
Abstract
Studies in experimental traumatic brain injury (TBI) suggest both deleterious and protective effects of inducible nitric oxide synthase (iNOS). Early after injury, iNOS may be detrimental via formation of peroxynitrite and iNOS inhibitors are protective. In contrast, we reported impaired long-term functional outcome after TBI in iNOS knockout (ko) versus wild-type (wt) mice. To elucidate potential neuroprotective and neurotoxic mechanisms for iNOS, we studied nitric oxide formation by electron paramagnetic resonance (EPR) spectroscopy using diethyldithiocarbamate—iron (DETC—Fe) as a spin trap and markers of nitrosative (S-nitrosothiol (RSNO, Fluorescent assay); nitrotyrosine (3NT, ELISA)) and oxidative stress (ascorbate, HPLC) at 72 h after controlled cortical impact (CCI) in iNOS ko and wt and in uninjured iNOS ko and wt mice. 3NT immunostaining with macrophage and myeloperoxidase (MPO) dual labeling was also assessed in brain sections. Brain DETC—Fe—NO low-temperature EPR signal intensity was ∼2-fold greater in wt versus iNOS ko at 72 h after CCI. Ascorbate levels decreased in injured hemisphere in wt and iNOS ko versus uninjured —this decrease was more pronounced in iNOS ko. In wt mice, RSNO and 3NT levels were increased after CCI versus uninjured (50% and 400%, respectively, P<0.05). RSNO levels were not increased in iNOS ko after CCI. Nitrotyrosine levels increased after CCI in wt and ko versus respective uninjured —this increase was more pronounced in wt (2.34±0.95 versus 1.27±0.49 pmol/mg protein, P<0.05). Increased 3NT immunoreactivity was detected in wt versus iNOS ko at 72 h after CCI, and colocalized with macrophage marker and MPO. Our data support a role for iNOS-derived NO as an endogenous antioxidant after CCI. iNOS also contributes protein nitrosylation and nitration. Colocalization of 3NT with macrophages and MPO suggests generation of nitrating agents by macrophages and/or phagocytosis of nitrated proteins.
Introduction
Oxidative stress plays a central role in the pathophysiology of many central nervous system (CNS) diseases, including traumatic brain injury (TBI), cerebral ischemia, Alzheimer's disease, and Parkinson's disease. The basis of this contention in TBI stems from findings in both animal models and patients showing production of free radicals leading to oxidation of lipids, proteins, and depletion of antioxidants mainly ascorbate (Tyurin et al, 2000; Bayır et al, 2002). Although oxidative stress is detrimental after TBI, nitric oxide (NO) is linked both to secondary damage and neurologic recovery. Detrimental versus beneficial effects of NO may be critically governed by timing, cellular localization and linked to the level of oxidative stress in the injured tissue.
Nitric oxide is poorly reactive with most biomolecules but highly reactive with other free radicals. Reaction of NO with superoxide forms peroxynitrite with resultant protein nitration, DNA damage, PARP activation and energy failure (Beckman et al, 1990). Peroxynitrite-mediated damage is caused by its decomposition products that possess potent free radical characteristics. Peroxynitrite can generate nitrogen dioxide (•NO2) either by protonation to peroxynitrous acid (ONOOH) and subsequent decomposition to •NO2 and hydroxyl radical (•OH), or through reaction with carbon dioxide (CO2) to generate nitrosoperoxocarbonate (ONOOCO2) and subsequent decomposition to •NO2 and carbonate radical (Hall et al, 2004). Alternatively, NO can be oxidized to nitrite, which can be converted to •NO2 by myeloperoxidase (MPO) in neutrophils and macrophages, or by other pseudoperoxidases (Schopfer et al, 2003).
As a result of its high reactivity with other free radicals, NO also acts as a potent antioxidant. NO can scavenge reactive propagating free radicals and terminate damaging free radical processes. Interaction of NO with lipid peroxyl radicals results in effective inhibition of lipid peroxidation (Kagan and Laskin, 2001). Evidence for an antioxidant role of NO in CNS is largely derived from work in experimental models of Parkinson's disease. Nitric oxide and NO donors have been shown to scavenge •OH radical and reverse nigrostriatal dopamine depletion caused by ferrous citrate and 1-methyl-4phenylpyridinium (Rauhala et al, 1996; Mohanakumar et al, 2002). However, support for an antioxidant role for NO in TBI is lacking.
Nitric oxide is generated from
Inducible nitric oxide synthase (iNOS) is not constitutively present, but its expression is induced during development and in pathologic states, characteristically those associated with inflammation such as ischemia or TBI. Inducible nitric oxide synthase produces large amounts of NO limited only by substrate and cofactor availability. The time course for iNOS expression differs according to species and insults studied. Typically, iNOS is not found in the brain until 6 to 12 h after TBI (Clark et al, 1996a). Inducible nitric oxide synthase mRNA expression peaks at 12 to 48 h in rats and 48 to 96 h in mice after middle cerebral artery (MCA) occlusion (Yoshida et al, 1995; Iadecola et al, 1995). Inducible nitric oxide synthase is expressed at 3 to 7 days after TBI in adult rats, predominantly in macrophages and blood vessels (Wada et al, 1998). A recent study showed that iNOS is also expressed in human brain at 2 to 8 days after TBI (Orihara et al, 2001). As in rodents, iNOS immunoreactivity was observed in neutrophils, macrophages, and in blood vessels in areas surrounding the injured region (Orihara et al, 2001; Gahm et al, 2002). Furthermore, cerebrospinal fluid (CSF) levels of nitrate/nitrite peaked at 30 to 42 h after TBI and this increase was greater in nonsurvivors (Clark et al, 1996b). The time course of iNOS expression in humans after TBI mirrors the time course of S-nitrosothiol (RSNO) accumulation and ascorbate depletion reported in our previous studies (Bayır et al, 2002, 2003).
After experimental focal cerebral ischemia iNOS-derived NO exhibits detrimental effects. These effects are proposed to occur as a result of NO production in an oxidant milieu, with subsequent production of peroxynitrite, although some beneficial effects of protein nitration have been suggested to occur (Greenacre and Ischiropoulos, 2001). Several lines of evidence support detrimental effects of iNOS after cerebral ischemia. First, administration of a nonselective iNOS inhibitor, aminoguanidine, reduces infarct volume by 30 to 40% with improvement in neurologic deficit (Nagayama et al, 1998). Second, treatment with a selective iNOS inhibitor, 1400W, reduces infarct volume in rats after MCA occlusion (Parmentier et al, 1999). Third, iNOS knockout (ko) mice have smaller infarcts and better neurologic outcomes than wild-type (wt) after MCA occlusion (Zhao et al, 2000; Iadecola et al, 1997).
Unlike cerebral ischemia, there is controversy regarding detrimental effects of iNOS derived NO after TBI. Administration of aminoguanidine pre- or post-injury decreases cortical necrotic neuron counts (Wada et al, 1998), lesion volume, and improves functional outcome early after lateral fluid percussion injury in rats (Lu et al, 2003). Similar results were obtained with aminoguanidine treatment pre- and post-injury early after cortical freeze lesion in rats (Stoffel et al, 2000, 2001; Gorlach et al, 2000). In contrast, prolonged aminoguanidine treatment exacerbates brain injury at 21 days after controlled cortical impact (CCI) in rats. Furthemore, iNOS ko mice show markedly worse functional outcome after CCI than do wt mice (Sinz et al, 1999).
Many of the beneficial mechanisms proposed for NO have implicated an important role for S-nitrosylation. Transfer of NO groups to csyteine sulfydryls on proteins results in the formation of RSNO that are widespread in vivo (Stamler et al, 1992). A parallel dichotomous effect of iNOS is seen in experimental allergic encephalomyelitis in mice, with early benefit from iNOS inhibitors but detrimental effects in the iNOS ko (Fenyk-Melody et al, 1998; Sahrbacher et al, 1998), which may result from an RSNO-mediated increase in antiinflammatory T-helper-2 cytokine production (Fenyk-Melody et al, 1998; Kahl et al, 2004). Furthermore, activity of a number of enzymes and regulatory proteins including caspases 1, 3, 8, 9, and NMDA receptor has been shown to be regulated by S-nitrosylation (Kim et al, 1998, 2000; Mannick et al, 1999, 2001; Lipton et al, 1993). Other possible late beneficial effects of iNOS-derived NO are promotion of cerebral blood flow and neurogenesis (Rauhala et al, 1996; Okamoto et al, 1998; Zhu et al, 2003).
Thus, the current study was designed to assess the possible mechanisms involved in the late beneficial effects of iNOS by evaluating contribution of iNOS to NO production by electron paramagnetic resonance (EPR), oxidative stress by quantification of brain ascorbate levels, nitrosative stress by quantification of brain RSNO, and 3-nitrotyrosine (3NT) levels when iNOS induction is maximal (72 h) after CCI in mice. We chose to evaluate ascorbate as the marker of oxidative stress since ascorbate is the primary sacrificial antioxidant and it regulates RSNO levels. To evaluate regional and cellular distribution of 3NT and possible role for MPO in 3NT formation, immunohistochemical studies were also performed in brain sections after CCI.
Methods
Mice
All experiments were approved by the University of Pittsburgh Institutional Animal Care and Use Committee and complied with the NIH Guide for the Care and Use of Laboratory Animals. Mice were given free access to food and water and were housed in laminar flow racks in a temperature-controlled room with 12-h day/night cycles. Mice in which iNOS was deleted by homologous recombination were backcrossed by at least 10 generations to the C57BL/6. These iNOS ko mice were used at 12 to 13 weeks of age (23 to 38 g). Wild-type C57BL/6 mice were purchased from Charles Rivers Laboratories (Washington, DC, USA) and matched for both age and weight to serve as controls for all studies.
Controlled Cortical Impact
A previously described mouse model of CCI was used (Whalen et al, 1999). Anesthesia was induced in male mice (12 weeks old, weighing 30 to 35 g) with 4% isoflurane in O2. Mice were then positioned in a stereotaxic frame, spontaneously breathing 2% isoflurane, 66% N2O, and 32% O2. A temperature probe was inserted through a burr hole into the left frontal cortex. A craniotomy was made over the left parietal cortex. Once a brain temperature of 37°C±0.5°C was reached, it was maintained at that level for 5 mins before a vertically directed CCI was delivered, using a 3-mm flat-tipped impounder with a depth of 1.2 mm and a velocity of 5.0±0.2 m/s. After injury, the bone flap was replaced, sealed with cement, and the scalp incision was closed, and the anesthetic was discontinued. Mice were observed for 30 mins in supplemental O2 and then returned to their cages. At 72 h after CCI mice were reanesthetized with 4% isoflurane in O2 via nose cone, perfused transcardially with ice-cold saline and brains were quickly removed, divided into ipsilateral and contralateral hemispheres and snap frozen in liquid nitrogen. For immunohistochemical studies mice were perfused with ice-cold saline and then 4% paraformaldehyde (PFA) at 72 h after CCI. Brains were left in 4% PFA until paraffin embedding process and sectioned with a microtome.
Electron Paramagnetic Resonance Measurments of Nitric Oxide
Model paramagnetic mononitrosyl iron complex of NO with diethyldithiocarbamate (DETC) (DETC—Fe—NO) was prepared by mixing an NO donor PAPANONOate (1 mmol/L) with FeSO4 (0.1 mmol/L) and DETC (0.5 mmol/L) in DMSO. For the detection of NO in brain by spin trapping technique, mice received intraperitoneal injections of DETC (500 mg/kg) in 0.2 mL of normal saline and iron sulfate (37.5 mg/kg) with sodium citrate (187.5 mg/kg) in 0.2 mL of normal saline 30 mins before decapitation. EPR measurements of the DETC—Fe—NO signals were performed from whole brain hemispheres on a JEOL-RE1X spectrometer under the following conditions: 9.078 GHz, microwave frequency; 10 mW, microwave power; 320.0 mT, center field; 50 and 20 mT, sweep width; 0.5 mT, field modulation; 2 mins, time scan; 0.3 secs, time constant; 77 K, temperature. Electron paramagnetic resonance spectra of several homogenized samples were also recorded at room temperature using instrumental settings as follows: 330.0 mT, center field; 15 mT, sweep width; 1 mT, field modulation; 100 mW, microwave power.
High-Performance Liquid Chromatography Assay of Ascorbate
Supernatants obtained after precipitation of proteins in brain homogenates by 10% trichloroacetic acid and sedimentation (2,000g × 10 mins) were utilized for high-performance liquid chromatography (HPLC) measurements. A Hypersil column (5 μm particle size, 4.6 × 200 mm, Hewlett Packard) and a mobile phase of 1:24 methanol—water adjusted to pH 3.0 by acetic acid at a flow rate of 1.0 mL/min was used. A Shimadzu LC-10A HPLC system was used with an LC-600 pump and an SPD-10A UV detector (detection was by absorbance at 264 nm).
Fluorometric Assay for S-nitrosothiol
The content of RSNOs in brain homogenates was determined using 4,5-diaminofluorescein (DAF-2), that specifically reacts with NO (but not with stable oxidized forms of NO such as NO X and NO2−, NO3−, nor with other reactive oxygen species) yielding fluorescent DAF-2 triazole (DAF-2T) (Kojima et al, 1998). The assay includes decomposition of nitrosothiols by UV-irradiation (>330 nm). Aliquots of brain homogenates were mixed with DAF-2 (5 μmol/L) in 2.5 mL phosphate-buffered saline (PBS) and exposed to UV-irradiation (15 mins) using an Oriel UV light-source (model 66002) (Oriel Instruments, Stanford, CT, USA) and a cutoff filter (Balzers, >330 nm). After UV-irradiation, aliquots of mixture were heated (80°C, 4 mins) and centrifuged at 14,000g, 5 mins. Fluorescence emission intensity of DAF-2T was determined at 515 nm with excitation at 495 nm (slits: excitation 1.5 nm, emission 5.0 nm) in 2.5 mL cuvettes using a Shimadzu RF-5301 PC spectrofluorophotometer (Kyoto, Japan). The data obtained were exported and processed using RF-5301 PC Personal Software. A standard curve was established using nitrosoglutathione as the standard.
Nitrotyrosine ELISA
Mouse brain homogenates were centrifuged at 10,000g for 5 mins and 3NT measurements were performed in the supernatants. Samples and standards were incubated in microtiter wells coated with antibodies recognizing protein bound and free 3NT (Cell Sciences, Norwood, MA, USA). Biotynilated second antibody, then streptavidine-peroxidase conjugate and tetramethylbenzidine were added to the wells in the subsequent steps. The enzyme reaction was stopped by addition of citric acid and the absorbance at 450 nm was measured using a plate reader (Molecular Devices, USA). A standard curve was obtained by plotting the absorbances versus the corresponding concentrations of standards.
Immunohistochemistry
Nitrotyrosine was detected immunohistochemically in brain sections through hippocampus as an indicator of the presence of peroxynitrite and other nitrosating agents, as previously described (Whalen et al, 1999). Endogenous peroxidase was quenched for 15 mins with 0.3% hydrogen peroxide in methanol. Nonspecific binding was minimized by incubating the sections in 3% normal goat serum in PBS for 1 h. Sections were then incubated overnight with 1:500 dilution of primary anti-nitrotyrosine antibody (Upstate Biotechnology, Lake Placid, NY, USA), and specific labeling was detected with a biotin conjugated goat antirabbit IgG and avidin—biotin peroxidase complex (Vectastain Elite ABC kit, Vector Laboratories, Burlingame, CA, USA). For 3NT and macrophage double staining, sections were incubated overnight with 1:500 dilution of primary anti-nitrotyrosine antibody (Upstate Biotechnology, Lake Placid, NY, USA) and 1:500 dilution of primary anti-macrophage antibody (F4-80, Serotec, Oxford, UK), then specific labeling was detected with a biotin-conjugated goat anti-rabbit IgG and avidin—biotin peroxidase complex (Vectastain Elite ABC kit, Vector Laboratories, Burlingame, CA, USA). For 3NT and MPO double staining, sections were incubated overnight with 1:200 dilution of primary anti-nitrotyrosine antibody (Upstate Biotechnology, Lake Placid, NY, USA) and 1:400 dilution of primary anti-MPO antibody (DakoCytomation California Inc., Carpinteria, CA, USA) then specific labeling was detected with a biotin-conjugated goat anti-rabbit IgG and sections were incubated with Fluorescein avidin D (green) or Texas Red Avidin D (Vector Laboratories, Burlingame, CA, USA) for 30 mins.
Assessment of Neuropatholgy
At 72 h after CCI, mice were anesthetized and perfused with 100 mL of 2% paraformaldehyde. Brains were removed and stored in 2% paraformaldehyde. Coronal brain sections (10 μm) approximately 2.6 mm posterior to bregma, for examination of the contused cortex and dorsal hippocampus structures, were used for analysis. This corresponds to the brain level immediately beneath the center of the impact tip. Sections were stained with cresyl violet. CA1 and CA3 neurons in dorsal hippocampus were counted. Only normal-appearing neurons (having a clearly defined cell body and nucleus) were counted. For TUNEL labeling, the method reported by Clark et al (1997) was applied. Sections were fixed in 10% neutral-buffered formalin for 10 mins at room temperature. Sections were then incubated in 3% Triton X-100 (Sigma, St Louis, MO, USA) in PBS at room temperature for 1 h, and placed in 3% H2O2 and 30% methanol in PBS for 20 mins. Sections were then incubated with 300 U/mL TdT and 20 nmol/mL biotin-16-dUTP (Boehringer Mannheim) in 1 mL 1 × buffer at 37°C for 90 mins, washed with PBS three times then washed in PBS containing bovine serum albumin (BSA, 0.5 mg/mL) for 5 mins. The slides were incubated in avidin—biotin complex (ABC standard kit; Vector Labs, Burlingame, CA, USA), and DNA strand breaks visualized in diaminobenzidine (DAB, Vector Labs). Slides were cover slipped for light-microscopic analysis.
Determination of Protein
Protein concentration in brain homogenates was determined by the Bio-Rad Protein Assay kit. A standard curve was established by addition of bovine serum albumin to Bio-Rad assay kit and protein content was calculated.
Statistical Methods
Data are expressed as mean±s.d. or median (range) where appropriate. Brain NO, 3NT and RSNO levels were compared between different groups using ANOVA with Tukey's posttest. Ascorbate loss between iNOS ko and wt mice after CCI was compared using t-test. Immunohistochemistry sections were assessed semiquantitatively by one of the authors (PMK) masked to the study groups. Number of TUNEL-positive hippocampal neurons was compared between groups by Mann—Whitney U-test.
Results
Assessment of Endogenous Nitric Oxide formation by Diethyldithiocarbamate—Iron
Chemically prepared DETC—Fe—NO complex has EPR signal with characteristic triplet hyperfine splitting on nitrogen atom of NO (AN=1.29 mT) and g=2.035 (Figure 1A, line 1). We used DETC—Fe as a spin trap to detect NO produced at 72 h after CCI. Mice were injected with DETC 30 mins before decapitation. Low-temperature EPR spectrum recorded from brain hemisphere represents a superposition of signals from DETC—Fe—NO complex and DETC—Cu complex (Figure 1A, lines 2 and 3). Electron paramagnetic resonance spectra from wt and iNOS ko mice were different only in the DETC—Fe—NO complex region. Wild-type mice had a greater amount of DETC—Fe—NO, since difference between signals from wt and iNOS ko mice gave a pure DETC—Fe—NO spectrum (Figure 1A, line 4). Relative amount of NO in samples was estimated based on the amplitude between peak at g=2.047 and trough at g=2.025, which have the least interference with DETC—Cu signal (Sjakste et al, 1999). NO production was dramatically enhanced after CCI versus uninjured mice (Figure 1B); two times greater NO level was detected in wt versus iNOS ko animals. Surprisingly an increase in NO levels was observed in both ipsilateral and contralateral hemispheres in wt mice at 72 h after CCI. Similar results were obtained using EPR spectroscopy of brain homogenates at room temperature—conditions, at which the DETC—Cu signal does not interfere with DETC—Fe—NO but the sensitivity of EPR measurements is greatly decreased.
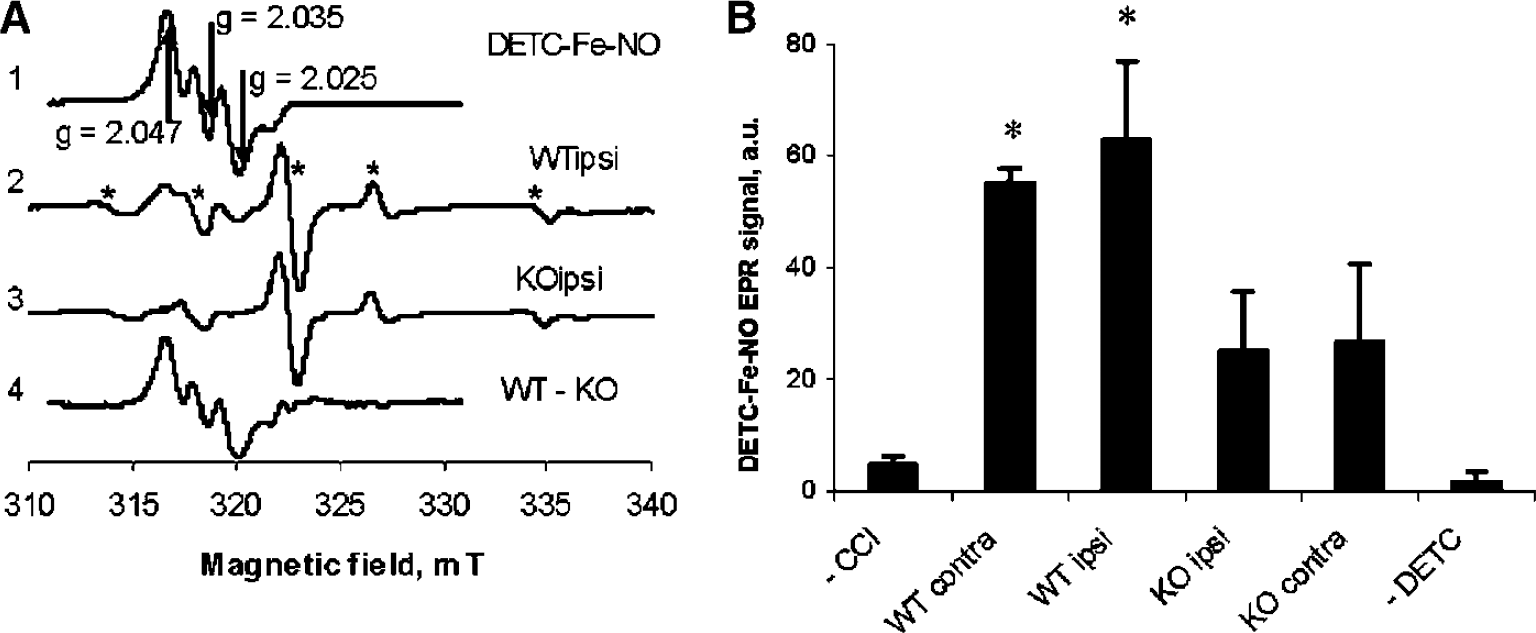
Detection of nitric oxide (NO) in mouse brain using diethyldithiocarbamate—iron (DETC—Fe) as a trap. Spectra of ipsilateral brain homogenates from wild-type (wt) and inducible nitric oxide synthase (iNOS) knockout (ko) mice, respectively, at 72 h after controlled cortical impact (CCI). (
Assessment of Oxidative Stress
To quantify oxidative stress after CCI in wt and iNOS ko mice, we measured brain ascorbate levels. Brain ascorbate levels were decreased in ipsilateral hemispehere in wt and iNOS ko mice versus normal values at 72 h after CCI (25.67±2.63 and 23.15±2.53 nmol/mg protein, respectively). The decrease in brain ascorbate content was more pronounced in iNOS ko mice compared with wt mice at 72 h after CCI (Figure 2). In contrast, ascorbate content in the ipsilateral brain of normal mice did not differ between wt and iNOS ko (38.65±1.99 and 33.41±3.24 nmol/mg protein, respectively).
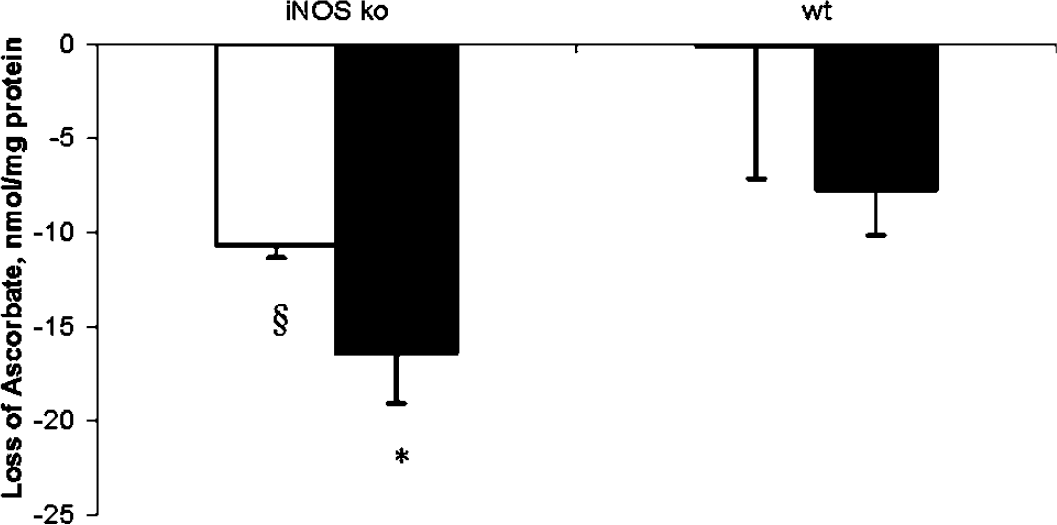
Amount of ascorbate loss in mouse brain at 72 h after controlled cortical impact (CCI). Brain ascorbate levels were decreased in wild-type (wt) and inducible nitric oxide synthase (iNOS) knockout (ko) mice versus normal values at 72 h after CCI. The decrease in brain ascorbate content was more pronounced in iNOS ko mice compared with wt mice at 72 h after CCI. Ascorbate content in the ipsilateral brain of uninjured mice were 38.65±1.99 and 33.41±3.24 nmol/mg protein in wt and iNOS ko mice, respectively. *P<0.05 iNOS ko ipsilateral hemisphere versus wt ipsilateral and contralateral hemispheres, §P<0.05 iNOS ko contralateral hemisphere versus wt contralateral hemisphere, ANOVA. (▪:ipsilateral hemisphere and □: contralateral hemisphere).
Assessment of Nitrosative Stress
To determine the content of RSNO in brain homogenates, we used fluorometric assay. This method has been shown to permit reliable and quantitative measurements of RSNO levels in plasma at concentrations as low as 50 nmol/L (Tyurin et al, 2001). We found that normal mice regardless of their genetic background in relation to iNOS, had detectable and comparable amounts of RSNO both in left and right hemispheres. However, at 72 h after CCI, total RSNO concentration was increased in wt but not in ko mice versus respective uninjured mice (Figure 3). Specifically, wt mice had ∼1.5-fold higher RSNO concentration in ipsilateral hemisphere versus wt uninjured (0.52±0.08 versus 0.33±0.04 nmol/mg protein). This increase in S-nitrosothiol after CCI was not seen in iNOS ko mice.
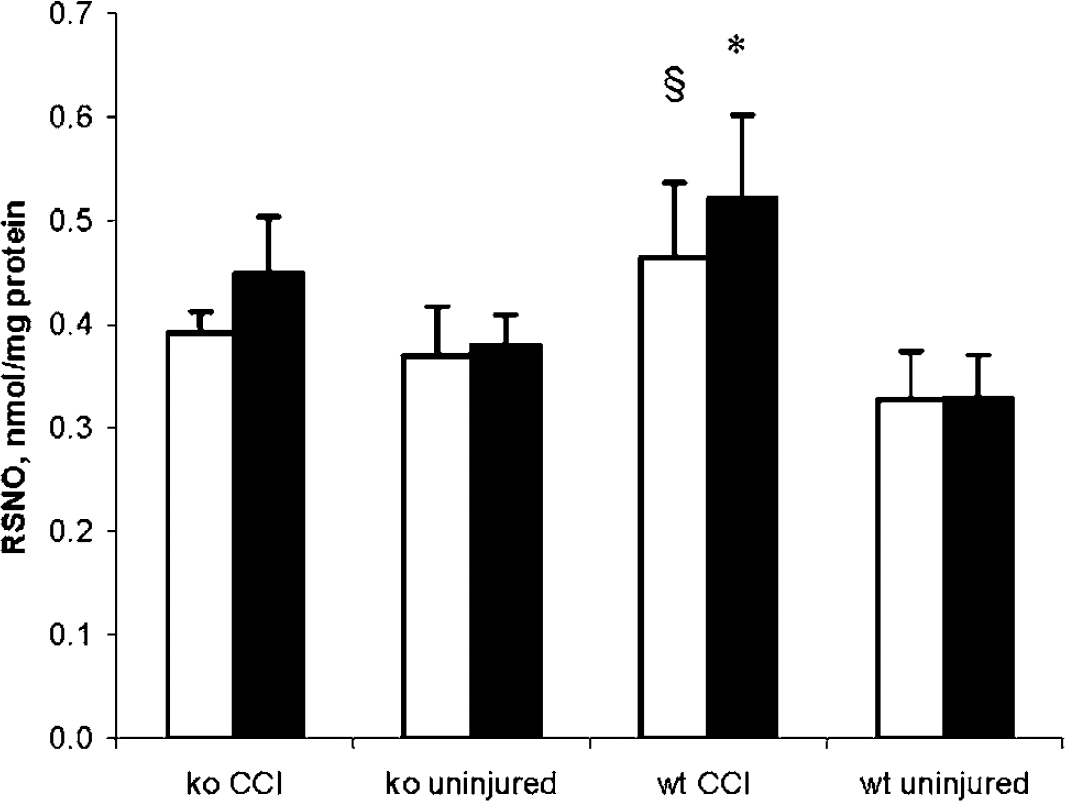
Content of S-nitrosothiol (RSNO) in mouse brain at 72 h after controlled cortical impact (CCI). Total RSNO concentration was increased in wild-type (wt) but not in knockout (ko) mice versus respective uninjured at 72 h after CCI. *P<0.05 wt CCI versus wt uninjured and inducible nitric oxide synthase (iNOS) ko uninjured, ANOVA. §P<0.05 wt CCI versus wt uninjured, ANOVA. (▪:ipsilateral hemisphere and □: contralateral hemisphere).
Next, we quantified 3NT levels in brain homogenates from wt and iNOS ko mice at 72 h after CCI. We found that uninjured mice, regardless of their genetic background in relation to iNOS, had detectable and comparable amounts of 3NT both in left and right hemispheres (Figure 4). 3NT levels were increased in both wt and iNOS ko mice at 72 h after CCI (P<0.05) in ipsilateral and contralateral hemispheres. However, the increase in wt mice was higher versus iNOS ko mice. Specifically, wt mice had ∼2-fold higher 3NT level in ipsilateral hemisphere versus iNOS ko mice at 72 h after CCI (2.24±0.95 versus 1.27±0.49 nmol/L, P<0.05).
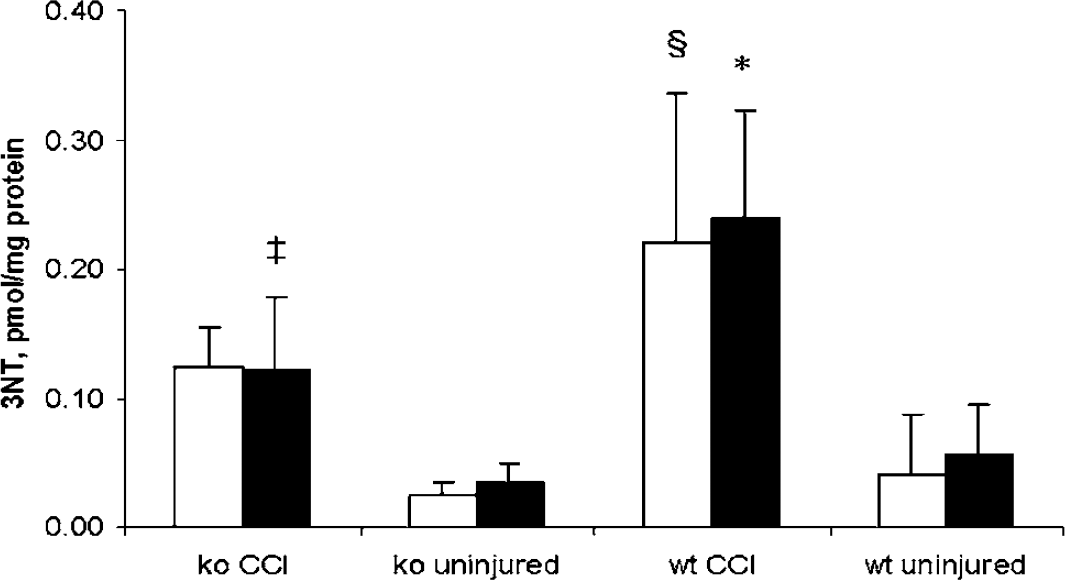
Concentration of nitrotyrosine (3NT) in mouse brain at 72 h after controlled cortical impact (CCI). 3NT levels were increased at 72 h after CCI. The increase was higher in wild-type (wt) mice versus inducible nitric oxide synthase (iNOS) knockout (ko) mice. ‡P<0.05 iNOS ko CCI versus iNOS ko uninjured, ANOVA. *P<0.05 wt CCI versus iNOS ko CCI, iNOS ko uninjured and wt uninjured, ANOVA. §P<0.05 wt CCI versus uninjured and iNOS ko uninjured, ANOVA. (▪:ipsilateral hemisphere and □: contralateral hemisphere).
Regional and Cellular Distribution of Nitrotyrosine Formation
Immunohistochemical detection of 3NT in brain at 72 h after CCI in representative wt and iNOS ko mice is shown in Figure 5. Corroborating the ELISA data, 3NT staining was present in both injured and uninjured hemispheres, but was most evident in the pericontusional area in wt versus iNOS ko mice. Cells with morphology of neutrophils and macrophages were positively stained. We further performed double staining for 3NT and macrophages in brain sections taken through hippocampus at 72 h after CCI (Figure 6). Many of the pericontusional 3NT-stained cells were also labeled with the macrophage marker (F4-80). Macrophages are known to contain MPO, which can form •NO2 resulting in nitration of the 3-position tyrosine residues in proteins. Supporting this, dual labeling revealed a close association between MPO and 3NT immunoreactivity in the pericontusional area, as the merged image shows (Figure 7).
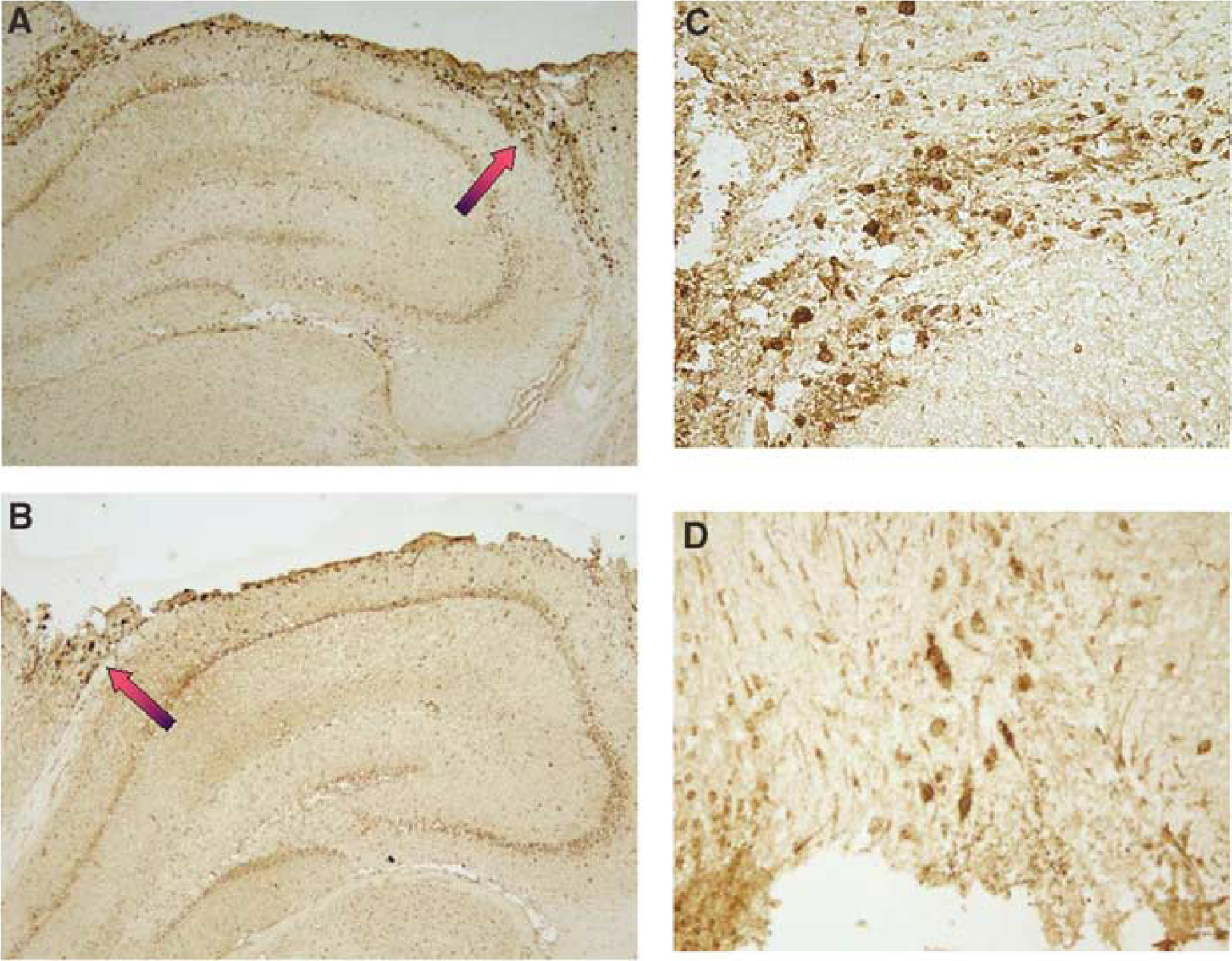
Nitrotyrosine (3NT) staining in mouse brain at 72 h after controlled cortical impact (CCI). (
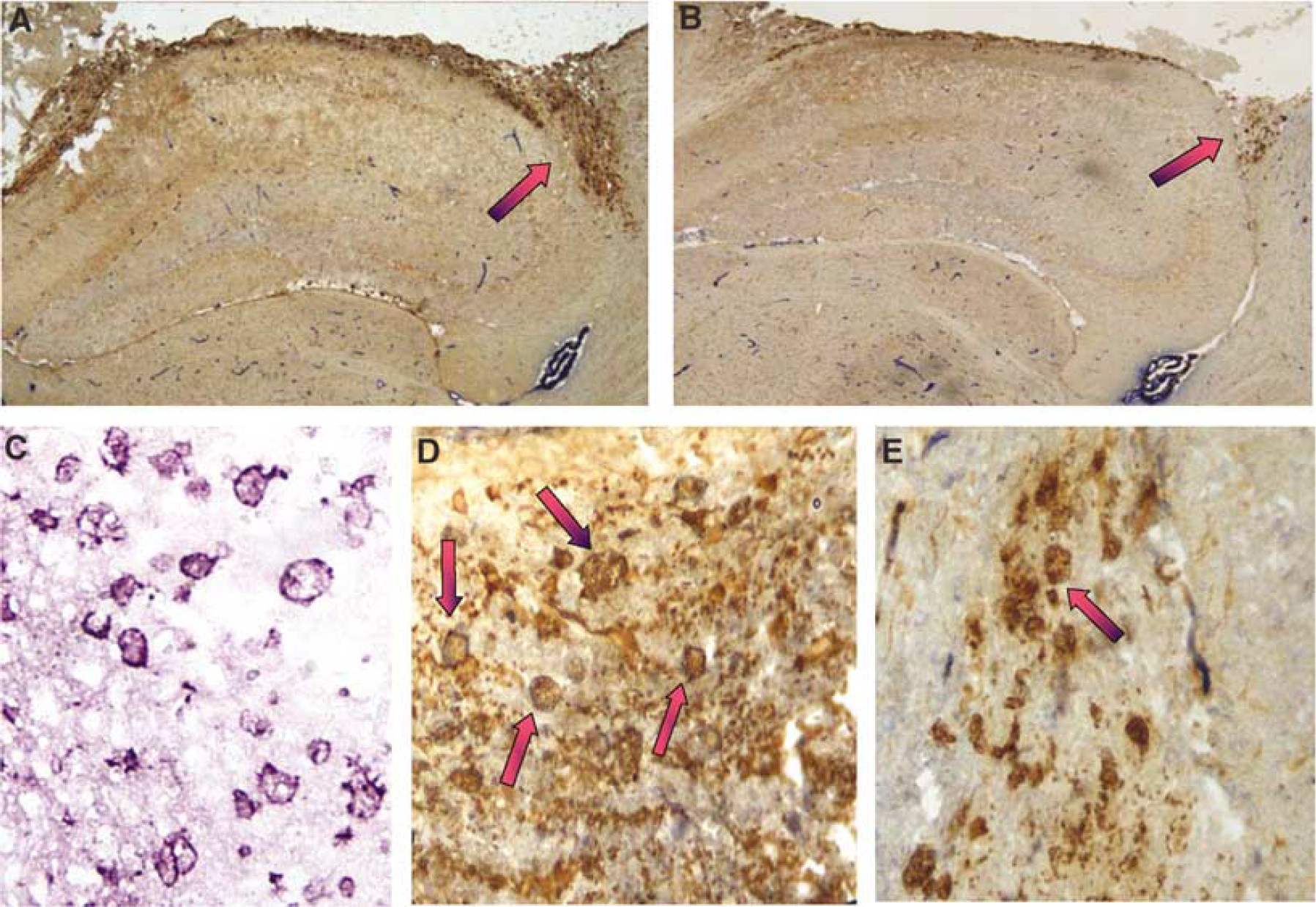
Nitrotyrosine (3NT) and macrophage double staining at 72 h after controlled cortical impact (CCI). (
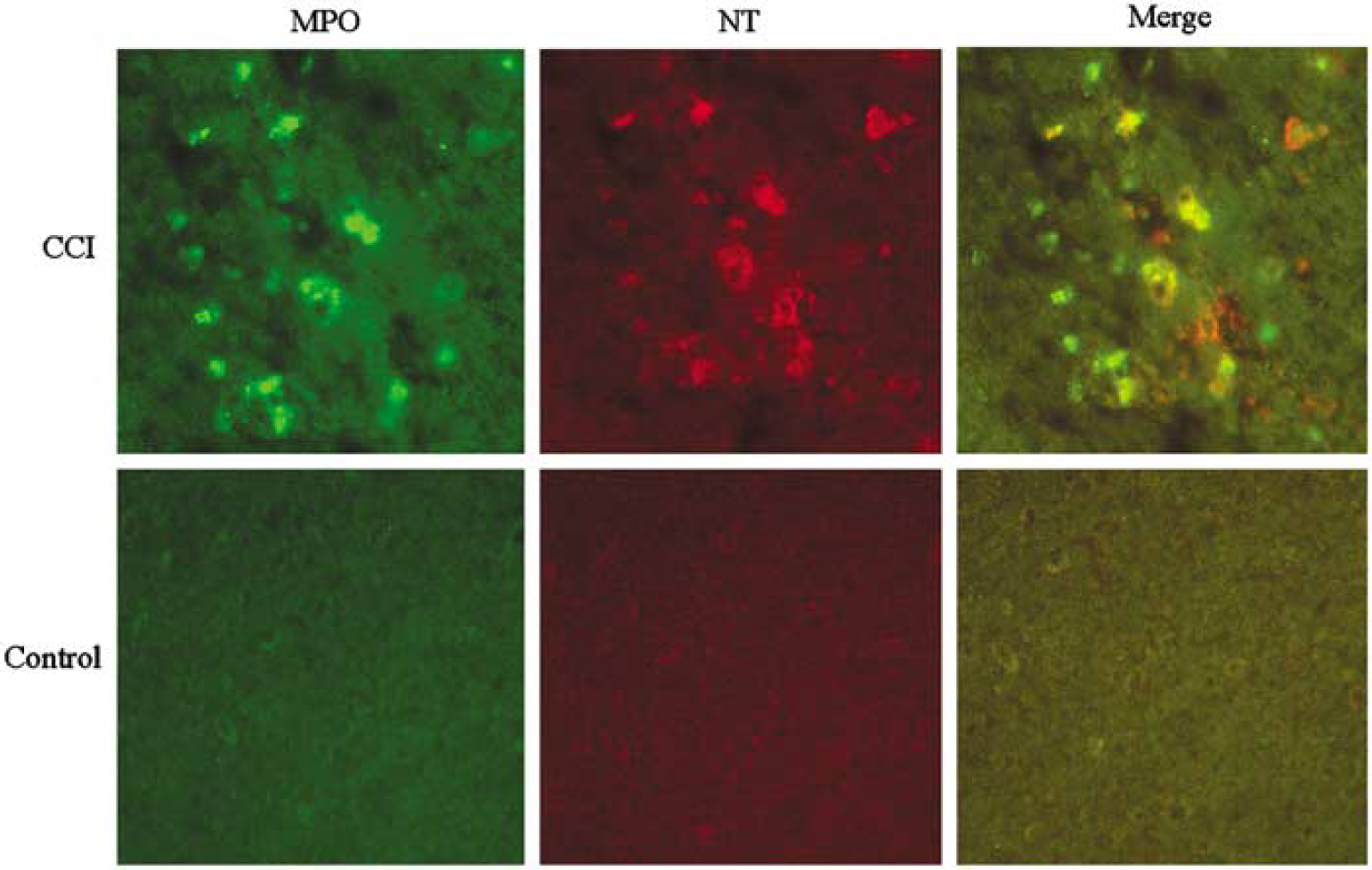
Nitrotyrosine (3NT) and myeloperoxidase (MPO) double staining at 72 h after controlled cortical impact (CCI). Representative sections through ipsilateral cortex (× 20) were dually stained with Ab versus MPO (green) and 3NT (red). Immunoreactivity shows colocalization of 3NT and MPO (yellow) in phagocytic cells at 72 h after CCI. Immunostaining was largely absent in sections from uninjured mice. Some 3NT immunostaining (orange) was also seen in parenchyma.
Assessment of Neuropathology
We found no difference in hippocampal neuronal cell counts between wt and iNOS ko mice at 72 h after CCI. This injury level breached the hippocampus in some of the mice precluding the assessment of hippocampal neuronal counts in those mice. The number of TUNEL-positive neurons was also not different between the two groups (17 (0 to 46) and 9 (6 to 31) for CA1 and CA3, respectively, in wt and 7 (0 to 30) and 7 (0 to 11) for CA1 and CA3, respectively, in iNOS ko mice, n=6/group, P=0.33 for CA1 and P=0.66 for CA3).
Discussion
The present results have, for the first time, defined the degree of contribution of iNOS to NO production, ascorbate depletion, RSNO, and 3NT formation and at a time point (72 h) when iNOS expression is at its peak level after experimental TBI. Our results show that NO production is increased in wt versus iNOS ko mice and ascorbate loss was greater in iNOS ko mice, supporting a role for iNOS-derived NO as an antioxidant after TBI. We also observed that RSNO levels are increased in wt but not in iNOS ko mice, and 3NT levels are markedly increased in wt and modestly increased in iNOS ko mice, supporting a role for iNOS, protein nitrosylation, and nitration after experimental TBI. Furthermore, our study showed that 3NT immunoreactivity is seen in the pericontusional area colocalized with macrophage marker and MPO suggesting that macrophage derived MPO may represent an important source for 3NT formation.
Effect of Inducible Nitric Oxide Synthase on Posttraumatic NO Levels Assessed by Electron Paramagnetic Resonance
We used EPR to directly quantify the amount of NO produced after TBI. Using the property that NO reacts with iron—sulfur complexes, Vanin et al developed the NO trapping method using DETC—Fe complex (Kubrina et al, 1992). The EPR study of NO—metal complexes has several decades of history behind its use (Drapier et al, 1991). However, NO trapping in traumatized brain tissue is a novel approach. Using EPR NO trapping technique, Tominaga et al (1994) found that the amount of trapped NO during forebrain ischemia increased up to seven times that of the control. The other direct method of showing NO production is using NO electrochemical probes. Using this technique, Cherian et al (2000) reported increased NO production early after CCI in rats; NOS inhibitors blunted this increase.
Effect of Inducible Nitric Oxide Synthase on Oxidative Stress after Traumatic Brain Injury
Our previous study showed that iNOS ko mice had worse long-term neurologic outcome and sustained inhibition of iNOS increased hippocampal neuronal death. Similarly delayed regeneration was observed after peripheral nerve injury in iNOS ko mice (Levy et al, 2001) suggesting iNOS-derived NO as a critical factor in the repair of injured tissue. The beneficial effects of NO in TBI are speculated to be secondary to a variety of mechanisms. Augmentation of cerebral blood flow after TBI by NO (Cherian et al, 2000) is thought to be one of them; however, evidence for other mechanisms is lacking. Our results show that in the absence of NO there is increased consumption of ascorbate suggesting a potent antioxidant role for iNOS-derived NO. NO reacts slowly with biological molecules (such as hemoglobin and other heme proteins, cellular thiols, and metals) but is very reactive with other free radicals, particularly with peroxyl radicals during lipid peroxidation in biomembranes. Thus, it can inhibit lipid peroxidation (O'Donnell et al, 1997). NO binds to iron complexes such as ferrous citrate and thiyl-iron, which in turn may reduce redox reactions, minimizing generation of reactive oxygen species (Rauhala et al, 1996). Central nervous system injury can lead to formation of free radicals and iron complexes (Hall and Braughler, 1982; Chan et al, 1987), thus NO may play an important role as an antioxidant after TBI.
We have previously shown that tissue concentration of ascorbate decrease after TBI (Tyurin et al, 2000) in rats. Similarly, marked and sustained decreases in CSF ascorbate concentration have been reported after clinical TBI in infants and children and, in adults (Bayır et al, 2002; Brau et al, 1984). Our current study confirms this finding in mice. Ascorbate acts as a primary defense against aqueous oxygen, nitrogen, and sulfydryl radicals. There is a high concentration of ascorbate in both the gray and white matter of the brain (Spector and Eells, 1984). Ascorbate is highly effective in trapping free radicals and preventing the formation of lipid hydroperoxides (Frei et al, 1989). Ascorbate is the primary antioxidant essential for protection of brain against TBI-induced oxidative stress. Even in the presence of Fe(II)/H2O2, DMPO—OH adducts in Electron paramagnetic resonance spectra are observed only after pretreatment of brain homogenates with ascorbate oxidase. Similar results were also shown for glutathionyl DMPO-adducts (Tyurin et al, 2000). Hence, monitoring ascorbate concentration and ascorbate radical production is regarded as a valuable method for the assessment of degree of oxidative stress in TBI.
Effect of Inducible Nitric Oxide Synthase on Nitrosative Stress after Traumatic Brain Injury
Our results also show that iNOS is responsible from increased formation of RSNO after TBI since the wt but not the iNOS ko mice had a 1.5-fold increase in RSNO levels after CCI. Another factor increasing steady-state RSNO concentration in biological fluids is the depletion of reductants such as ascorbate, which was decreased after CCI in both wt and iNOS ko mice. Ascorbate stimulates decomposition of RSNO via conversion of contaminating transition metal ions to their reduced forms (Xu et al, 2000). S-nitrosothiols may have direct neuroprotective effects after TBI by either sustained nitrosylation of NMDA receptor (Lipton et al, 1993) thereby decreasing excitotoxicity or nitrosylation of caspases (Mannick et al, 1999) leading to inhibition of apoptosis. We have previously shown that increased RSNO levels are associated with decreased intracranial pressure after TBI in infants and children, accordingly, RSNO—an NO sink—may attenuate cerebral edema leading to decreased ICP after TBI (Bayır et al, 2003). Additionally, accelerated RSNO breakdown has been implicated in pathopysiology of amytrophic lateral sclerosis supporting a beneficial role for RSNO in the CNS (Johnson et al, 2001).
Detrimental effects of iNOS after TBI are mostly thought to be secondary to peroxynitrite-mediated generation of •NO2 with resultant protein nitration. Hall et al, (2004) evaluated nitrosative (3NT) and oxidative (4-hydroxynonenal) stress by immunohistochemistry in a mouse model of diffuse closed TBI. 3NT immunoreactivity peaked in brain sections at 24 h and was sustained to 120 h. The time courses of 3NT and 4-hydroxynonenal immunostaining were similar, supporting a link between nitration and oxidative stress. Although a role for iNOS was implicated in that study, its contribution was not assessed. Our data using the CCI model in the iNOS ko mouse mirror these findings and support an important role for iNOS in nitration at 72 h. However, the magnitude of 3NT formation at 72 h after CCI in our model is substantially lower than in the initial 24 h (unpublished data) arguing against a critical role for iNOS in nitration early after injury.
Nitration of protein tyrosine residues by •NO2 results in diverse pathologies. This reaction adds a bulky substituent that decreases the pKa of the tyrosine hydroxyl group, altering protein structure and function. Protein nitration also results in cytoskeletal damage and disturbances in energy production and cell signal transduction (Schopfer et al, 2003). Important protein targets including actin, enolase,
Regional and Cellular Distribution of Nitrotyrosine Formation
Along with peroxynitrite, MPO in neutrophils and macrophages is implicated as a putative important source for highly reactive •NO2 generation resulting in wasteful consumption of NO and antioxidant (including ascorbate) depletion (Eiserich et al, 2002). In line with these data, our results show colocalization of 3NT immunostaining with a macrophage marker and MPO suggesting generation of nitrating agents by macrophages. However, we cannot rule out the possibility that nitrated proteins are phagocytosed by infiltrating macrophages. Neutrophils are the major source of MPO and neutrophil accumulation predominates in the initial 24 h after CCI (Clark et al, 1994). Macrophages have approximately 3-fold less MPO than neutrophils (Bos et al, 1978) and macrophages are abundant at 72 h after CCI (unpublished results), suggesting the possibility of a lesser role for MPO-mediated protein nitration at 72 h than 24 h after CCI. Further studies at earlier time points are needed.
Assessment of Neuropathology
We did not see any difference in hippocampal neuronal cell counts and TUNEL at 72 h between iNOS ko and wt mice. However, by 14 to 21 days, the iNOS ko mouse is markedly impaired functionally after injury versus the wt. Despite an obvious difference in function even at 14 to 21 days, we were not able to show a parallel detrimental effect using histopathologic assessments at 21 days in our model (Sinz et al, 1999). Our current studies were performed to examine a spectrum of biochemical pathways that are initiated in the subacute period. These studies (along with the aforementioned work of Zhu et al (2003)) are necessary to elucidate mechanisms responsible for the delayed beneficial effects of iNOS.
Limitations of the Study
There are several limitations to this study. Region-specific changes in ascorbate content and protein nitrosylation in the brain were not assessed. The 72 h time point was chosen to focus on the effect of iNOS on NO formation, oxidative and nitrosative stress during the period of peak expression after CCI. Since treatment with iNOS inhibitors early after injury has been shown to be beneficial (Wada et al, 1998; Lu et al, 2003; Stoffel et al, 2000, 2001; Gorlach et al, 2000), a comprehensive assessment of the time course of biomarkers of nitrosative and oxidative stress in iNOS ko and wt mice may be important for better temporal characterization of their potentially critical role. Further study is needed to identify the specific proteins posttranslationally modified by nitrosylation and nitration—along with identification of their consequences. Further studies with a comprehensive analysis of biochemical data as they relate to associated mechanisms of secondary damage and repair such as apoptosis (Clark et al, 1997), excitotoxicity (Ruppel et al, 2001), and inflammation (Kochanek et al, 2000) could also be revealing.
Conclusions
Our results show that NO levels are decreased and ascorbate loss is greater in iNOS ko mice versus wt mice suggesting a role for iNOS-derived NO as an endogenous antioxidant at 72 h after CCI, when iNOS induction is maximal. iNOS is responsible from RSNO formation at 72 h after injury, which could confer beneficial effects. iNOS also contributes to some of the increase in 3NT formation at 72 h after CCI in mice. Pericontusional 3NT immunoreactivity is increased in wt versus iNOS ko mice at 72 h and colocalized with macrophage marker and MPO, which could be because of the generation of nitrating agents by macrophages or result from active phagocytosis of nitrated proteins in an effort to remove them from sites of inflammation. Detrimental versus beneficial effects of NO are critically governed by timing, cellular localization, and linked to the level of oxidative stress after TBI.