
Abstract
Administration of lactate during hypoglycemia suppresses symptoms and counterregulatory responses, as seen in patients with type 1 diabetes and impaired awareness of hypoglycemia (IAH), presumably because lactate can substitute for glucose as a brain fuel. Here, we examined whether lactate administration, in a dose sufficient to impair awareness of hypoglycemia, affects brain lactate levels in patients with normal awareness of hypoglycemia (NAH). Patients with NAH (n = 6) underwent two euglycemic-hypoglycemic clamps (2.8 mmol/L), once with sodium lactate infusion (NAH w|lac) and once with saline infusion (NAH w|placebo). Results were compared to those obtained during lactate administration in patients with IAH (n = 7) (IAH w|lac). Brain lactate levels were determined continuously with J-difference editing 1H-MRS. During lactate infusion, symptom and adrenaline responses to hypoglycemia were considerably suppressed in NAH. Infusion of lactate increased brain lactate levels modestly, but comparably, in both groups (mean increase in NAH w|lac: 0.12 ± 0.05 µmol/g and in IAH w|lac: 0.06 ± 0.04 µmol/g). The modest increase in brain lactate may suggest that the excess of lactate is immediately metabolized by the brain, which in turn may explain the suppressive effects of lactate on awareness of hypoglycemia observed in patients with NAH.
Introduction
Hypoglycemia is the most frequent side-effect of insulin therapy in patients with type 1 diabetes. When blood glucose levels drop, this normally initiates a hierarchically organized counterregulatory response, which includes release of counterregulatory hormones and the appearance of hypoglycemic symptoms. It has been demonstrated that the administration of lactate considerably diminishes symptomatic and hormonal responses to hypoglycemia and simultaneously mitigates cognitive dysfunction during hypoglycemia.1–3 These effects are suggested to be the result of lactate acting as an alternative fuel for the brain, when glucose supply is low. 2 Clinically, the administration of lactate mimics the situation seen in patients with type 1 diabetes and impaired awareness of hypoglycemia (IAH). In IAH, the threshold for the onset of hypoglycemic symptoms and counterregulatory hormone responses is shifted to lower glucose values, which substantially increases the risk for severe hypoglycemia. 4
Most studies that investigated the effects of lactate administration during hypoglycemia have focused on clinical effects (i.e. symptoms, hormonal responses and cognitive function), and did not examine physiological changes in the brain. Whether elevated plasma lactate levels lead to an accumulation of lactate in the brain is subject of debate. In a 13C-MR study, performed under euglycemic conditions in non-diabetic subjects, it was calculated that brain lactate levels and lactate oxidation increase almost linearly with the increase in plasma lactate levels. 5 In contrast, other studies have reported direct oxidation of lactate after its transport across the blood–brain barrier, with limited accumulation in the brain.6,7 Thus, lactate oxidation may even spare glucose as a cerebral fuel under euglycemic conditions. 8
The only study that has investigated the effect of exogenous lactate on human brain lactate concentrations during hypoglycemia, calculated increased brain lactate concentrations without increased oxidation, in patients with type 1 diabetes compared to healthy volunteers. 9 Such elevated brain lactate levels are in line with studies that demonstrated an enhanced capacity to transport lactate into the brain in patients with diabetes and IAH.10,11 The lack of a euglycemic control and a placebo arm (i.e. no lactate infusion) precluded conclusions about lactate accumulation during euglycemia versus hypoglycemia, or about the effect of lactate infusion on awareness of hypoglycemia.
In the current study, we aimed to show that infusion of lactate, in a dose sufficient to impair awareness of hypoglycemia in patients with type 1 diabetes and normal awareness of hypoglycemia (NAH), increases brain lactate levels. Secondly, we aimed to compare the effect of lactate administration on brain lactate levels during euglycemia and hypoglycemia between patients with NAH and those with IAH.
Material and methods
Participants
We enrolled seven patients with type 1 diabetes and NAH and seven patients with type 1 diabetes and IAH. The classification of awareness state was based on the Dutch modified version of the Cox questionnaire.12,13 Patients with type 1 diabetes were eligible if they were younger than 50 years, had an HbA1c below 9.0% (75 mmol/mol) and were free from microvascular complications, except for background retinopathy. Exclusion criteria included contraindications to the MRI examination, a history of brain injury, epilepsy, liver or cardiovascular diseases, anxiety disorders, and the use of drugs other than insulin interfering with glucose metabolism. One participant with NAH moved too much during data acquisition, which hampered MR data quality and was consequently excluded from data analysis. The study was approved by and performed in accordance with the ethical standards of the institutional review board of the Radboud university medical center (Commissie Mensgebonden Onderzoek Arnhem-Nijmegen) and with the Helsinki declaration of 1975/1983. All participants gave written informed consent.
Study protocol
Patients with NAH underwent two hyperinsulinemic euglycemic-hypoglycemic clamp studies, once with sodium lactate infusion (NAH w|lac) and once with saline infusion (NAH w|placebo), as described below. Experiments were carried out in a single-blind fashion (i.e. participants were blinded for the infusions) and in randomized order, scheduled at least two weeks apart. In female participants, both experiments were performed during equal phases of the menstrual cycle. Patient with IAH participated only in the sodium lactate infusion study arm (IAH w|lac).
Participants came to the research facility in the morning after an overnight fast, having abstained from smoking, alcohol and caffeine containing substances for 24 h prior to the experiment, and from strenuous exercise for at least two days before the experiment. In addition, participants were instructed to prevent hypoglycemia and to check their blood glucose levels regularly in the 24 h before the experiment.
The brachial artery of the non-dominant arm was cannulated under local anesthesia (Xylocaine 2%) for frequent blood sampling. Blood was sampled every 5 min for the determination of plasma glucose and plasma lactate levels (Biosen C-line; EKF Diagnostics). An intravenous catheter was inserted in the antecubital vein of the contralateral arm for infusion of glucose 20% (Baxter B.V., Deerfield, IL), insulin (insulin aspart; Novo Nordisk, Bagsvaerd, Denmark) and sodium lactate (600 mmol/L; Spruyt Hillen, Ijsselstijn, The Netherlands and prepared by the Department of Pharmacy, Radboud university medical center, Nijmegen, The Netherlands) or normal saline (sodium chloride; 0.9%).
After cannulations, a two-step hyperinsulinemic (60 mU/m2/min) euglycemic (5.0 mmol/L)-hypoglycemic (2.8 mmol/L) glucose clamp was initiated, and participants were placed in the MR scanner for baseline MR spectroscopy (MRS) data acquisition. At 15 min after the start of the euglycemic clamp, infusion of sodium lactate, or an equivalent volume of saline, was started, while MRS data acquisition continued. We aimed for plasma lactate levels of approximately 3.5 mmol/L and initially used a primed (50 µmol/kg/min for 15 min) continuous (30 µmol/kg/min) infusion of sodium lactate. Since achieved plasma lactate levels were slightly above target levels, the dose was lowered after the first two participants (both patients with NAH) to 40 µmol/kg/min for 15 min and 25 µmol/kg/min for the remainder of the experiment. After ∼25 min, blood glucose levels were allowed to drop to 2.8 mmol/L and were maintained at that level for 45 min. Hypoglycemic symptoms were quantified with a validated questionnaire just prior to positioning the participant in the MR scanner and at the end of the stable hypoglycemic phase. Participants were asked to score from 0 to 6 (none to most severe) for each of 18 symptoms which included six autonomic symptoms, six neuroglycopenic symptoms, four general and two dummy symptoms. Additional blood samples were taken for determination of plasma insulin, pH, catecholamines, cortisol and growth hormone prior to the start of the sodium lactate or saline infusion (baseline), at the end of the euglycemic phase and at the end of the hypoglycemic phase. During hypoglycemia, counterregulatory hormone responses were determined at 15-min intervals (see Figure 1(a) for a schematic overview of the study protocol). After completing both experimental days, patients with NAH were asked on which day the hypoglycemic phase was most evident or felt more intense.
Study protocol, plasma glucose and plasma lactate levels. (a) Schematic overview of the study protocol. MRS measurements were performed continuously during euglycemia and hypoglycemia. Counterregulatory hormone responses (CH) and hypoglycemic symptoms (Symp) were determined at several time points during euglycemia and hypoglycemia, as indicated. (b) Time courses of plasma glucose and (c) plasma lactate levels. The dashed lines represent the start of the lactate or saline infusion (T = 0 min), the end of euglycemia, and the start of hypoglycemia. Open circles: patients with type 1 diabetes and NAH with saline infusion (NAH w|placebo); black circles: patients with type 1 diabetes and NAH with lactate infusion (NAH w|lac); black triangles: patients with type 1 diabetes and IAH with lactate infusion (IAH w|lac).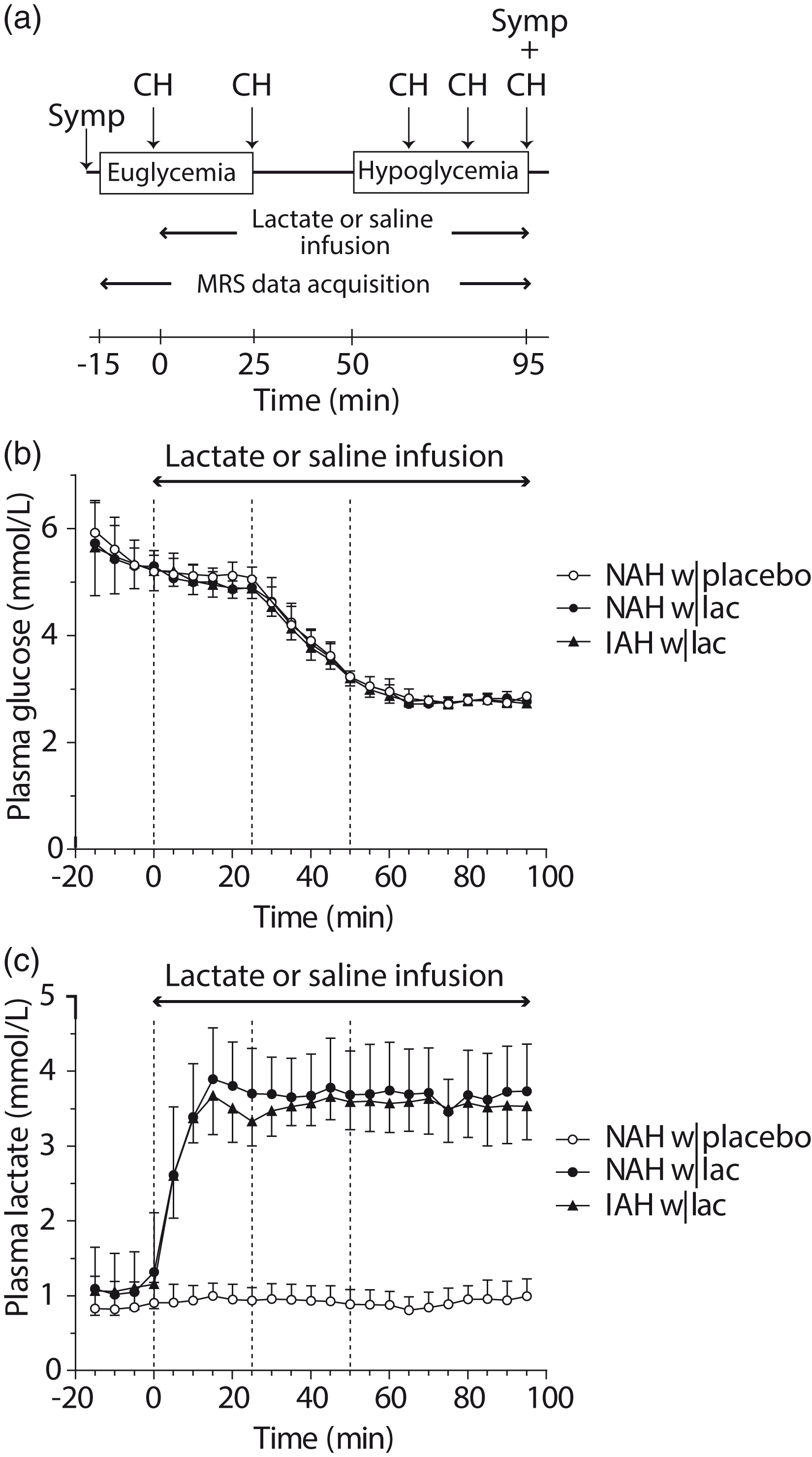
MRS protocol and data analysis
To measure brain lactate, we performed MR spectroscopy (MRS) on a 3 T MR system (MAGNETOM Prisma, Siemens, Erlangen) using a body coil for excitation and a 12-channel receive-only head coil. A T1-weighted anatomical image (MPRAGE, echo time (TE) 2.4 ms, repetition time (TR) 1900 ms, and a field of view of 256 × 256 mm2) was acquired, and a 22.5 cm3 voxel for MRS data acquisition was placed in the periventricular and supraventricular region (Figure 2(a)). Brain lactate levels were then determined continuously with an interleaved J-difference editing semi-LASER
14
spectroscopy sequence (TE 144 ms, TR 3000 ms and 8 averages). J-difference editing was performed with frequency selective inversion MEGA pulses
15
(30 ms, 75 Hz bandwidth). Unsuppressed water spectra (TE 33 ms, TR 5000 ms and 8 averages) were acquired from the same voxel for signal quantification.
Example of difference spectra. (a) Typical location of the MRS voxel on a T1-weighted anatomical image. (b) Representative examples of difference spectra of one subject NAH (NAH w|placebo and NAH w|lac) and one subject with IAH (IAH w|lac). The baseline difference spectra were recorded before the start of lactate or placebo infusion. Lac: lactate; MM: macromolecules.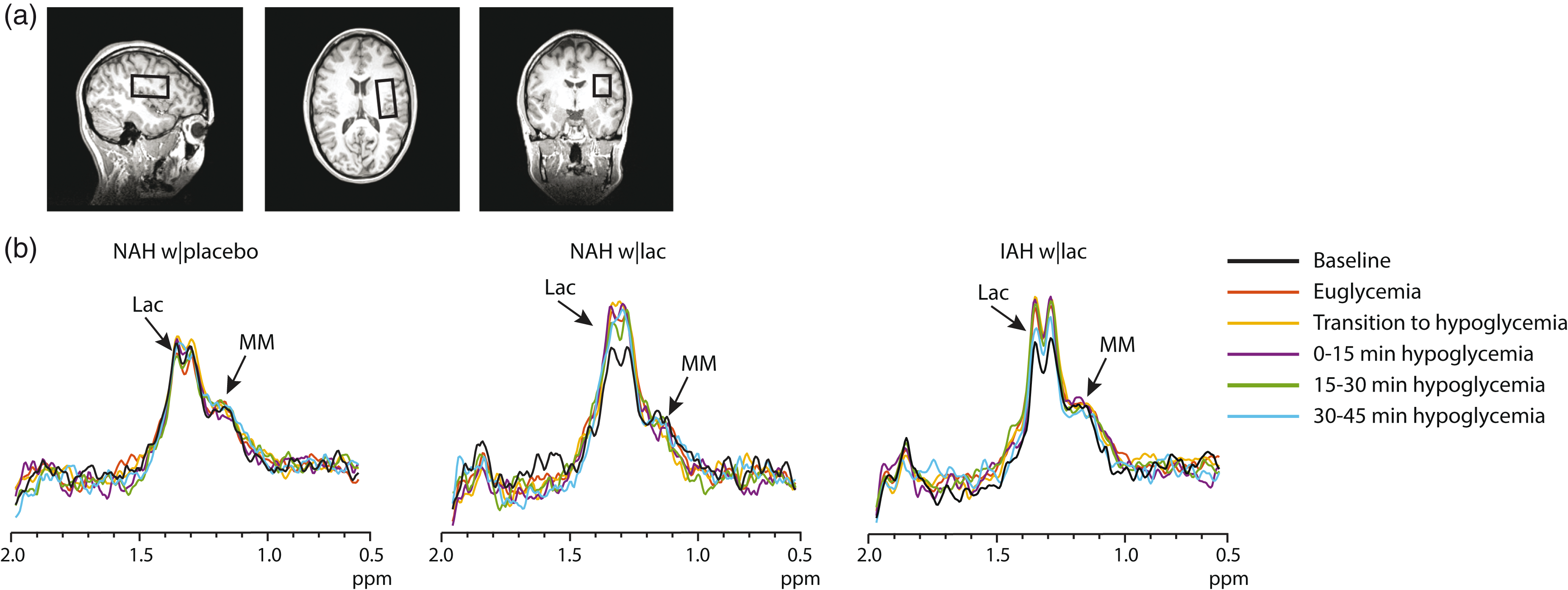
Additionally, at the end of euglycemia and at the end of hypoglycemia, we acquired MR spectra without J-difference editing from the same voxel (sLASER, TE 33 ms, TR 3000 ms, 32 averages) to explore differences in other major brain metabolites.
The J-difference edited spectra were zerofilled from 1024 to 2048 data points and phase and frequency aligned. 16 Motion-corrupted spectra, identified as a spectrum which deviated more than 2.6 standard deviations from the average spectrum of that participant, 17 were discarded. Subsequently, spectra were subtracted pairwise. The difference spectra were averaged within participants across six different phases: baseline (i.e. before the start of lactate or placebo infusion), euglycemia, transition to hypoglycemia, and each 15-min block of hypoglycemia. From these final difference spectra, the cerebral lactate doublet at 1.3 ppm was quantified using the AMARES algorithm in jMRUI. 18 Visual inspection of the difference spectra showed co-edited macromolecules at ∼1.2 ppm, which were included in the fitting routine in AMARES to avoid overestimation of the lactate signal. 19 The unsuppressed water signal was used as an internal standard for absolute quantification and we took the voxel composition and contribution of plasma lactate into account. 20 We assumed a T2 of 240 ms for cerebral lactate, 21 and a T2 of 80 ms and 110 ms for water in gray and white matter, respectively. 22 The voxel composition (i.e. percentages white matter, grey matter and cerebrospinal fluid) was determined by segmenting the T1-weighted anatomical images using SPM8 (Wellcome Trust Centre for Neuroimaging, UCL, London, UK) and subsequently co-registering the segmented image with the MRS voxel location. As the voxel mainly contained white matter, we assumed a vessel volume of 2%.23,24
Spectra acquired without J-difference editing were analyzed using the LCModel software. 25 A basis set was created in Bruker TopSpin, and extended with a measured macromolecular baseline. Metabolites quantified with Cramér-Rao lower bounds >50% were not included in the analysis (as recommended by the LCModel manual 26 ). The unsuppressed water signal was used as a reference for absolute quantification of aspartate (Asp), total choline (tCho), total creatine (tCre), glutamate (Glu), glutamine (Gln), myo-inositol (mI), total N-acetylaspartate (tNAA), scyllo-inositol (scyllo) and taurine (Tau).
Analytical methods
Plasma insulin was determined by an in-house radioimmunoassay (RIA). 27 Plasma adrenaline and noradrenaline were analyzed by high-performance liquid chromatography combined with fluorometric detection. 28 Plasma growth hormone and cortisol were determined using a routine analysis method with an electrochemiluminescent immunoassay on a Modular Analytics E170 (Roche Diagnostics GmbH, Mannheim, Germany). pH was measured by routine arterial blood gas analysis on the RapidPoint 500 (Siemens Nederland B.V., Den Haag, the Netherlands).
Statistical analysis
Differences in means or medians within the NAH group (NAH w|lac vs. NAH w|placebo) and differences within groups between euglycemia and hypoglycemia were statistically tested with paired Student t tests or Wilcoxon signed rank tests when data were not normally distributed. Differences between NAH w|lac and IAH w|lac were compared with two-sided Student t tests or Mann–Whitney U tests, when appropriate. Serial data were compared between NAH w|lac and NAH w|placebo with a two-way repeated measure ANOVA. To analyze effects over time in patients with IAH, a one-way repeated measure ANOVA was performed. Since patients with IAH were only studied with lactate infusion, comparison to a placebo condition is not possible. Thus, to determine whether there was an effect of lactate infusion on brain lactate levels in patients with IAH, the area under the curve (AUC) of the baseline-corrected brain lactate levels was calculated by trapezoidal numerical integration, and a one-sample t test was performed. Differences in means at baseline across the three groups were analyzed by one-way ANOVA followed by pairwise Bonferroni post hoc tests. All data are expressed as mean ± SD unless otherwise indicated. Significance levels of Student t tests were not corrected for multiple testing. A P value <0.05 was considered statistically significant. Statistical analyses were performed with IBM SPSS Statistics 20.
Results
Participant characteristics.
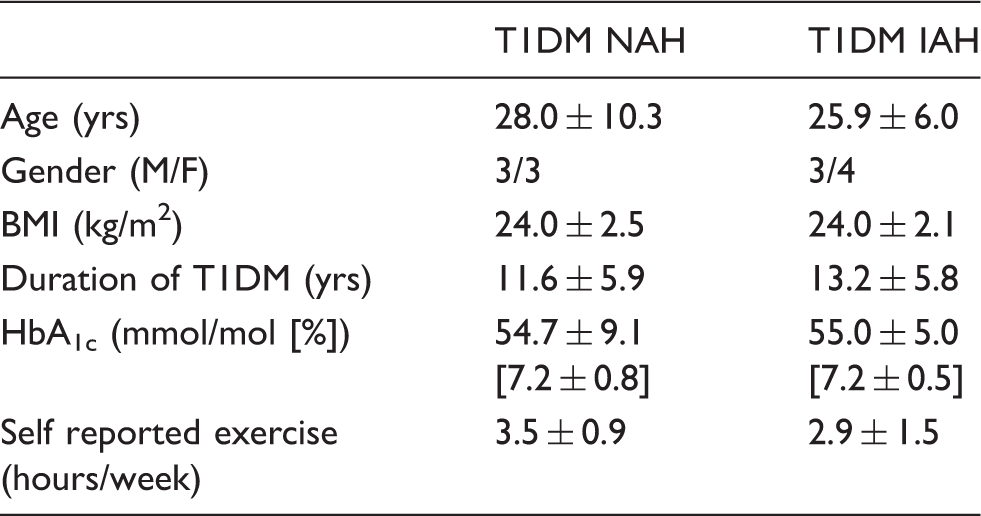
Note: Data are presented as number or mean ± SD.
M: male; F: female; BMI: body mass index; T1DM NAH: type 1 diabetes with normal awareness of hypoglycemia; T1DM IAH: type 1 diabetes with impaired awareness of hypoglycemia.
Hypoglycemic symptoms and counterregulatory hormones
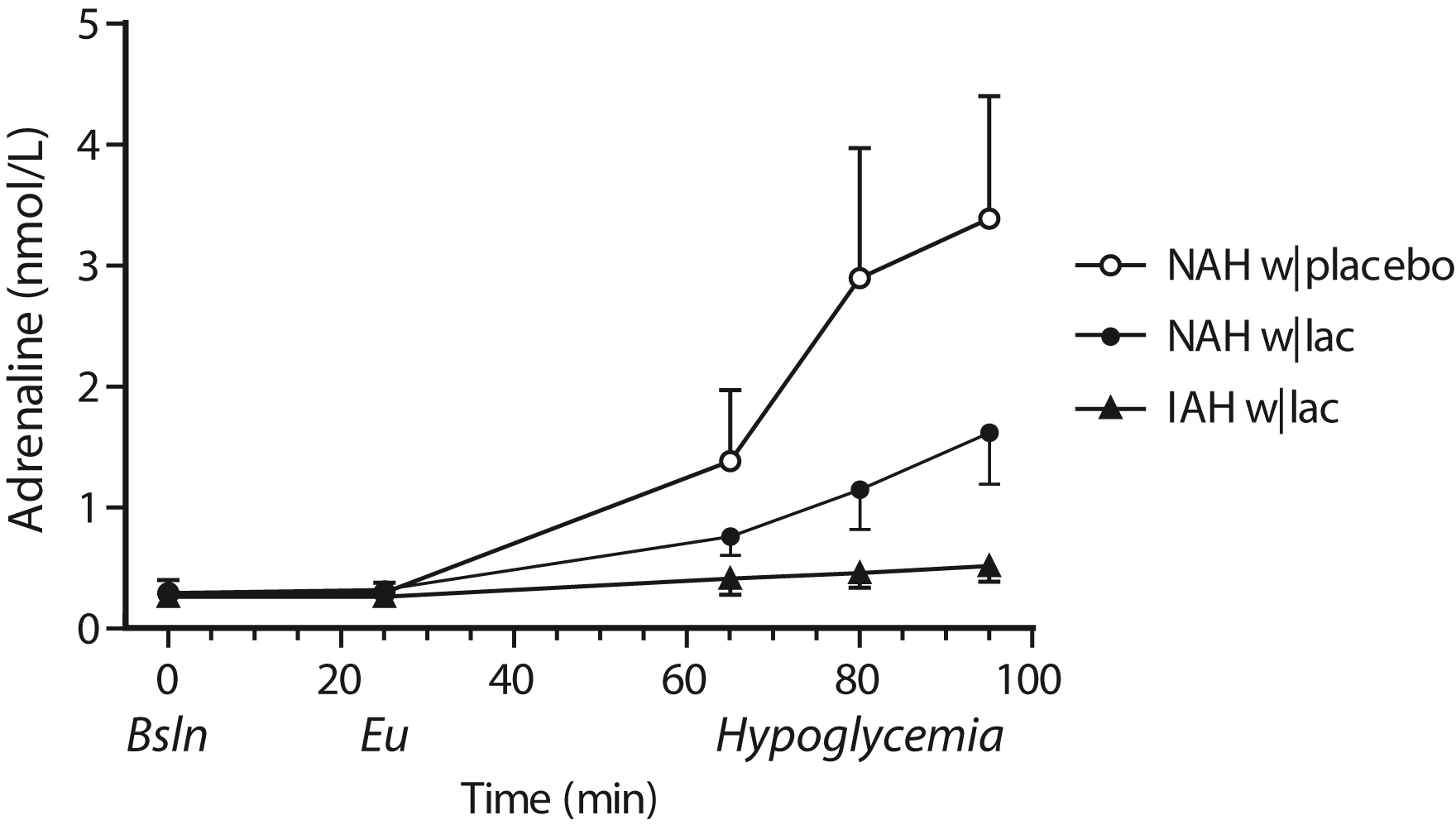
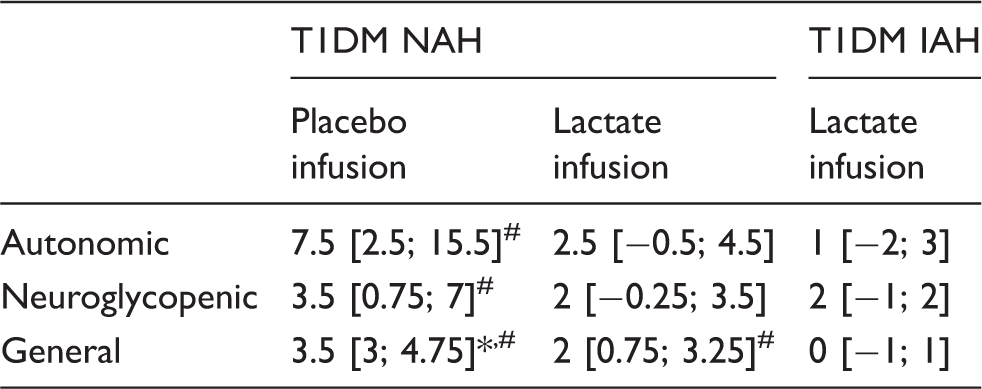
Lactate infusion clearly suppressed total symptom scores during hypoglycemia in patients with NAH, as compared to placebo (p = 0.03, Figure 3), and resembled symptom score of patients with IAH when lactate was infused (p = 0.18 between groups). Symptom subcategories are depicted in Table 2. Five out of six patients with NAH indicated that hypoglycemia was less severe on the day of the lactate infusion, one patient experienced no difference between both study days. In patients with IAH, neither the total nor the subcategory symptom score changed significantly in response to hypoglycemia. Lactate infusion considerably suppressed the adrenaline response to hypoglycemia in patients with NAH as compared to placebo infusion (p < 0.01; Figure 4). Lactate infusion also suppressed cortisol and hGH responses to hypoglycemia in patients with NAH (both p < 0.01), but did not significantly reduce noradrenaline responses (p = 0.18) (Supplementary Figure 2).
Difference in symptom scores. Total symptoms scores in response to hypoglycemia in patients with type 1 diabetes and NAH with saline infusion (NAH w|placebo), patients with NAH with lactate infusion (NAH w|lac) and patients with IAH with lactate infusion (IAH w|lac). *p < 0.05 **p < 0.01. Adrenaline responses. Adrenaline levels were determined at baseline (Bsln; i.e. before the infusion of lactate or saline), during euglycemia (Eu) and at 15-min intervals during hypoglycemia. Open circles: patients with type 1 diabetes and NAH with saline infusion (NAH w|placebo); black circles: patients with type 1 diabetes and NAH with lactate infusion (NAH w|lac); black triangles: patients with type 1 diabetes and IAH with lactate infusion (IAH w|lac). Hypoglycemia-induced changes in symptom subscores (median [interquartile range]). p < 0.05 vs. NAH w|lacp < 0.05 vs. euglycemia. T1DM NAH: type 1 diabetes with normal awareness of hypoglycemia; T1DM IAH: type 1 diabetes with impaired awareness of hypoglycemia.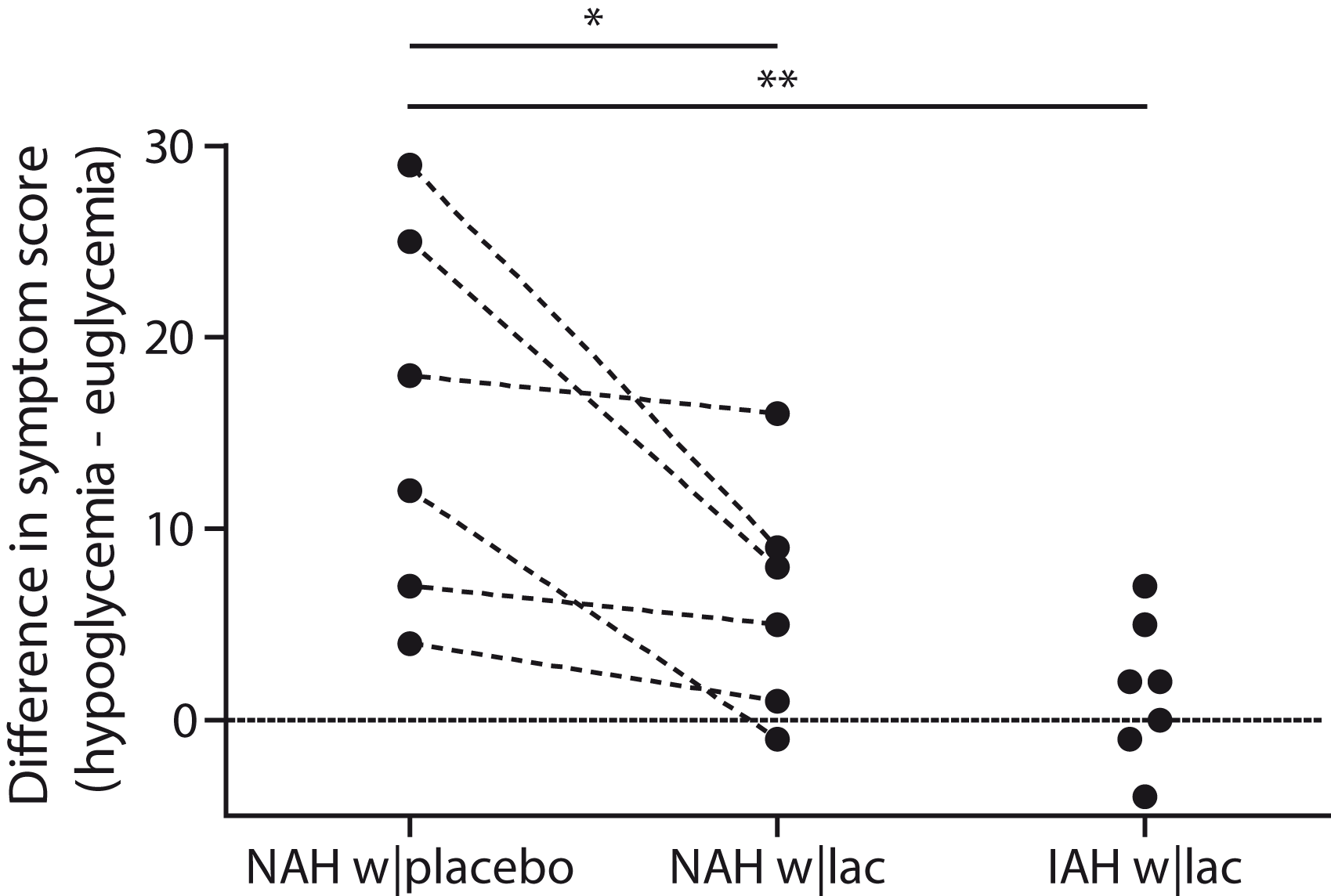
Brain lactate
The tissue content of the MRS voxels was not different between groups and contained on average 64.1 ± 11.1% white matter, 32.8 ± 10.5% grey matter and 3.2 ± 1.6% cerebrospinal fluid. Baseline brain lactate levels (i.e. before the infusion of lactate or placebo) were similar across groups (0.58 ± 0.08, 0.64 ± 0.1 and 0.59 ± 0.05 µmol/g in NAH w|lac, NAH w|placebo and IAH w|lac, respectively; p = 0.42). J-edited difference MR spectra, recorded during infusion of lactate, showed an increase of the lactate signal in the brain (Figure 2(b)). In patients with NAH, brain lactate levels were higher after infusion of lactate than after infusing saline (Figure 5(a); mean difference: 0.12 ± 0.05 µmol/g, equivalent to an increase of 20.7 ± 6.6%; p = 0.048). There was no significant interaction effect (time x infusion) or main effect of time. Although brain lactate levels in NAH w|lac tended to be elevated compared to placebo values at each time point, post hoc analysis revealed that the difference in brain lactate levels reached significance during the final 30 min of the experiment.
Brain lactate levels. Mean (±SEM) baseline corrected and percentage change in brain lactate levels in (a) patients with type 1 diabetes and NAH (NAH w|placebo and NAH w|lac) and (b) patients with type 1 diabetes and IAH (IAH w|lac). The dashed line represents the start of hypoglycemia. P values indicate significance of time-series analysis.
In patients with IAH, lactate infusion resulted in an increase in brain lactate levels compared to baseline values (Figure 5(b); mean increase: 0.06 ± 0.04 µmol/g, equivalent to an increase of 10.2 ± 4.2%; AUC of baseline-corrected brain lactate levels: 4.4 ± 4.3 µmol·min/g; p = 0.03). In these patients, brain lactate levels showed a trend towards an increase at each time point compared to baseline values, but the trend was stronger at the beginning of lactate infusion and disappeared at the end of hypoglycemia. When comparing brain lactate time curves with lactate infusion between patients with IAH and NAH, the increase in brain lactate levels in IAH w|lac seemed less pronounced compared to NAH w|lac, but this difference was not statistically significant (p = 0.36). Brain lactate levels did not change in patients with NAH in response to hypoglycemia when saline was infused. Notably, there was no correlation, whatsoever, between brain lactate levels and plasma lactate levels (R2 = 0.01, p = 0.27).
Other brain metabolites
Brain glutamate levels decreased in response to hypoglycemia in patients with NAH, both during lactate infusion (from 7.5 ± 0.7 to 7.1 ± 1.0 µmol/g, p = 0.03) and while infusing placebo (from 7.6 ± 0.6 to 7.0 ± 0.8 µmol/g, p = 0.01). In contrast, hypoglycemia did not change brain glutamate levels in patients with IAH (8.0 ± 0.7 versus 7.8 ± 0.9 µmol/g, p = 0.59). Additionally, we found an increase in myo-inositol in IAH w|lac and a slight decrease in myo-inositol in NAH w|lac. None of the other major brain metabolites (Asp, tCho, tCre, Gln, tNAA, scyollo and Tau) was significantly altered by the hypoglycemic condition in any of the groups (Supplementary Figure 3).
Discussion
The results of the present study confirm that lactate infusion considerably suppresses counterregulatory hormone responses to and symptomatic awareness of hypoglycemia in patients with NAH. While lactate infusion did increase brain lactate content, the increase was modest and not clearly different between patients with NAH and IAH. The lack of pronounced brain lactate accumulation suggests that the excess of lactate is immediately oxidized by the brain which may explain the suppressive effects of lactate on awareness of hypoglycemia in patients with NAH.
Our results are in line with those of several other studies reporting that elevated plasma lactate levels result in an increase in cerebral lactate transport and oxidation, preventing substantial accumulation of brain lactate upon infusion.6–8 However, an earlier study reported fivefold higher brain lactate concentrations in the brain of patients with type 1 diabetes who were exposed to frequent hypoglycemia, as compared to healthy controls, when 13C-labelled lactate was administered during hypoglycemia. 9 Another 13C study that used the similar methodology in healthy volunteers, found a linear correlation between plasma and brain lactate concentrations during euglycemia. 5 According to these calculations, brain lactate levels should have been as high as ∼2 µmol/g rather than ∼0.7 µmol/g with the plasma lactate levels of 3.5 mmol/L that we achieved. It should be noted that with 13C-MRS brain, lactate levels are calculated rather indirectly. These calculations require a number of critical assumptions, which may result in an overestimation of brain lactate levels. Here we used 1H-MRS to measure the signal intensity of brain lactate, which is directly proportional to the tissue concentration of lactate, thus providing a direct measurement of brain lactate levels.
Lactate is taken up by the brain through proton-linked facilitated diffusion by monocarboxylate transporters (MCTs), and its uptake is driven by a concentration gradient from blood to brain and further enhanced by neuronal activation.29,30 Therefore, it is likely that elevated plasma lactate levels result in an increased uptake of lactate by the brain. Furthermore, it has repeatedly been shown that lactate can serve as a metabolic substrate for the brain,5,7–9,31 especially when plasma lactate levels are elevated.7,32,33 Lactate taken up by the brain needs to be converted to pyruvate before it enters the citric acid cycle, a process catalyzed by lactate dehydrogenase (LDH). Even relatively small increases in local brain lactate concentration can drive lactate utilization by shifting the LDH reaction to pyruvate formation and to subsequent oxidation. 34 As we found only a small increase in brain lactate levels, we posit that lactate taken up by the brain is largely oxidized, both during euglycemia and hypoglycemia. Increased brain lactate concentrations during hypoglycemia are likely responsible for the suppressive effects on counterregulatory responses in patients with NAH, presumably because of increased brain lactate oxidation. Alternatively, lactate may exert its suppressive effects via alterations in cerebral blood flow, redox signaling or neuronal activity.35,36
Since brain lactate transport capacity is reported to be upregulated in patients with IAH,10,11 one can expect higher brain lactate levels in patients with IAH than in patients with NAH. Brain lactate concentrations are the result of a balance between its uptake, production, oxidation and export. The absence of a difference in brain lactate concentrations between NAH w|lac and IAH w|lac at the currently elevated plasma lactate levels either indicates comparable brain lactate uptake in both groups, or upregulation of both lactate uptake and lactate oxidation in patients with IAH. Without the use of exogenous lactate infusion, we previously showed that brain lactate levels fall in response to hypoglycemia in patients with IAH, but not in those with NAH. 37 This may indeed indicate upregulated capacity to oxidize lactate in patients with IAH. Analogously, the trend towards increased brain lactate levels in the current study disappeared at the end of hypoglycemia in patients with IAH, again hinting at upregulated capacity to oxidize lactate. When we used intense exercise to raise endogenous plasma lactate to much higher levels, we observed higher brain lactate levels in patients with IAH than in those with NAH. 38 Cerebral lactate uptake is not only dependent on plasma lactate concentrations but also on pH levels in plasma and in brain. 30 The lactate-induced increase in plasma pH value (i.e. a decrease in H+ concentration) observed in the current study may have limited brain lactate uptake in comparison to an exercise-induced decrease in plasma pH. However, these effects are generally small when pH values remain within the physiological range.
We found brain glutamate levels to fall in response to hypoglycemia in patients with NAH, which is in line with previous observations 39 and has been attributed to glutamate oxidation.39–41 This decrease in brain glutamate was also present in patients with NAH when lactate was infused, which suppressed adrenaline responses to hypoglycemia. This is in line with the absence of a relation between the hypoglycemia-induced decrease in glutamate and the adrenaline response to hypoglycemia, as reported by Terpstra et al., 39 and may indicate that even though lactate is abundantly available, it is not sufficient to compensate for the decrease in glucose supply to the brain in patients with NAH, or alternatively, that lactate oxidation is already saturated in these patients. In contrast, but again in line with previous data without lactate infusion, 39 brain glutamate did not decrease in patients with IAH, possibly because of their greater capacity to use (the excess of) lactate, thus eliminating the need to oxidize glutamate.
Our study has a small sample size, though based on power calculations. However, the study population was homogenous in terms of age, HbA1c and diabetes duration, and plasma glucose and lactate levels during the clamps were stable and almost identical in all groups. The homogeneity of the study population enables efficient comparison, but on the other hand, may limit the generalisability of the results. Importantly, to quantify the cerebral lactate content, we took the contribution of plasma lactate (ranging from ∼0.8 mmol/L to ∼3.5 mmol/L) to the MR signal of brain lactate into account. Since the voxel consisted of mainly white matter, we estimated a 2% vessel volume, based on literature.23,24 Deviations from this estimation (within the physiological range) may affect the magnitude of the outcome, but will not affect the main conclusion of the study.
In conclusion, we show that intravenous lactate administration reduces awareness of hypoglycemia and translates into a small increase in brain lactate levels, with no differences between patients with NAH and IAH. The rather limited increase in brain lactate suggests that the excess of lactate is immediately used by the brain both under euglycemic and hypoglycemic conditions. An increase in brain lactate oxidation during hypoglycemia may explain the suppressive effects of lactate on awareness of hypoglycemia observed in patients with NAH.
Supplemental Material
Supplemental material for Effect of lactate administration on brain lactate levels during hypoglycemia in patients with type 1 diabetes
Supplemental material for Effect of lactate administration on brain lactate levels during hypoglycemia in patients with type 1 diabetes by Evita C Wiegers, Hanne M Rooijackers, Cees J Tack, Bart WJ Philips, Arend Heerschap, Marinette van der Graaf and Bastiaan E de Galan in Journal of Cerebral Blood Flow & Metabolism
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Research support from the Dutch Diabetes Research Foundation (DFN 2012.00.1542), the European Foundation for the Study of Diabetes and by an unrestricted research grant from Sanofi, is gratefully acknowledged.
Acknowledgments
We thank all the volunteers for their participation in this work. We are indebted to Karin Saini and Adrianne Hofboer-Kapteijns (research nurses, Radboud university medical center) for assistance during the glucose clamps, and to Sjaak van Asten for his assistance with the LCModel analysis.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
EW, HR, BdG and MvdG designed the study with input from CT, AH and BP. HR recruited the participants and performed the glucose clamps. BP and EW developed/implemented the MR methods and MRS sequence. EW and HR collected the data. EW analyzed the MR data, HR and EW were both responsible for all other data analysis. All authors discussed the results and implications and commented on the manuscript at all stages. All authors approved the final version of the manuscript.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.