
Abstract
Uncontrolled oxidative stress contributes to the secondary neuronal death that promotes long-term neurological dysfunction following traumatic brain injury (TBI). Surprisingly, both NADPH oxidase 2 (NOX2) that increases and transcription factor Nrf2 that decreases reactive oxygen species (ROS) are induced after TBI. As the post-injury functional outcome depends on the balance of these opposing molecular pathways, we evaluated the effect of TBI on the motor and cognitive deficits and cortical contusion volume in NOX2 and Nrf2 knockout mice. Genetic deletion of NOX2 improved, while Nrf2 worsened the post-TBI motor function recovery and lesion volume indicating that decreasing ROS levels might be beneficial after TBI. Treatment with either apocynin (NOX2 inhibitor) or TBHQ (Nrf2 activator) alone significantly improved the motor function after TBI, but had no effect on the lesion volume, compared to vehicle control. Whereas, the combo therapy (apocynin + TBHQ) given at either 5 min/24 h or 2 h/24 h improved motor and cognitive function and decreased cortical contusion volume compared to vehicle group. Thus, both the generation and disposal of ROS are important modulators of oxidative stress, and a combo therapy that prevents ROS formation and potentiates ROS disposal concurrently is efficacious after TBI.
Introduction
Traumatic brain injury (TBI) is the major cause of mortality and morbidity affecting millions of individuals globally with a significant economic burden. 1 The secondary neuronal death is a known proponent of long-term neurological dysfunction following TBI. Many pathophysiological mechanisms including ionic imbalance/edema, inflammation, apoptosis, endoplasmic reticulum (ER) stress and oxidative stress synergistically mediate the secondary brain damage after TBI. In particular, oxidative stress after TBI kills neurons if uncontrolled. 2 Oxidative stress starts within hours and continues for days after TBI3,4 and is known to contribute to the activation of other secondary injury mechanisms including inflammation and ER stress.5–8 Paradoxically, pathways that promote as well as fight the oxidative stress are induced concurrently after CNS insults. One of the subunits of the nicotinamide adenine dinucleotide phosphate oxidase-2 (NOX2), gp91phox promotes oxidative stress by generating reactive oxygen species (ROS) that kills neurons.9–11 On contrary, activation of transcription factor Nrf2 induces several downstream neuroprotective genes including antioxidant enzymes and protein chaperones that can mitigate oxidative stress to protect neurons.12,13 Hence, therapeutic interventions that prevent formation and promote disposal of ROS can be highly effective in improving the outcome following TBI.3,14
As both NOX2 and Nrf2 are known to be induced after TBI,15,16 we currently tested if mono or combo therapy with a NOX2 inhibitor (apocynin) and/or a Nrf2 activator (tert-butylhydroquinone; TBHQ) decreases the secondary brain damage and promotes motor and cognitive functional recovery after TBI. We further tested the effect of TBI in knockout mice that lack either NOX2 or Nrf2.
Materials and methods
TBI
A moderate grade TBI was induced with a controlled cortical impact (CCI) device in adult male mice (12 weeks; 25–30 g; Jackson Labs USA) that lack NOX2 (B6.129SCybbtm1Din/J; Stock Number: 002365) or Nrf2 (B6.129X1Nfe2l2tm1Ywk/J, Stock No: 017009) and the wild-type controls (C57BL/6 J) as described earlier. 17 This is primarily a focal injury with minimal diffuse effects.18,19 We used adult mice for all studies as they respond to TBI and drug treatments in a consistent manner.
All animal experiments were performed according to the principles of the Guide for the Care and Use of Laboratory Animals (U.S. Department of Health and Human Services Publication 86-23, revised) and were approved by the Research Animal Resources and Care Committee of the University of Wisconsin-Madison. In brief, mice were anesthetized with isoflurane and were injured through a craniotomy (2.5 mm in diameter, 1 mm lateral and 1 mm caudal to bregma) at a velocity of 3 m/s and 1 mm deformation. After TBI procedure, the exposed cortex was covered with Surgicel, the skull piece was replaced, skin wound was sutured, and 0.5% bupivacaine (100 μl) was applied to the incision. During the surgery and the recovery, body temperature was maintained at 37℃ using a heating pad and an infrared lamp. After recovering from anesthesia, mice were returned to their cages with free access to food and water. Mice were randomly assigned to study groups. Group identities were blinded during analysis. All the animal experiments in this study have been reported according to the ARRIVE guidelines.
Drug administration
Apocynin (10 mg/kg; Sigma Chemicals USA) and/or TBHQ (25 mg/kg; Sigma Chemicals USA) dissolved in 1% DMSO or vehicle (1% DMSO) were injected intraperitoneally at 5 min or 2 h followed by the second dose at 24 h post-TBI.
Motor function and hippocampal-dependent memory evaluation
Post-TBI motor function was evaluated both by Rotarod (time to stay on an accelerating Rotarod from 4 to 40 rpm/min for 5 min) and beam walk tests (number of foot faults while crossing a 5 mm × 60 cm beam) on days 1, 3, 5 and 7 after TBI as described earlier.20–22 Hippocampal-dependent memory and learning were assessed by the Morris Water Maze (MWM) test at 30 days following TBI. Prior to testing, mice were trained to locate a hidden escape platform in a circular pool full of water for three consecutive days, as described previously.23–26 Mice were trained twice daily to locate a hidden platform using visual cues and then subjected to a probe trial on day 30. The frequency of platform crossings and the time spent (seconds) in the training quadrant were recorded.
Lesion volume estimation and hippocampal neuronal counting
After functional testing, mice were euthanized by transcardiac perfusion with 4% phosphate-buffered paraformaldehyde. Each brain was post-fixed, cryoprotected, sectioned (coronal; 40 µm thick at an interval of 480 µm) and serial sections covering the cortical lesion (from 0 to −3.0 mm from Bregma as per the Mouse Brain Atlas of Paxinos and Franklin 27 ) were stained with cresyl violet, scanned using NIH ImageJ software and cortical lesion volume was computed by numeric integration of data from six serial sections in respect to the sectional interval as described earlier. 17 Cresyl violet stained sections from the combo therapy and vehicle groups were scanned at 20 × magnification using EVOS XL microscope (Thermo Fisher Scientific) and neurons were counted in the CA1, CA2, CA3 and dentate gyrus (DG) regions of the hippocampus using ImageJ software.
Immunostaining of cleaved caspase-3 and 3-nitrotyrosine (3-NT)
Brain sections from mice subjected to TBI were first blocked with 5% BSA inTBST (0.3% Triton X-100) and probed with the primary antibodies against NeuN (1:200; monoclonal; Millipore), 3-NT (1:200; monoclonal; Abcam) and cleaved caspase-3 (1:400; monoclonal; Cell Signaling Technology). Alexa Fluor 488 (green) and 594 (red) conjugated secondary antibodies (1:200, Molecular Probes) were used according to the combination of primary antibodies as described previously.22,28
Statistics
For analyzing the Rotarod and beam walk test data at different time points, we performed two-way repeated-measures ANOVA with Sidak’s multiple-comparisons test. For the lesion volume, water maze data and hippocampal cell counting, we used the two-tailed Mann–Whitney U test. In all experiments, wild-type mice were used as controls for comparing the knockout mice and vehicle-treated mice were used as controls for comparing the drug-treated mice. GraphPad Prism 6 software was used for the statistical analysis.
Results
Genetic deletion of NOX2 decreased the post-TBI motor dysfunction and lesion volume
At seven days after TBI, the percent of time stayed on the Rotarod was significantly higher in NOX2−/− mice compared to wild-type controls (Figure 1(a)). The number of foot faults in the beam walk test was also significantly lower in NOX2−/− mice compared to wild-type control mice at three, five and seven days after TBI (Figure 1(b)). The cortical lesion volume evaluated at day 7 after TBI was also significantly smaller in NOX2−/− mice compared to wild-type control mice (Figure 1(c) and (d)). This indicates a neurotoxic role of NOX2 (which forms ROS) in the post-TBI brain.
NOX2−/− mice showed a significant improvement in motor function indicated by performance in the Rotarod test (a) and beam walk test (b) compared to the wild-type control (NOX2+/+) mice. NOX2−/− mice also showed significantly smaller cortical lesion volume compared to wild-type control cohort (c and d). Values are mean ± SEM (n = 5–6/group). *p < 0.05 compared with respective control by repeated-measures two-way ANOVA followed by Sidak’s multiple-comparisons post-test for the behavioral data. *p < 0.05 compared with respective control by Mann–Whitney U test for lesion volume data.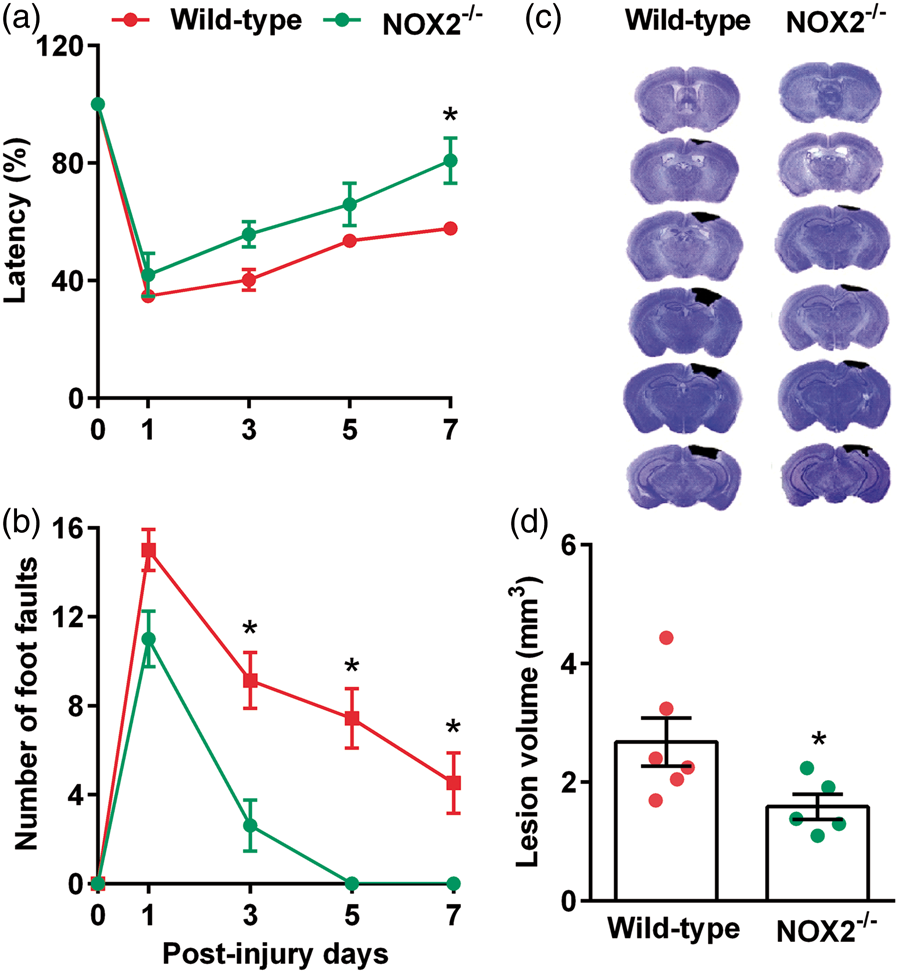
Genetic deletion of Nrf2 worsened the post-TBI motor function and lesion volume
At five and seven days after TBI, the Nrf2−/− mice stayed significantly less time on the Rotarod compared to wild-type control mice (Figure 2(a)). Nrf2−/− mice also showed a significantly higher number of foot faults in the beam walk test compared to wild-type control mice at three, five and seven days after TBI (Figure 2(b)). The cortical lesion volume measured at seven days after TBI was significantly larger in the Nrf2−/− mice compared to wild-type control mice (Figure 2(c) and (d)). This indicates a neuroprotective role of Nrf2 and the antioxidant enzymes downstream to Nrf2 in the post-TBI brain.
Nrf2−/− mice showed a decreased recovery of post-TBI motor function as indicated by performance in the Rotarod test (a) and beam walk test (b) compared to the wild-type control (Nrf2+/+) mice. Nrf2−/− mice also showed a significantly larger cortical lesion volume compared to wild-type control cohort (c and d). Values are mean ± SEM (n = 9/group). *p < 0.05 compared with respective control by repeated-measures two-way ANOVA followed by Sidak’s multiple-comparisons post-test for the behavioral data. *p < 0.05, compared with respective control by Mann–Whitney U test for lesion volume.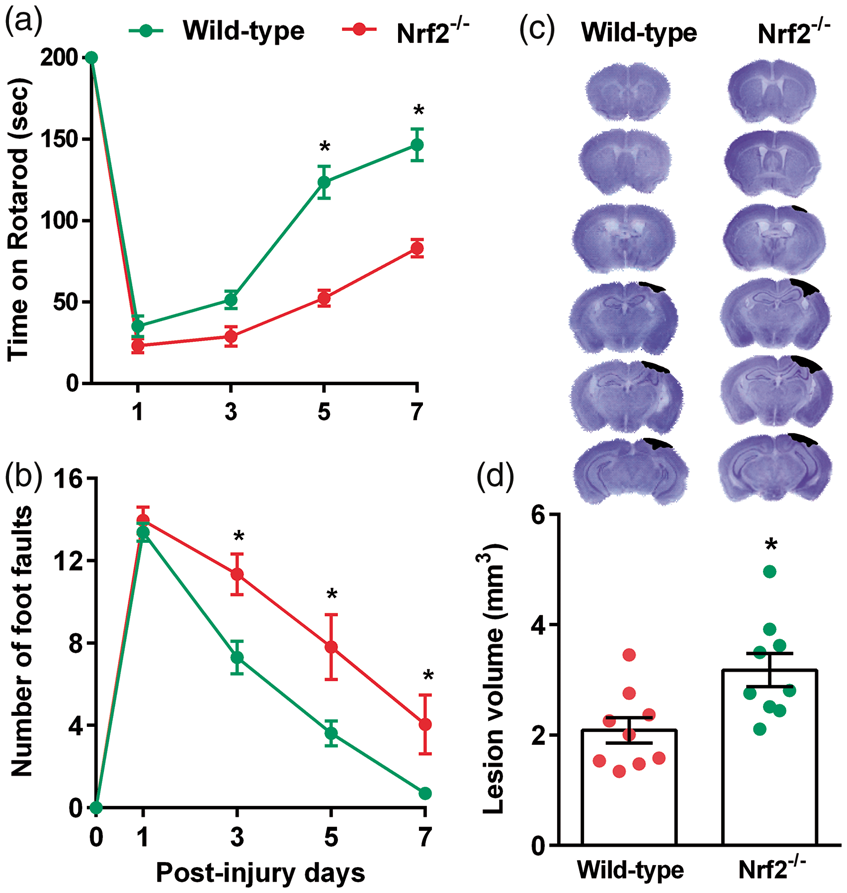
Genetic deletion of Nrf2 worsened cognitive performance
At 30 days after TBI, Nrf2−/− mice showed a significantly decreased memory function compared to wild-type control mice as assessed by the MWM test (Figure 3). The time spent in the platform quadrant was 3-fold lower (Figure 3(a) and (b)), and the frequency of crossing the platform quadrant was 4-fold lower (Figure 3(a) and (c)) in the probe test in Nrf2−/− mice compared to wild-type control mice. This indicates that failure to dispose of the ROS in the brain after TBI leads to long-term detrimental consequences.
At 30 days after TBI, Nrf2−/− mice showed a significant decrease in cognitive function indicated by performance in the MWM test compared to the wild-type control (Nrf2+/+) cohort. Representative track plots and heat maps show lower memory retention in the Nrf2−/− mice compared to wild-type control mice (a). Nrf2−/− mice showed significantly decreased time spent (b) and frequency of crossing the platform quadrant (c) during the probe test. Values are mean ± SEM (n = 8/group). *p < 0.05 compared with wild-type control mice by Mann–Whitney U test.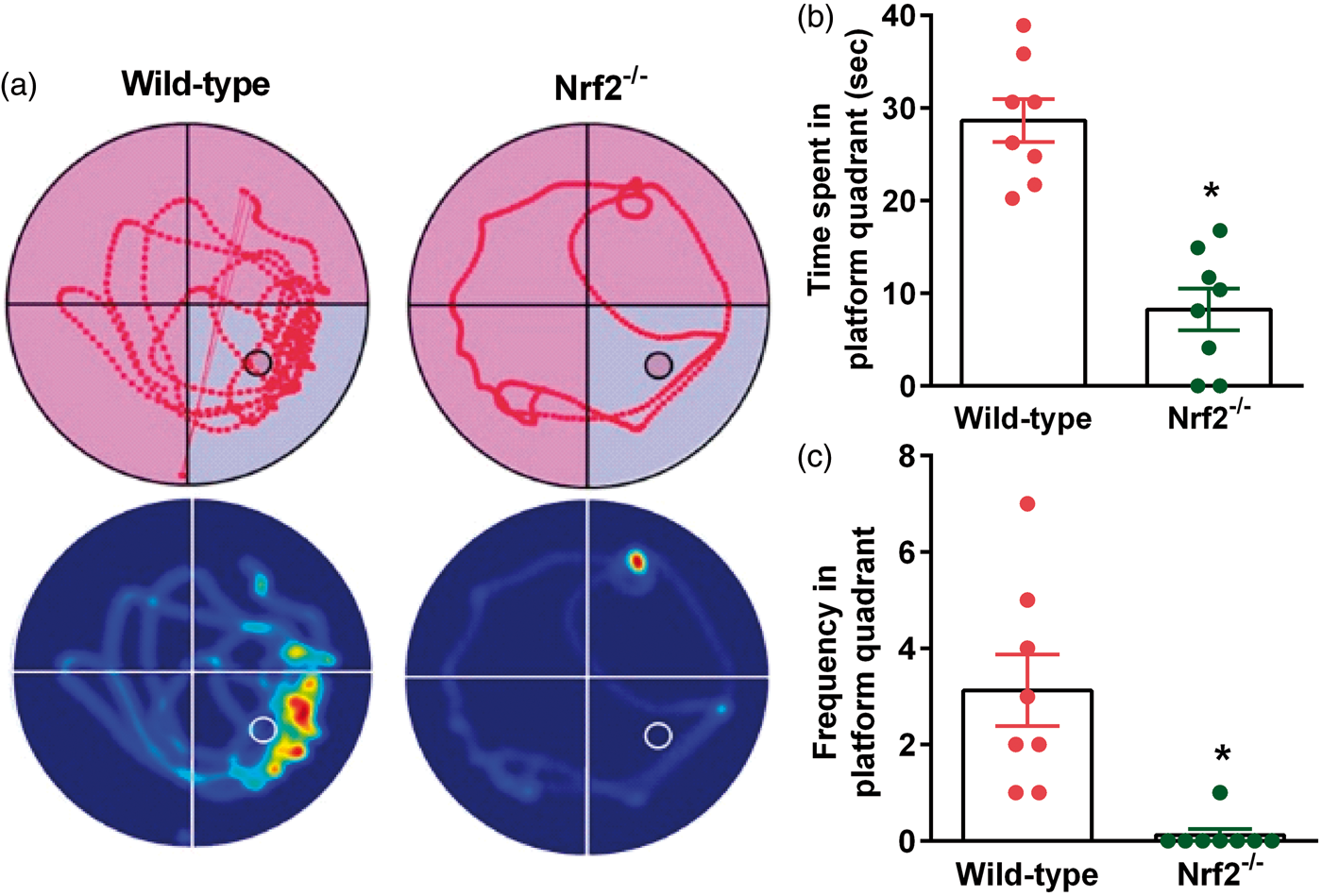
Administration of apocynin and TBHQ individually reduced TBI-induced motor deficits
Based on the knockout studies presented above, we tested if pharmacologic inhibition of NOX2 with apocynin and activation of Nrf2 with TBHQ leads to better outcomes after TBI. We tested two dose regimens for each drug (the first dose was given either at 5 min or 2 h and a second dose at 24 h after TBI). Both apocynin and TBHQ-treated groups showed the significantly longer length of stay on the Rotarod and fewer foot faults in beam walk test compared to respective vehicle-treated control groups between day 1 and 7 after TBI (Figure 4(a) and (b)). However, the cortical contusion volume was not significantly different in either apocynin or TBHQ-treated group regardless of the timing of the first dose compared to vehicle control (Figure 4(c) and (d)).
Apocynin (APO) and TBHQ monotherapies improved the motor function recovery, but had no effect on the lesion volume after TBI. Each drug was given as two doses with the first dose at either 5 min or 2 h and the second dose at 24 h after TBI. The panels show Rotarod test and beam walk test for either apocynin (a) or TBHQ- (b) treated mice. Panel C shows representative cresyl violet sections of each group and panel D shows the lesion volume computed from serial brain sections in each case. Values are mean ± SEM (n = 5 or 6/group). *p < 0.05 and #p < 0.05 denotes 5 min/24 h group and 2 h/24 h group, respectively, compared with the vehicle control group by repeated-measures two-way ANOVA followed by Sidak’s multiple-comparisons post-test.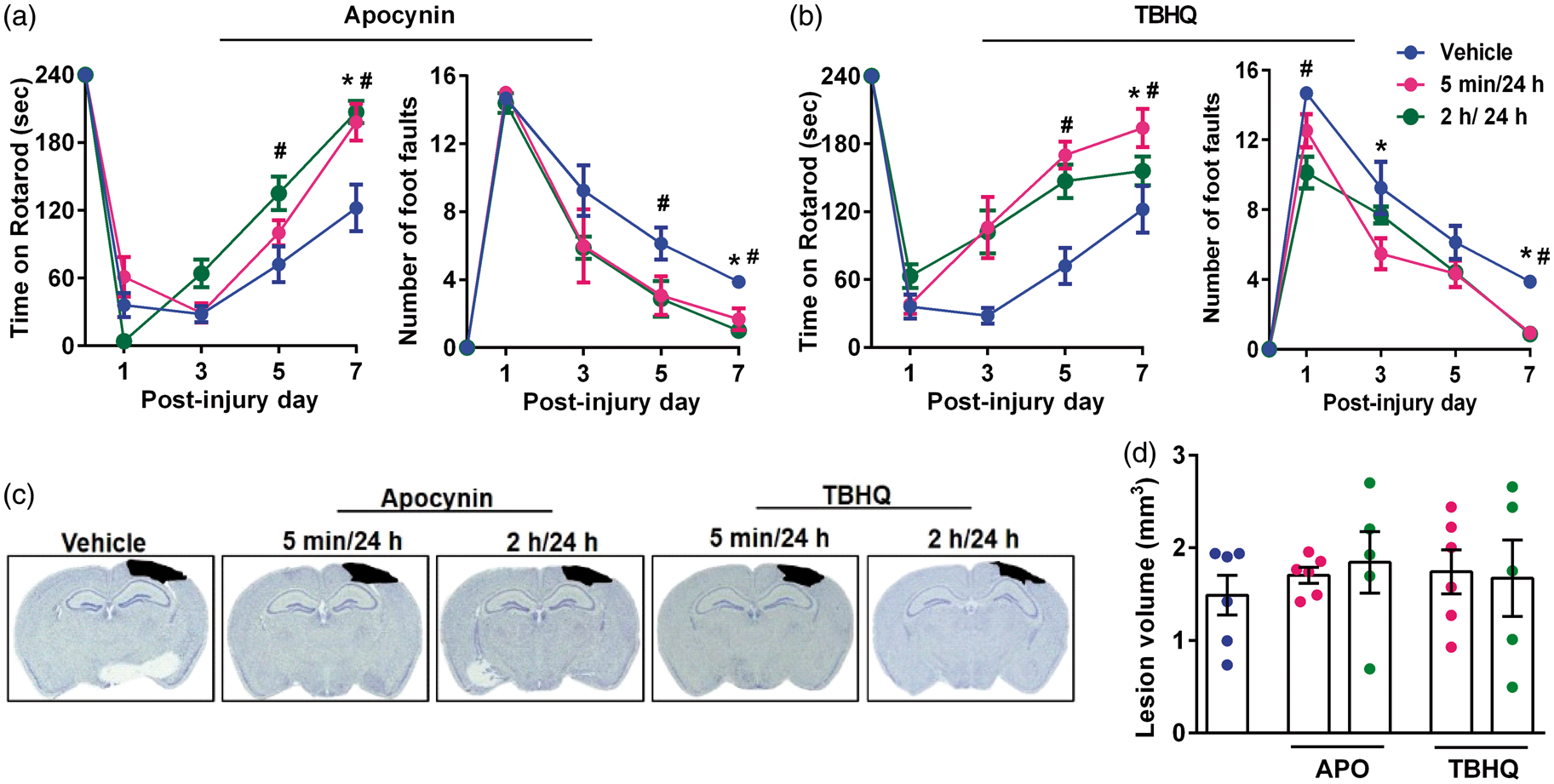
Combo treatment reduced the lesion volume, motor deficits and improved memory and learning after TBI
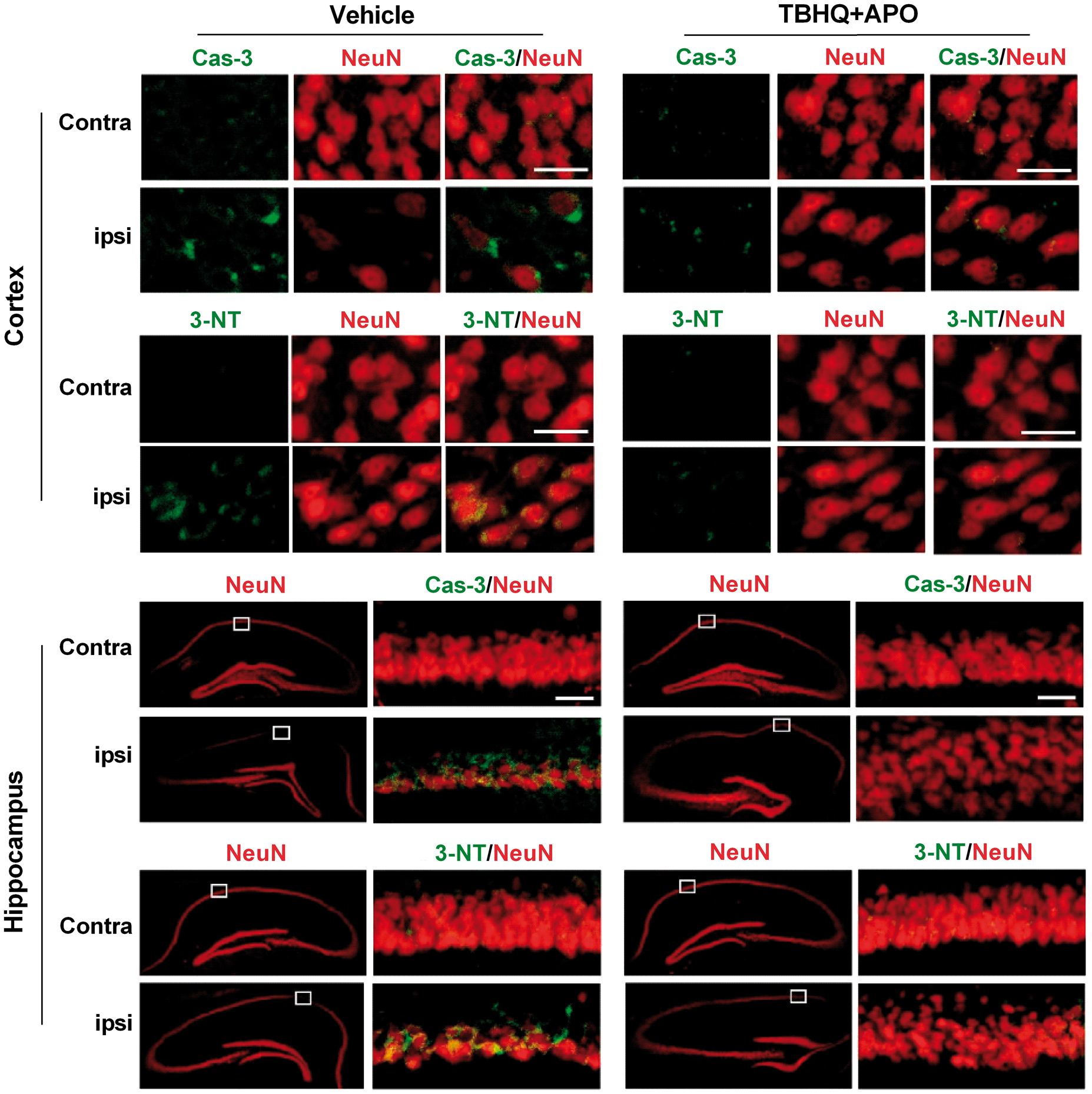
When mice were treated with a combination of apocynin and TBHQ at 5 min or 2 h (first dose) and 24 h (second dose) after TBI, there was a significant improvement in motor function assessed by Rotarod test (Figure 5(a)) and beam walk test between days 3 to 7 after TBI compared to vehicle control (Figure 5(b)). MWM test performed at 30 days after TBI in the 2 h/24 h apocynin + TBHQ group showed a significantly increased time spent in the platform quadrant (Figure 5(c) and (d)) and increased frequency of platform crossings (Figure 5(c) and (e)) compared to vehicle control. In addition, the combo therapy significantly decreased the secondary cortical contusion volume when the drugs were given at 5 min/24 h or 2 h/24 h after TBI compared to vehicle control (Figure 5(f) and (g)). Mice treated with the drug combo showed the significantly higher number of neurons in the hippocampal CA1 and CA2 regions compared to vehicle control group (Figure 5(h)). The combo therapy also decreased the levels of markers for neuronal apoptosis (cleaved caspase-3) and oxidative stress (3-NT) compared to the vehicle-treated group in the ipsilateral cerebral cortex and hippocampus compared to vehicle control (Figure 6).
Combo treatment with apocynin (APO) and TBHQ given in two doses (first dose at 5 min or 2 h and second dose at 24 h) after TBI reduced motor deficits as assessed by Rotarod test (a) and beam walk test (b), improved memory performance as evaluated by MWM test (c, d and e), decreased lesion volume (f and g) and increased the total number of neurons in the hippocampal CA1 and CA2 regions (h) compared to vehicle control. Values are mean ± SEM (n = 5 to 6/group except for hippocampal cell counting where n = 4/group). *p < 0.05 and #p < 0.05 denote 5 min/24 h group and 2 h/24 h group, respectively, compared with the vehicle control group by repeated-measures two-way ANOVA followed by Sidak’s multiple-comparisons post-test for the behavioral data. *p < 0.05, compared with the respective vehicle control group by Mann–Whitney U test for lesion volume and hippocampal cell counts. Combo treatment with apocynin (APO) and TBHQ after TBI decreased the levels of markers for apoptosis (cleaved caspase-3; Cas-3) and oxidative stress (3-nitrotyrosine; 3-NT) compared to vehicle-treated animals in the ipsilateral cerebral cortex and hippocampus. Scale bar is 30 µm.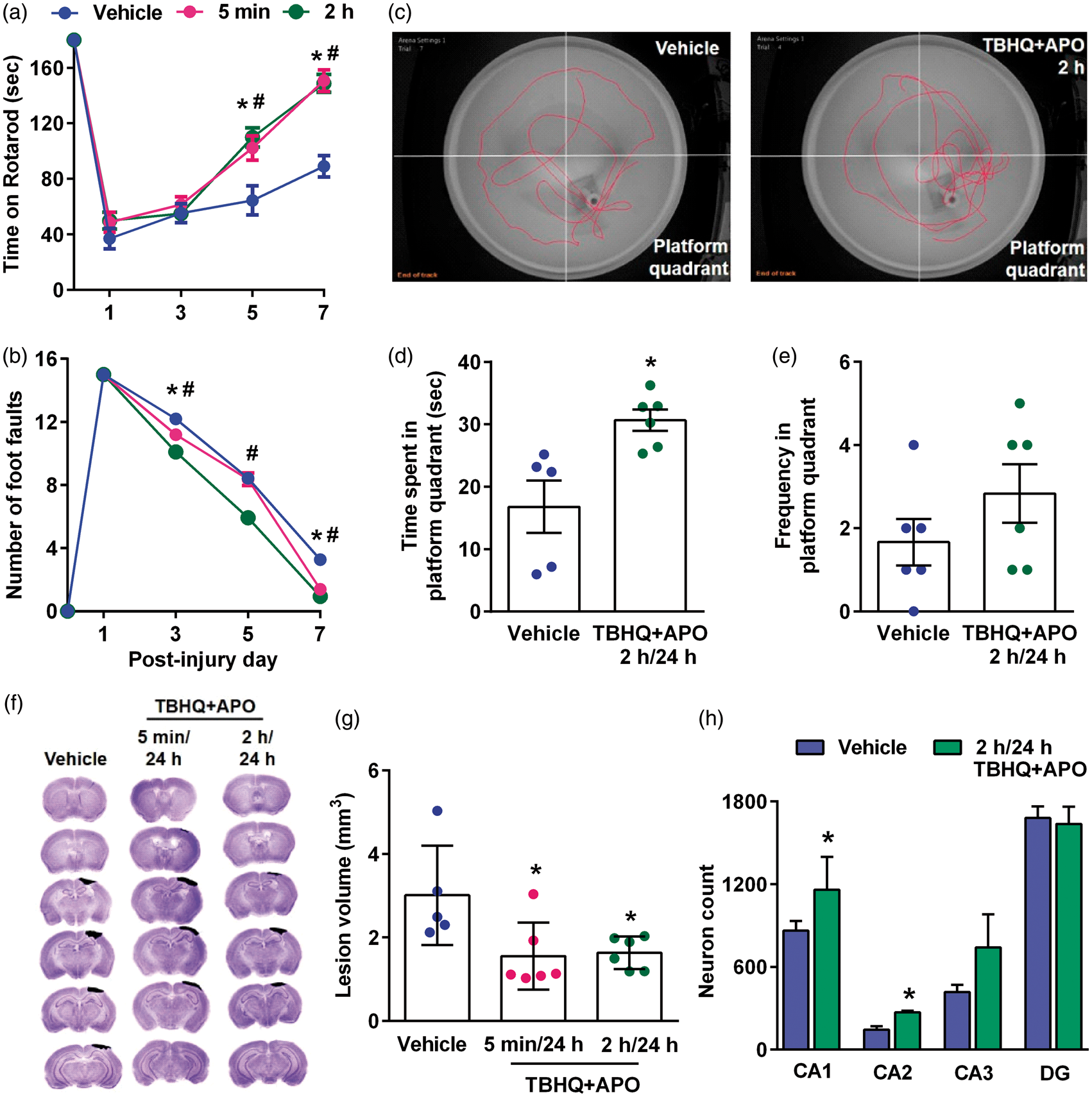
Discussion
Oxidative stress that starts within minutes and continues for hours to days is the major contributor to the secondary brain damage after TBI. ROS also potentiates both inflammation and ER stress that synergistically kill neurons after brain injury. It was previously shown that TBI induces both NOX2 which is the major producer of ROS and Nrf2 which is the major antioxidant transcription factor.15,16,29 Hence, we currently tested their role after TBI using gene knockouts as well as pharmacologic inhibitors. NOX2 knockout mice showed better, and Nrf2 knockout mice showed worsened functional outcome indicating the benefit of reducing oxidative stress after TBI. Furthermore, treatment with apocynin (NOX2 inhibitor) or TBHQ (Nrf2 activator) improved the post-TBI functional outcome, but the combo treatment with both drugs showed better therapeutic efficacy (improved functional outcome and decreased secondary contusion volume).
NOX2 is composed of membrane-associated subunits gp91phox and p22phox, and cytosolic subunits p47phox, p67phox and p40phox. 30 Of these, gp91phox is the rate-limiting catalytic subunit that is thought to be the major contributor of ROS after an injury.31,32 The genetic deletion of gp91phox decreases the production of NADPH oxidase-mediated superoxide indicating its contribution to oxidative stress. 33 Furthermore, NOX2 genetic ablation or pharmacologic inhibition (by apocynin) was shown to block NMDA-induced ROS production and neuronal death in mice. 34 Furthermore, NOX2 was implicated in microglial activation that results in oxidative damage to neuronal cells.29,35 Following TBI, induction of plasma membrane Ca2+ pumps increases intracellular Ca2+ that activates CaMKII which in turn induces NOX2.36,37 Apocynin is known to be a robust NOX2 inhibitor 38 that was shown to decrease oxidative stress and induce neuroprotection following intracerebral hemorrhage, 39 global ischemia 40 and focal ischemia.33,39,41,42 A previous study demonstrated that apocynin decreases ischemia-induced ROS production and infarction in wild-type, but not NOX2−/− mice showing the specificity of the drug. 43 Apocynin was also shown to be neuroprotective in other experimental models of TBI.15,44–49 Our current studies show that either lack or inhibition of NOX2 promotes better recovery of a neurological function indicating its role in promoting secondary brain damage after TBI. Our studies are also corroborative with the previous reports that show that NOX2 is the major inducer of oxidative stress after TBI.30,50 Previous studies also showed that apocynin treatment improves cognitive performance after CCI injury, weight drop injury and fluid percussion injury in rodents.46–48
While NOX2 induces oxidative stress, Nrf2 helps cells to fight oxidative stress by inducing antioxidant genes and other neuroprotective genes.51,52 These include those that maintain glutathione homeostasis, mediate phase-II detoxification, protein chaperoning and degradation of misfolded proteins. Under normal condition, the E3 ubiquitin ligase Keap1 binds to Nrf2 leading to its constant ubiquitination and degradation; and hence Nrf2 levels remain low. 51 ROS and other toxins induce dissociation of Nrf2 from Keap1 leading to its activation. 53 Once activated, Nrf2 translocates to nucleus and induces the expression of genes that includes glutathione-S-transferase, γ-glutamylcysteine ligase catalytic and modifying subunits, glutathione reductase, glutathione peroxidase, γ-glutamylcysteine synthase, peroxiredoxin, NAD(P)H:quinone oxidase-1, aldehyde dehydrogenase, catalase, thioredoxin, heme oxygenase-1, HSP70, sequestosome-1 and ubiquitin C.54–56 Furthermore, Nrf2 also trans-repress the expression of pro-inflammatory genes like cyclooxygenase-2, tumor necrosis factor-α, interleukin-6 and interleukin-1β.52,57,58 Treatment with Nrf2 activators TBHQ and sulforaphane was shown to induce neuroprotection following TBI, cerebral ischemia and intracerebral hemorrhage.59–63 TBHQ was also shown to protect neurons in vitro from stretch injury. 64 Oral pretreatment with TBHQ was shown to decrease NF-kB activation in mice after closed head injury. 65 Our present studies show that lack of Nrf2 worsens post-TBI outcome and treatment with Nrf2 activator TBHQ decreases motor and cognitive dysfunction after TBI. TBHQ was also known to be efficacious in controlling oxidative stress in other animal models of TBI.64–66
While both apocynin that prevents the formation of ROS and TBHQ that increases disposal of ROS were observed to promote better neurological recovery, neither decreased the lesion volume after TBI. Secondary brain damage after TBI is multifactorial. While drugs that target a single pathway are useful as specific therapeutics, it is often important to use a combination therapy to achieve neuroprotection by targeting multiple interactive pathways. To efficiently control oxidative stress, it is essential to curtail the formation of ROS and at the same time increase the disposal of ROS. To achieve this, we tested a combination of apocynin and TBHQ and observed that the combo therapy is more efficacious to promote neurological recovery and also decreased the secondary cortical lesion volume after TBI than monotherapies. The combination therapy also decreased the post-TBI cognitive dysfunction. Importantly, we show that neuroprotection can be achieved even if the first dose of apocynin and TBHQ is delayed by 2 h after TBI. This is relevant for translating the antioxidant therapies to humans in future.
The drug combo used in this study has a high possibility to be translated for human use as apocynin is a plant-derived organic compound and TBHQ is an FDA-approved food additive.38,67 But there might be a few potential limitations for administering this drug combo including a less efficient drug response and/or side effects in humans due to a possible dose mismatch. A detailed preclinical pharmacological study of the dose combinations is needed to identify an efficacious dose with no toxicity. Another confounding issue is the potential physicochemical incompatibility of the drugs in the combination which might lead to a decreased brain bioavailability of one or both of them. Hence, a detailed PK/PD analysis of the combination is needed before translating to clinics. Overall, our studies show that a strategy to use a combination of drugs that prevent ROS formation and promote ROS disposal is a good option to fight the oxidative stress and thereby promote the neuroprotection after TBI.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was partially supported by the National Institutes of Health Grants – NS083007 and NS082957, US Veterans Administration Merit Review Grant (BX001638), K08 NS088563-01A1 from NINDS (Cengiz P) and NIH P30 HD03352 (Waisman Center).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
RC, SLM, THK, HK and CK performed the animal surgeries, drug administrations, motor behavioral testing, histopathology and immunostaining experiments. RV, RC, SLM, THK designed the experiments, analyzed the data, wrote and edited the manuscript. EU, VC, and PC performed the cognitive testing.