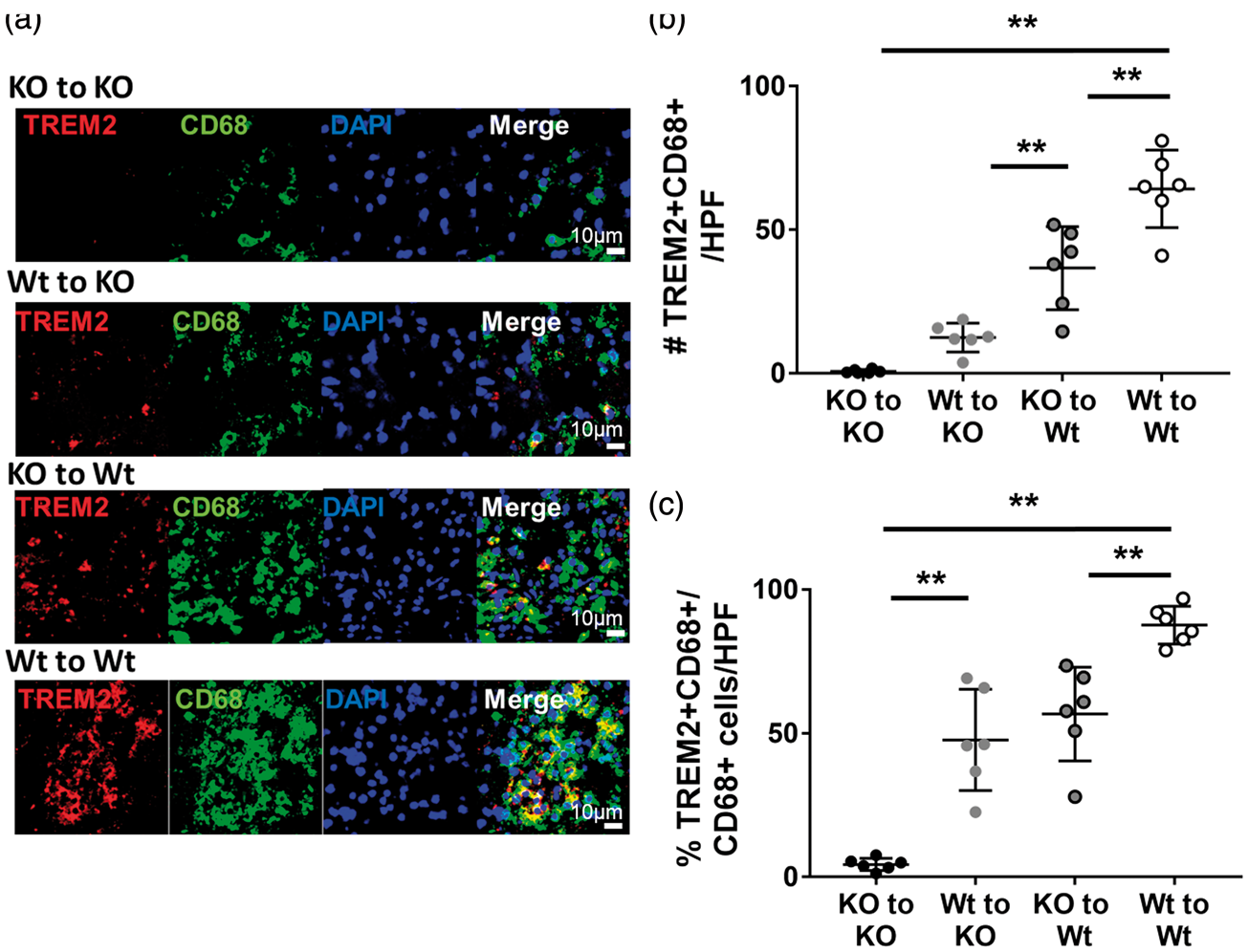
We then analyzed colocalization of TREM2 (red) and CD68 (green). TREM2 positive cells colocalized exclusively to CD68 positive cells (Figure 7(a)). Both the total numbers of double-labeled cells (Figure 7(b)) and the proportion of double labeled (TREM2+ CD68+) cells to all CD68+ cells (Figure 7(c)) were significantly decreased in the order of Wt to Wt, KO to Wt, Wt to KO, and KO to KO groups (n = 6/group). These results indicate that intact TREM2 in the brain contributes more to phagocytosis compared to TREM2 on circulating microphage. That there are significant differences between the KO to Wt and Wt to KO with more oil red and double positive cells amongst the KO to Wt group, suggests that brain resident cells with intact TREM2 contribute more to phagocytosis than those from the circulation.
Discussion
Immune responses accompanying stroke involve both circulating leukocytes and microglia, the brain’s endogenous immune cell. Recent work has demonstrated the complexity of this response, which has proven to be both damaging and beneficial depending on the cell type and the time period after the insult.1,29,30 A beneficial property of this immune response is that phagocytosis and removal of necrotic tissue are essential prior to repair.31,32 One immune receptor involved in this phagocytosis appears to be TREM2. TREM2 is a single-spanning membrane receptor belonging to the immunogloblin-lectin-like superfamily and is expressed exclusively on myeloid cells, including macrophages and microglia.13,14,22,33 It was first described on the surface of circulating macrophages and brain resident microglia, where it bound various pathogens and participated in immune surveillance function. 12 Once activated, TREM2 engages its adapter protein, DAP12 (DNAX-activating protein of 12 kDa) ultimately triggering phagocytosis. 22
To date, work concerning TREM2 in the brain has focused largely on neurodegenerative diseases, especially Alzheimer’s. 34 In humans, TREM2 gene variants have been linked to an increased risk of the development of Alzheimer’s disease, 35 and genetic disruptions of TREM2 or its adapter protein DAP12 have been associated with a rare degenerative brain disease (Nasu-Hakola disease). 36 In models of Alzheimer’s, TREM2 has been shown to promote microglial phagocytosis of amyloid and amyloid-induced proinflammatory reactions.37,38 Overexpression of TREM2 prevented neuron loss, preserved synapses and improved cognitive function, while decreasing expression of proinflammatory cytokines.37,38 Although the precise mechanisms underlying TREM2’s loss of function due to these mutations are unclear, these findings support the notion that TREM2 is necessary for the maintenance of normal brain function.
The role of TREM2 in acute brain insults has been less studied, but we and others showed that TREM2 deficiency worsened outcome in experimental stroke.8,9,11 In a previous paper from our group, we showed TREM2 upregulation in a tMCAO model of mice treated with therapeutic hypothermia.9,39 Furthermore, we demonstrated worsened neurological recovery and larger infarct volumes in TREM2 deficient mice. 8 In this experimental model, TREM2 expression peaked around seven days post stroke. We also observed that phagocytes deficient in TREM2 could not engulf apoptotic/necrotic cells, which led to the impairment of resorption of ischemic tissue. 8 We considered that this inability to clear damaged tissue may have led to increased damage to neighboring tissue through continued elaboration of DAMPs (damage-associated molecular patterns) and/or a crowding effect. Although it seems that TREM2 plays a crucial role in post-stroke recovery, there are clearly questions that remain.
Here, we reproduced our prior findings that TREM2 deficiency worsened outcomes from experimental stroke and impeded phagocytosis. We further demonstrate here through the use of a BM chimera model that this effect is mediated mostly through brain resident microglia, rather than BM-derived circulating macrophages, although nonsignificant differences between the KO to Wt and Wt to Wt suggest that there is a minor contribution from the circulating macrophages. TREM2 is unique to myeloid cells and has been documented in macrophages, monocytes, microglia and dendritic cells.13,40 It is not expressed in other brain cell types such as neurons, oligodendrocytes or astrocytes.11,14,16 TREM2 KO mice transplanted with either Wt or KO BM had worse outcomes than Wt mice, even amongst Wt mice transplanted with KO BM. Thus, it is reasonable to conclude that this effect is a result of TREM2 deficiency in brain resident myeloid cells, of which the majority is microglia.
However, whether TREM2 is beneficial or detrimental to neurological conditions is still somewhat unclear. The results presented here are in line with that previously reported by our group,8,9 as well as a few reports from other groups. Wu et al. 11 demonstrated that genetic knock down of TREM2 exacerbated stroke outcomes, while Zhai et al. 10 showed that TREM2 overexpression was protective. Although it seems that TREM2 confers beneficial effects in experimental ischemic stroke, there is a previous report that showed no effect of TREM2 deficiency in a related stroke model. 41 There are also differing reports of how TREM2 deficiency affects outcome from other experimental neurological conditions. A few reports indicate that its deficiency is actually associated with improved outcome from experimental brain trauma 42 and Alzheimer’s. 14 Reasons for these discrepancies are unclear, but could be explained by differences in the experimental models and approaches. The study by Sieber and colleagues 41 that failed to demonstrate a beneficial effect of TREM2 applied 30 min transient MCA occlusion followed by reperfusion, compared to the present study, which applied permanent distal MCA occlusion. One possibility is that their model, with a shorter duration of ischemia, may not have sufficiently activated the endogenous immune response or, that because they utilized a model of reperfusion, their model led to more infiltration of BM-derived immune cells compared to our model of permanent dMCAO which leads to incomplete reperfusion through collateral vessels. 8 It is possible that immune cells in the brain in our model may have been more microglia predominant, whereas BM-derived macrophages that infiltrated the brain may have predominated in their model. Our present results may be consistent with this speculation, as we observed a greater effect of TREM2 expressed in the brain resident microglia, rather than BM. However, an earlier chimeric study by Schilling et al. 18 similarly showed that phagocytosis in a model of transient MCAO was mediated more by brain resident microglia, rather than in BM-derived macrophages, and supports the notion that it is more likely to be microglia mediating phagocytosis, rather than whether MCAO is transient versus permanent.
Interestingly, others have shown that TREM2 deficiency leads to a potentiation of the immune responses in related disease models.11,43 TREM2 overexpression has similarly been shown to inhibit the production of pro-inflammatory cytokines, but increasing the production of the anti-inflammatory cytokine IL-10. 11 Zhai et al. 10 also reported its beneficial role through the regulation of microglial phenotypes. They found that suppression of TREM2 enhanced microglial pro-inflammatory responses while increasing neuronal apoptosis, whereas upregulation of TREM2 ameliorated these deleterious effects and led to mostly anti-inflammatory responses. In contrast, we found a relative deficiency of myeloid cells among TREM2KO animals in our stroke model both here and in our prior studies.8,9 This cannot be explained by a baseline deficiency in microglia, as counts of microglia in TREM2KO and Wt nonischemic brains are similar (Supplemental Figure 1). To explain this, we speculated that TREM2 has the effect on microglia/macrophage’s mobilization capacity. It is widely known that ischemic 41 stroke induces accumulation of microglia/macrophages toward the peri-ischemic lesion. We considered that TREM2 may also regulate microglial migration capacity, and not only phagocytic function. In support of this, a recent study by Mazaheri et al. 44 showed that loss of TREM2 impaired microglial responses to injury and suppressed signals that normally evoke chemotaxis towards the injury site. Thus, we conclude that, in stroke pathology, fewer accumulated microglia/macrophages in TREM2 deficient animals may be explained by the reduced migration capacity owing to the lack of TREM2 function. Such observations were seen not only in the present study but also several previous papers.8,41 However, to clarify the precise underlying mechanism, further investigation is required. Nevertheless, TREM2 function in stroke pathophysiology seems to be crucial to improving neurological outcome.
One important point of this study relates to whether TREM2 induction or replacement may be a viable therapeutic option for treatment of ischemic stroke. We demonstrated better neurological recovery and reduced infarct volume in mice with intact brain TREM2 on microglia, compared to mice lacking brain TREM2, regardless of TREM2 expression on circulating macrophages. This observation implies that novel therapy should be developed and designed to target ‘brain resident microglia’ rather than circulating macrophages.
We should also point out that we do not know the consequences of life long TREM2 deficiency in this mouse model. In a related condition, Nasu Hakola disease, patients develop an early dementia along with bony deformities. In our mouse model, TREM2 KO mice did not have any gross abnormalities in brain anatomy, even out to two years of age and failed to show differences on cognitive assays (unpublished preliminary observations). However, more detailed studies in this mouse model may be needed, as well as the study of conditional knockouts or activation/downregulation of TREM2 in genetically intact adult mice. Second, TREM2’s ligand is not well known, and could impede the development of therapeutic strategies in patients. This has been an issue that has plagued the field in general, but a few candidate molecules have been studied. Although anionic moieties on bacteria and yeast 12 are known ligands of TREM2 in the circulation, the endogenous ligands in brain are still unclear. The 60 kDa heat shock protein (Hsp60) has been previously shown to ligate TREM210,45; however, we were unable to show that HSP60 could activate TREM2 an in vitro model (data not shown), and HSP60 was not colocalized to TREM2 positive cells in our stroke model. 9 Our prior study did show that ischemic and apoptotic neurons express and/or elaborate nucleic acids/nucleic acid fragments which bind TREM2, and leads to its activation in our stroke models, 8 and another group has shown that anionic and zwitterionic lipids can also activate TREM2. 46 Thus, it appears that like other innate immune receptors, TREM2 can be activated by typical danger-associated molecular patterns (DAMPs). This should not be particularly surprising, considering its role in innate immunity, but further investigation in this area is needed.
A limitation of the present study is that we were not able to completely identify the role and function of brain resident microglia and circulating macrophages individually. We demonstrated better recovery from stroke and more pronounced phagocytotic activity, when TREM2 is present in brain microglia compared to that of circulating macrophages. This indirectly indicates that TREM2 expression on brain microglia is more important than that of BM macrophage in stroke recovery. However, it is still unclear what the functional differences of these cells are, and how these differences affect post-stroke recovery. In order to clarify this question, the use of chimera models where TREM2 deficient cells could be tagged would be helpful, and should be addressed in future research.
In conclusion, we demonstrated that TREM2 expression on brain microglia and circulating macrophages led to better neurological recovery. The better neurological recovery and beneficial effect were predominantly conferred when TREM2 was present in brain microglia. We also show that not all microglia express TREM2. These observations would suggest an opportunity for clinical translation, whereby TREM2 expression should be targeted specifically to the brain. These novel findings might be useful in developing new therapeutic strategies for related neurological conditions as well.
Supplemental Material
Supplemental Figure1 - Supplemental material for Triggering receptor expressed on myeloid cells-2 expression in the brain is required for maximal phagocytic activity and improved neurological outcomes following experimental stroke
Supplemental material, Supplemental Figure1 for Triggering receptor expressed on myeloid cells-2 expression in the brain is required for maximal phagocytic activity and improved neurological outcomes following experimental stroke by Kota Kurisu, Zhen Zheng, Jong Youl Kim, Jian Shi, Atsushi Kanoke, Jialing Liu, Christine L Hsieh and Midori A Yenari in Journal of Cerebral Blood Flow & Metabolism
Supplemental Material
Supplemental Figure2 - Supplemental material for Triggering receptor expressed on myeloid cells-2 expression in the brain is required for maximal phagocytic activity and improved neurological outcomes following experimental stroke
Supplemental material, Supplemental Figure2 for Triggering receptor expressed on myeloid cells-2 expression in the brain is required for maximal phagocytic activity and improved neurological outcomes following experimental stroke by Kota Kurisu, Zhen Zheng, Jong Youl Kim, Jian Shi, Atsushi Kanoke, Jialing Liu, Christine L Hsieh and Midori A Yenari in Journal of Cerebral Blood Flow & Metabolism
Supplemental Material
Supplemental Figure3 - Supplemental material for Triggering receptor expressed on myeloid cells-2 expression in the brain is required for maximal phagocytic activity and improved neurological outcomes following experimental stroke
Supplemental material, Supplemental Figure3 for Triggering receptor expressed on myeloid cells-2 expression in the brain is required for maximal phagocytic activity and improved neurological outcomes following experimental stroke by Kota Kurisu, Zhen Zheng, Jong Youl Kim, Jian Shi, Atsushi Kanoke, Jialing Liu, Christine L Hsieh and Midori A Yenari in Journal of Cerebral Blood Flow & Metabolism
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the NIH National Institute of Neurological Disorders and Stroke R01 NS40516 and Veteran’s Merit Awards I01 BX000589 (to MY), I01BX002690 (to CLH) and I01BX003335 and NIH ROI NS102886 to JL, and the Uehara Foundation (2016 Postdoctoral Fellowship, to KK). The grants to MY CLH, JL were administered by the Northern California Institute for Research and Education, and supported by resource of the Veterans Affairs Medical Center, San Francisco, California.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
KK: Acquisition of data, analysis and interpretation of data, and drafting the manuscript; ZZ, JYK, AK, JS: Acquisition of data; CH, JL: Analysis and interpretation of data, editing and approving the final manuscript; MAY: Conception and design, analysis and interpretation of data and editing and approving the final manuscript.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.