
Abstract
Nitric oxide (NO) is implicated in both secondary damage and recovery after traumatic brain injury (TBI). Transfer of NO groups to cysteine sulfhydryls on proteins produces S-nitrosothiols (RSNO). S-nitrosothiols may be neuroprotective after TBI by nitrosylation of N-methyl-D-aspartate receptor and caspases. S-nitrosothiols release NO on decomposition for which endogenous reductants (i.e., ascorbate) are essential, and ascorbate is depleted in cerebrospinal fluid (CSF) after pediatric TBI. This study examined the presence and decomposition of RSNO in CSF and the association between CSF RSNO level and physiologic parameters after severe TBI. Cerebrospinal fluid samples (n = 72) were obtained from 18 infants and children on days 1 to 3 after severe TBI (Glasgow Coma Scale score < 8) and 18 controls. Cerebrospinal fluid RSNO levels assessed by fluorometric assay peaked on day 3 versus control (1.42 ± 0.11 μmol/L vs. 0.86 ± 0.04, P < 0.05). S-nitrosoalbumin levels were also higher after TBI (n = 8, 0.99 ± 0.09 μmol/L on day 3 vs. n = 6, 0.42 ± 0.02 in controls, P < 0.05). S-nitrosoalbumin decomposition was decreased after TBI. Multivariate analysis showed an inverse relation between CSF RSNO and intracranial pressure and a direct relation with barbiturate treatment. Using a novel assay, the presence of RSNO and S-nitrosoalbumin in human CSF, an ∼1.7-fold increase after TBI, and an association with low intracranial pressure are reported, supporting a possible neuroprotective role for RSNO. The increase in RSNO may result from increased NO production and/or decreased RSNO decomposition.
Although the primary event in traumatic brain injury (TBI) is direct disruption of brain parenchyma, a significant amount of damage results from a secondary cascade of biochemical, cellular, and molecular events (Kochanek et al., 2000). Secondary insults contribute to evolution of damage, protection, and repair, thus providing a variety of therapeutic targets.
Nitric oxide (NO) is implicated in both injury (Dawson et al., 1994) and neurologic recovery (Holscher, 1997) after brain injury. The location, timing, and amount of NO production may determine its role, as well as the type of injury sustained (Iadocola et al., 1995; Lipton et al., 1993). Nitric oxide is synthesized by at least three known isoforms of NO synthase (NOS). Nitric oxide produced by endothelial NOS (eNOS) provides benefit in models of TBI (DeWitt et al., 1997). Nitric oxide produced by neuronal NOS (nNOS) contributes to neuronal damage after ischemic or excitotoxic insults (Huang et al., 1994; Schulz et al., 1995). Nitric oxide derived from inducible NOS (iNOS) may contribute to either secondary damage (Wada et al., 1998a) or recovery (Sinz et al., 1999) after experimental TBI.
Transfer of NO groups to cysteine sulfhydryls on proteins results in the formation of S-nitrosothiols (RSNO). S-nitrosothiols are a class of chemical compounds that are widespread in vivo, thus providing a reliable source of NO. S-nitrosothiols have been detected in human plasma, erythrocytes, platelets, and leukocytes, and in rat cerebellum (Stamler et al., 1992; Kluge et al., 1997; Gaston, 1999; Kelm, 1999; Gladwin et al., 2000). Steady-state concentrations of RSNO are tissue specific and depend on physiologic state (Gaston, 1998, 1999). For example, endogenous levels of S-nitrosoglutathione in human airways are elevated during pneumonia and are depleted in asthmatics (Gaston et al., 1993).
S-nitrosylation may control activity of a variety of enzymes and regulatory proteins (transcription factors, G-proteins, ion channels, receptors, and kinases) (Konorev et al., 2000; Hogg, 1999), modulate energy metabolism (Clancy et al., 1994), and inhibit apoptosis (Mannick et al., 1999). Specifically, neuroprotective and antioxidant effects of RSNO have been described (Rauhala et al., 1996). One potential mechanism of neuroprotection by RSNO involves S-nitrosylation of free thiols at the redox modulatory site of the N-methyl-D-aspartate receptor (Lipton et al., 1993). Several biochemical pathways yield RSNO. They can be formed by direct recombination of NO with thiyl radicals (Murphy, 1999). Activation of NOS itself may form RSNO through intermediate generation of peroxynitrite and its interactions with thiols. Finally, protein nitrosylation by nitrites may also take place in cells (Gaston, 1999).
Decomposition of RSNO occurs via heterolytic (in the presence of transitional metals) as well as homolytic (photolysis) mechanisms, forming NO+, NO·, and NO− (Hogg, 2002). Reducing agents (such as ascorbate) stimulate decomposition of RSNO via conversion of contaminating transition metal ions to their reduced forms (Xu et al., 2000).
The major RSNO in human plasma is S-nitrosoalbumin. Albumin is an abundant circulating protein, which in its nitrosated form acts as a sink for trans-nitrosylation reactions (Heuil et al., 2000). Albumin is also a major antioxidant defense against oxidizing agents generated by either endogenous processes or exogenous agents (Halliwell, 1988). Albumin treatment may improve outcome after experimental and clinical TBI (Belayev et al., 1999; Tomita et al., 1994).
After TBI, increased formation of RSNO could be anticipated because of increased production of NO by NOS isoforms and increased formation of peroxynitrite. We reported increased iNOS expression within first 2 days in experimental TBI in rats (Clark et al., 1996b). Similarly, excitotoxicity mediated production of NO by nNOS has been implicated in both experimental and clinical TBI (Wada et al., 1998b; Bullock et al., 1998). Our group has also reported increased CSF levels of nitrate and nitrite in patients (Clark et al., 1996a) and increased nitrotyrosine formation—indicative of peroxynitrite production—in mice after TBI (Whalen et al., 1999). Another factor increasing steady-state RSNO concentration in biologic fluids is the availability of reductants such as ascorbate. We reported marked ascorbate depletion in CSF from children in the initial 7 days after severe TBI (Bayir et al., 2002). Thus, we hypothesize that RSNO and S-nitrosoalbumin can be detected in human CSF and increase after TBI.
In the present study, we measured steady-state concentrations of RSNO and S-nitrosoalbumin in CSF from patients with TBI and controls, with special emphasis on decomposition of S-nitrosoalbumin. We report for the first time the presence of RSNO and S-nitrosoalbumin in human CSF and ∼1.7-fold increase after TBI. Moreover, RSNO decomposition is decreased after TBI. Increased total CSF RSNO concentration is associated with low intracranial pressure (ICP) and barbiturate use.
MATERIALS AND METHODS
Patient selection and data collection
This study was approved by Institutional Review Board of Children's Hospital of Pittsburgh. All patients with severe TBI (Glasgow Coma Scale [GCS] score < 8) admitted to the Children' s Hospital of Pittsburgh are treated with a ventricular catheter insertion, and CSF is drained continuously to limit intracranial hypertension. Cerebrospinal fluid samples (n = 72) from 18 infants and children with severe TBI, collected at the time of intraventricular catheter placement and daily for 3 days, and 18 controls were studied. Control CSF samples were obtained from patients who underwent lumbar puncture to rule out meningitis. None of these patients had evidence of meningitis (as determined by negative cultures and lack of pleocytosis), TBI, hypoxia-ischemia, or seizures. Samples were centrifuged for 10 minutes at 5,000g and were immediately frozen at −70°C until the time of analysis.
Demographic and clinical parameters (age, sex, mechanism of injury [accidental vs. abuse], admission GCS, outcome [survival vs. death]) were recorded for each patient (Table 1).
Clinical and demographic characteristics

TBI, traumatic brain injury; MVA, motor vehicle accident; GCS, Glasgow Coma Scale.
Fluorometric assay for S-nitrosothiols
The concentration of RSNO in CSF samples was determined fluorometrically using 4,5-diaminofluorescein (DAF-2), which specifically reacts with NO (but not with other NOx, nitrite, or ONOO-) yielding DAF-2 triazole (DAF-2T) (Kojima et al., 1998). The assay included decomposition of nitrosothiols by ultraviolet irradiation (>320 nm). Aliquots of CSF (50 μL) were mixed with DAF-2 (5 μmol/L) in 2.5 mL phosphate-buffered saline (PBS) and exposed to ultraviolet irradiation (10 minutes) using an Oriel ultraviolet light source (model 66002; Oriel Instruments, Stratford, CT, U.S.A.) and cut-off filter (>320 nm; Balzers, Pittsburgh, PA, U.S.A.). After ultraviolet irradiation, aliquots of mixture were heated (80°C, 5 minutes). Fluorescent emission intensity of DAF-2T was determined at 515 nm with excitation at 495 nm (slits: excitation 5.0 nm, emission 5.0 nm) in 2.5-mL cuvettes using a Shimadzu RF-5301 PC spectrofluorophotometer (Shimadzu, Kyoto, Japan). The data obtained were exported and processed using Shimadzu RF-5301 PC Personal Software. A standard calibration curve was established by using S-nitrosoglutathione as the standard. The detection limit for RSNO was 50 nmol/L.
Determination of S-nitrosylated albumin by the biotin switch method
Because this is the first report on S-nitrosoalbumin in human CSF, to confirm the presence and micromolar concentrations of RSNO in CSF measured by fluorescent assay, we quantified CSF RSNO levels by an alternative independent method (biotin switch assay) in two randomly selected patients with TBI. NitroGlo Nitrosylation detection kit (PerkinElmer Life Sciences, Boston, MA, U.S.A.) with minor modifications (Jaffrey and Snyder, 2001) was utilized for this purpose. Briefly, protein (40 μg) was added to 100 μL HENS (250 mmol/L HEPES-NaOH, 1 mmol/L ethylene diamine tetraacetic acid, 0.1 mmol/L neocuproine) with 5 mmol/L A-ethylmaleimide (NEM). Samples were incubated at 50°C in the dark for 30 minutes to block the free thiols. Protein was subsequently precipitated by acetone and dissolve in 60 μL solubilization buffer provide by PerkinElmer, 3 μL of 50 mmol/L ascorbate and 20 μL of HPDP-biotin were added to switch the nitrosylated albumin to biotinylated albumin. Samples were incubated at room temperature for 1 hour. At the end of incubation, the same volume of sodium dodecyl sulfate polyacrylamide gel electrophoresis sample buffer (2x, nonreducing) was added to samples and immediately loaded onto the gel. Electrophoresed samples were transferred to nitrocellulose membrane for immunoblotting. Biotinylated proteins were detected with antibiotin mouse monoclonal antibody according the instructions of the NitroGlo kit. Based on the calibration curve generated by the use of known amounts of S-nitrosoalbumin, concentration of nitrosylated albumin in CSF was calculated.
Purification of albumin
The albumin-enriched fraction of CSF was obtained by affinity column chromatography (Travis et al., 1976). CSF (100 μL) was loaded on a HiTrap column (1 mL; HiTrap Blue; Pharmacia Biotech, Piscataway, NJ, U.S.A.) and was collected in two fractions. Fraction 1 (albumin-poor fraction) of CSF was collected in 5 mL of buffer A (50 mmol/L phosphate buffer, 0.1 mol/L KCl, pH 7.0). Fraction 2 (albumin-enriched fraction) of CSF was collected in 5 mL of buffer B (50 mmol/L phosphate buffer, 1.5 mol/L KCl, pH 7.0). The elution rate was 0.5 mL/min. After column chromatography, the amount of RSNO was measured in each fraction as described above. Patients with highest RSNO levels from both TBI and control groups were chosen for S-nitrosoalbumin analysis to maximize the yield of the reaction product in light of the demanding nature of this assay. The same samples were used for the RSNO decomposition analysis described in “Detection of Nitric Oxide Release.”
Polyacrylamide gel electrophoresis
Native polyacrylamide gel electrophoresis was performed for electrophoretic separation of proteins in CSF and CSF fractions by the method of Sambrook et al. (1989) using an 8% acrylamide gel. Gels were stained with Coomassie blue (R-250) and scanned using Fluor-S Multilmager (Bio-Rad, Hercules, CA, U.S.A.).
Quantification of protein
The protein concentration in the CSF samples and chromatographic fractions of CSF was determined by Bio-Rad Protein Assay Kit. A standard curve was established by addition of bovine serum albumin to the Bio-Rad kit, and protein content was calculated.
Detection of nitric oxide release from S-nitrosoalbumin in cerebrospinal fluid
First, NO release in a model system with known concentrations of S-nitrosoalbumin (1 μmol/L), ascorbate (40–200 μmol/L) and copper (1 μmol/L) in PBS was measured amperometrically using a Clark-type NO electrode (Iso-NO with 2-mm shielded sensor: World Precision Instruments, Sarasota, FL, U.S.A.). Second, ascorbate was replaced with CSF—as the reductant source—from control subjects and patients with TBI in the aforementioned system, and NO release was measured the same way. The applied method is a modification of a previously published procedure (Pfeiffer et al., 1998) using copper ions to cleave the S-NO bonds. Cerebrospinal fluid was diluted 1:20 in 50 mmol/L PBS and incubated at pH 7.4 and room temperature in a reaction chamber under continuous stirring. Changes in current output (pA) in response to administration of CuSO4 were recorded, and NO release was quantified by comparison to a standard curve constructed by addition of increasing concentrations of NaNO2 in nitrite-free water under reducing conditions (KI/H2SO4).
Preparation of S-nitrosoalbumin
Human albumin (hSA) (50 mg/mL) was initially treated for 45 to 60 minutes at room temperature in the dark with 1 mmol/L dithiothreitol in PBS supplemented with 640 μmol/L diethylenetriaminepentaacetic acid to reduce the cystein-34 thiol group (hSA-SH). After this reaction time, reduced albumin was extensively washed out (four times with distilled water, and three times with Chelex [Sigma Chemical; St. Louis, MO, U.S.A.]-treated PBS, pH 7.4) by centrifugal ultrafiltration (14,000g, 25 minutes, 4°C) by using Microcon centrifugal filter devices with a 30,000 Da cut-off filter (Millipore, Bedford, MA, U.S.A.). Albumin concentration was then determined by its absorbance at 279 nm (0.609 absorbance units per 1 mg/mL) and sulfhydryl groups at 412 nm (∈ = 13.6 mmol/L−1 cm−1) by using the Ellman's reagent. Nitrosylated human albumin (hSA-SNO) was prepared using trans-nitrosylation reaction and S-nitroso-glutathione (GSNO) as a donor (Ji et al., 1999). GSNO was prepared in the reaction of acidified NaNO2 with glutathione. Briefly, 100 mmol/L glutathione was mixed with 100 mmol/L of NaNO2 in 200 mmol/L HCl at room temperature (Park, 1988). pH was adjusted to 7.40 with NaOH, and the solution was used for S-trans-nitrosylation of hSA-SH as soon as possible (was not stored for more than 30 minutes at 0°C). For S-trans-nitrosylation, hSA-SH was mixed with 50 times excess of GSNO and the solution was incubated in the dark for 20 minutes at room temperature. To prevent S-glutathiolation of hSA, the reaction was stopped with NEM (4:1, mol NEM to mol hSA-SH), and the samples were immediately extensively washed as described above to remove low-molecular-weight components (Ji et al., 1999; Konorev et al., 2000; Hogg, 1999). The resultant hSA-SNO preparation was checked for the level of residual hSA-SH as well as hSA-SNO using the Ellman's reagent and DAF-2 assays, respectively, as described previously. The molar ratio of SH/hSA was <0.01 and the molar ratio of NO/hSA was −0.55.
Electron paramagnetic resonance spectroscopy measurements of ascorbate radical formation
Electron paramagnetic resonance measurements of ascorbate radical were performed using a JEOL-RE1X spectrometer (JEOL, Kyoto, Japan) at 25°C in gas-permeable Teflon tubing (0.8-mm internal diameter, 0.013-mm thickness; obtained from Zeus, Raritan, NJ, U.S.A.). The tube (∼8 cm long) was filled with 60 μL of CSF, folded, and placed in an open 3-mm internal diameter electron paramagnetic resonance quartz tube in such a way that all of the sample was within the effective microwave irradiation area. The spectra were recorded at 3,351 G, center field; 10 mW, power; 0.79 G, field modulation; 50 G, sweep width; 4,000, receiver gain; 0.1 second, time constant. Spectra were collected using EPRware software (Scientific Software Services, Bloomington, IL, U.S.A.).
Statistical analysis
Data are shown as mean ± SD. The difference between CSF RSNO levels between patients with TBI and controls was analyzed by one-way analysis of variance. Post hoc comparisons were made using the Student-Newman-Keuls test. Differences between CSF S-nitrosoalbumin levels, ascorbate radical signal intensity, and half-life between patients with TBI (day 3 only) and controls were analyzed by Student's two-tailed t-test. The association between age, GCS score, CSF collection time after injury, ICP, cerebral perfusion pressure, vasopressor requirement, barbiturate treatment, core temperature at the time of CSF sampling, and CSF RSNO level was assessed by multivariate and dichotomous analyses. Statistical significance was set as P < 0.05.
RESULTS
Patient demographics
Demographic and clinical parameters of patients are shown in Table 1. Age ranged from 4 months to 16 years in patients with TBI, and 1 month to 15 years in control subjects (P > 0.05). Mechanism of injury included both motor vehicle accidents and child abuse. Initial GCS score ranged from 3 to 7. Fourteen of 18 patients survived.
Content of S-nitrosothiols in cerebrospinal fluid
To determine the content of RSNO in CSF, we used fluorometric assay based on ultraviolet-induced decomposition of RSNO (Stamler et al., 1992) and subsequent detection of released NO with a specific reagent, DAF-2 (Kojima et al., 1998), which produces DAF-2T with a characteristic fluorescence emission spectrum. Figure 1 demonstrates that the detectable levels of S-nitrosothiols in CSF were significantly different from the background fluorescence response from DAF-2 and are comparable with the responses from the standards (S-nitrosylated albumin and nitrosoglutathione). This method has been shown to permit reliable and quantitative measurements of RSNO in plasma at concentrations as low as 50 nmol/L (Tyurin et al., 2001, 2002). We found that CSF contained 0.86 ± 0.04 μmol/L RSNO in controls. S-nitrosothiols concentration was higher in patients with TBI versus control (P < 0.05, analysis of variance) (Fig. 2). Specifically, patients with TBI had ∼1.7-fold higher RSNO concentration versus controls on day 3 (1.43 ± 0.11 vs. 0.86 ± 0.04 μmol/L).
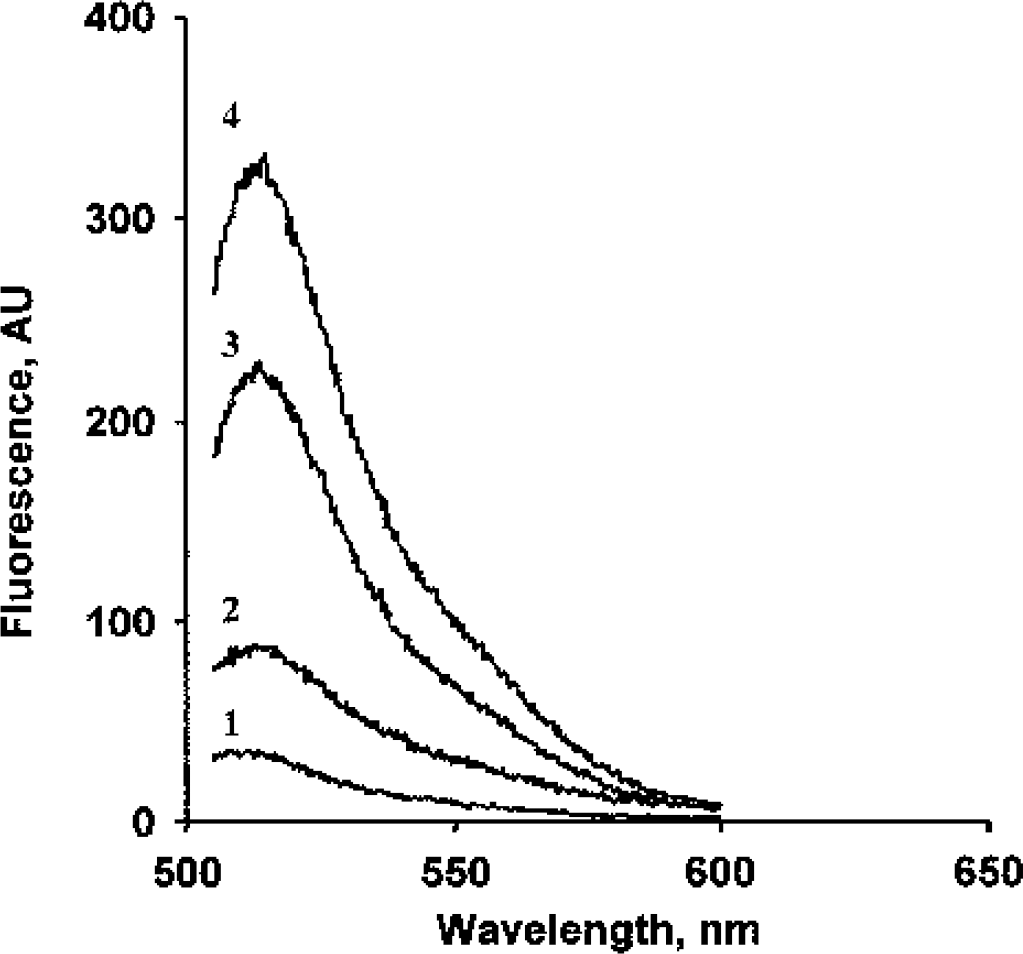
Typical fluorescence emission spectra of 4,5-diaminofluorescein triazole (DAF-2T) formed after ultraviolet-induced decomposition of S-nitrosothiols in the presence of DAF-2. Line 1 indicates DAF-2 alone; line 2, cerebrospinal fluid plus DAF-2; line 3, S-nitrosoalbumin plus DAF-2; and line 4, nitrosylated glutathione plus DAF-2. AU, arbitrary units.
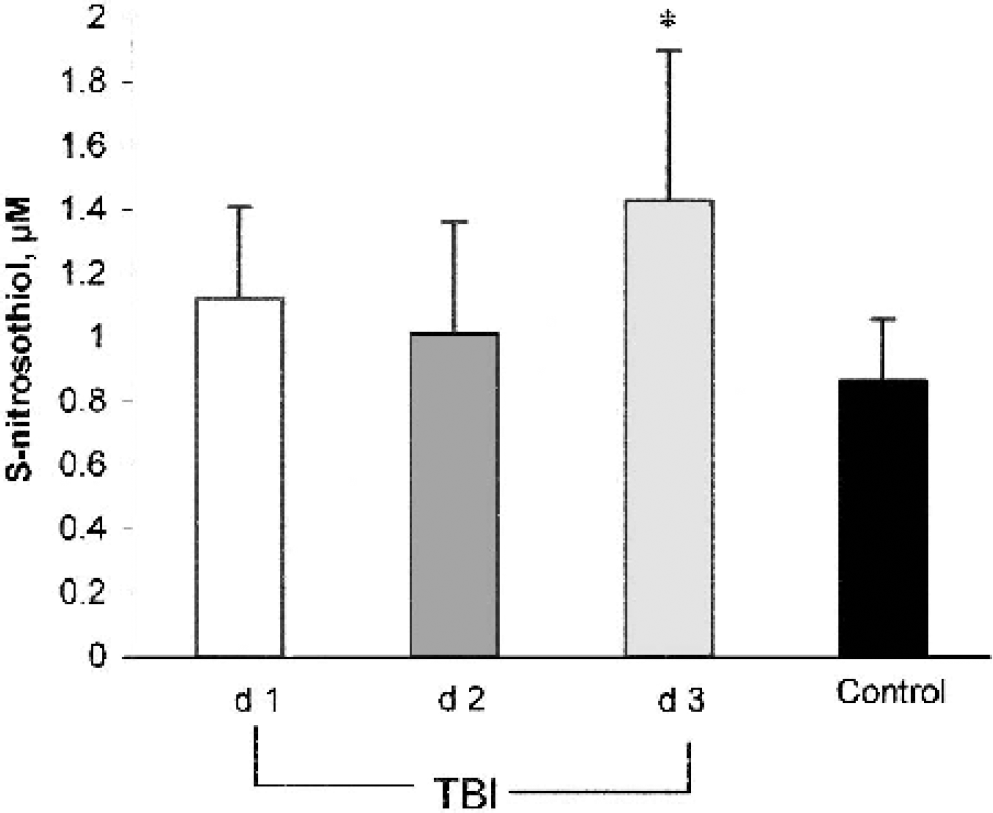
Concentration of S-nitrosothiols (RSNO) in cerebrospinal fluid (CSF) samples obtained from infants and children on days 1 to 3 after traumatic brain injury (TBI) and control subjects. Control CSF contained 0.86 ± 0.04 μmol/L RSNO. Total RSNO concentration was higher in TBI versus control subjects on day 3 after injury. (*P < 0.05, analysis of variance and Student-Newman-Keuls).
Content of S-nitrosoalbumin in cerebrospinal fluid
Because albumin is the most abundant protein in CSF (Illi et al., 1983) and the major source of S-nitrosylated thiols in plasma (Stamler et al., 1992), we generated albumin-enriched fractions of CSF in a subset of patients and determined the levels of nitrosylated thiols in them (n = 8, day 3 TBI and n = 6, control). Affinity column chromatography of CSF samples yielded two fractions (Fig. 3A). The first fraction was albumin free and the second fraction was albumin rich as evidenced by native polyacrylamide gel electrophoresis (Fig. 3B). In a separate series of control experiments, we showed that the recovery of RSNO from CSF fractions was 98% when compared with the direct measurements of RSNO in nonfractionated total CSF samples. The albumin fraction comprised 46% of total CSF RSNO in control samples and 59% in patients with TBI. Levels of S-nitrosoalbumin were −2.5-fold higher in the subset of patients with TBI versus controls. About 72% of the increase in total RSNO concentrations after TBI was detected as S-nitrosoalbumin (Fig. 4). Figure 5 represents the S-nitrosoalbumin concentration of 0.25 μmol/L from a patient with TBI on day 3, measured by biotin switch assay that is used as a confirmatory alternative method.
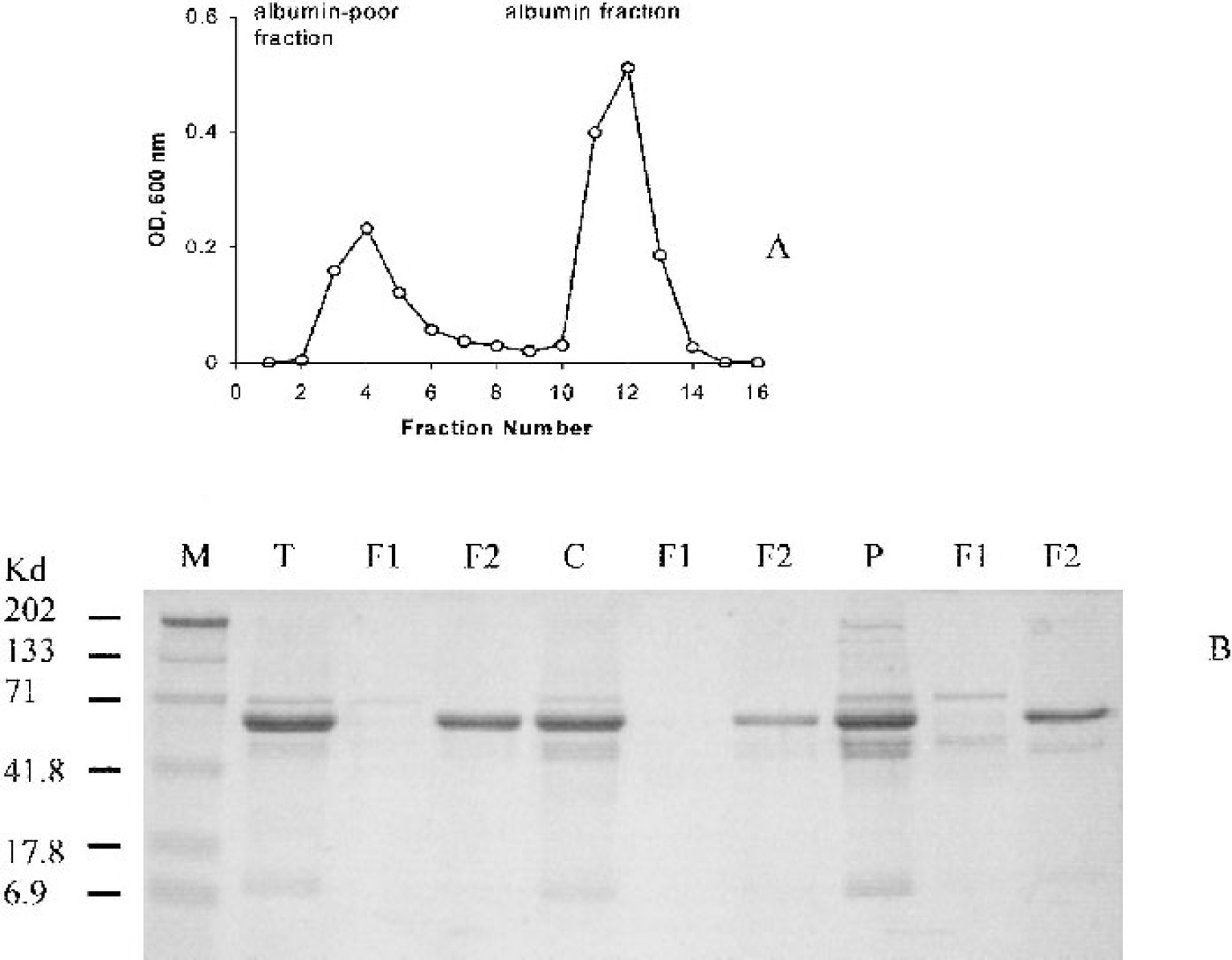
Typical affinity column chromatography of cerebrospinal fluid (CSF) illustrating isolation of albumin-poor and albumin-enriched fractions
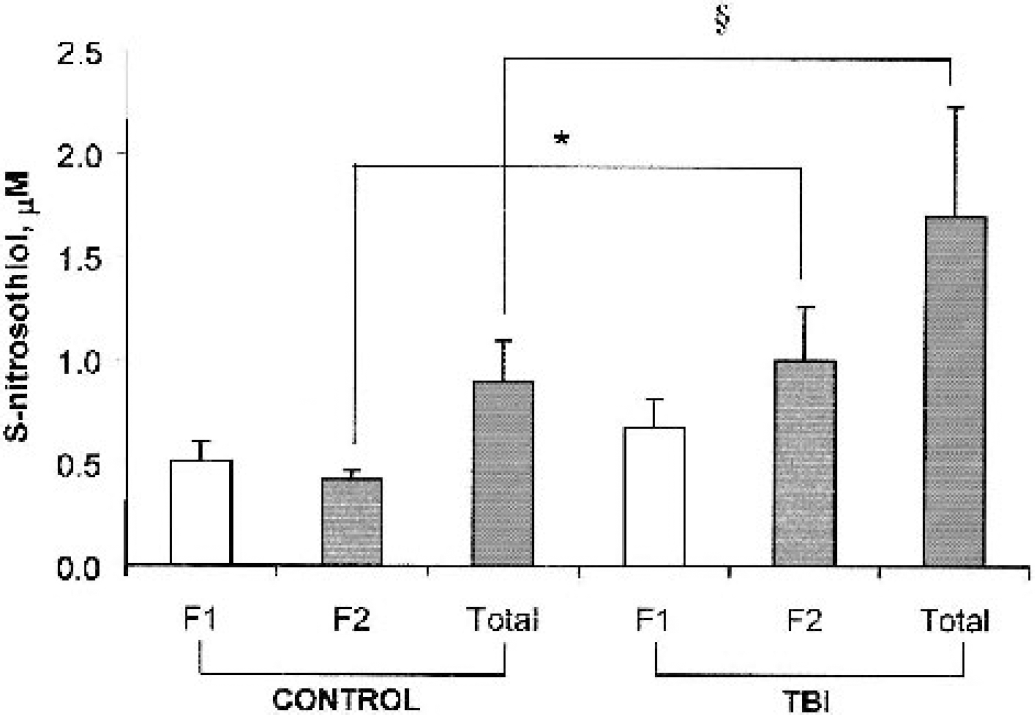
S-nitrosothiols (RSNO) in cerebrospinal fluid (CSF) fractions obtained from infants and children on day 3 after traumatic brain injury (TBI) and control subjects. F1, albumin poor fraction; F2, albumin fraction; total, total RSNO concentration. Levels of S-nitrosoalbumin were ∼2.5-fold higher in TBI versus control subjects. Albumin fraction formed 59% of total CSF RSNO in patients with TBI and 46% in control samples. (*Control F2 vs. trauma F2 P = 0.0019; §control total vs. trauma total P = 0.02, Student's two-tailed t-test.)

Determination of S-nitrosylated albumin in cerebrospinal fluid (CSF) by the biotin switch method. Lane 1 presents completely reduced albumin. Lanes 2, 3, 4, 5, and 6 are 250, 50, 10, 2, and 0.4 ng nitrosylated albumin, respectively. Lane 7 is normal plasma. Lane 8 is CSF from a patient with traumatic brain injury (TBI). Lane 9 is normal plasma after ultraviolet irradiation. Based on the calibration curve generated by the use of known amounts of S-nitrosoalbumin, concentration of nitrosylated albumin in CSF after TBI was calculated as 0.25 μmol/L.
Decomposition of S-nitrosothiols in cerebrospinal fluid
Electrochemical detection of NO release from S-nitrosoalbumin in PBS showed that addition of copper alone does not cause NO release. Presence of ascorbate and copper together, however, caused characteristic release of electrochemically detectable NO (Fig. 6A). Furthermore, when ascorbate in the system was replaced with CSF as the ascorbate source, NO release was greater in control (n = 6) versus patients with TBI (n = 8) (Figs. 6B and C), probably due to higher ascorbate concentrations in controls versus TBI (Bayir et al., 2002). Addition of ascorbate oxidase (10 U) to CSF samples from both the control subjects and patients with TBI completely abolished NO release from RSNO (Figs. 6D and E). To further demonstrate that ascorbate oxidation was occurring during decomposition of RSNO in CSF, we detected ascorbate radical formation by electron spin resonance spectroscopy (Figs. 7A and B). The intensity and half-life of ascorbate radical signals were greater in CSF from control versus TBI under these conditions (Figs. 7C and D). In addition, CSF ascorbate and RSNO levels were inversely correlated in patients with TBI (r2 = 0.495, P < 0.05) (Fig. 8). Ascorbate concentrations measured by high-performance liquid chromatography in each sample were previously reported (Bayir et al., 2002).
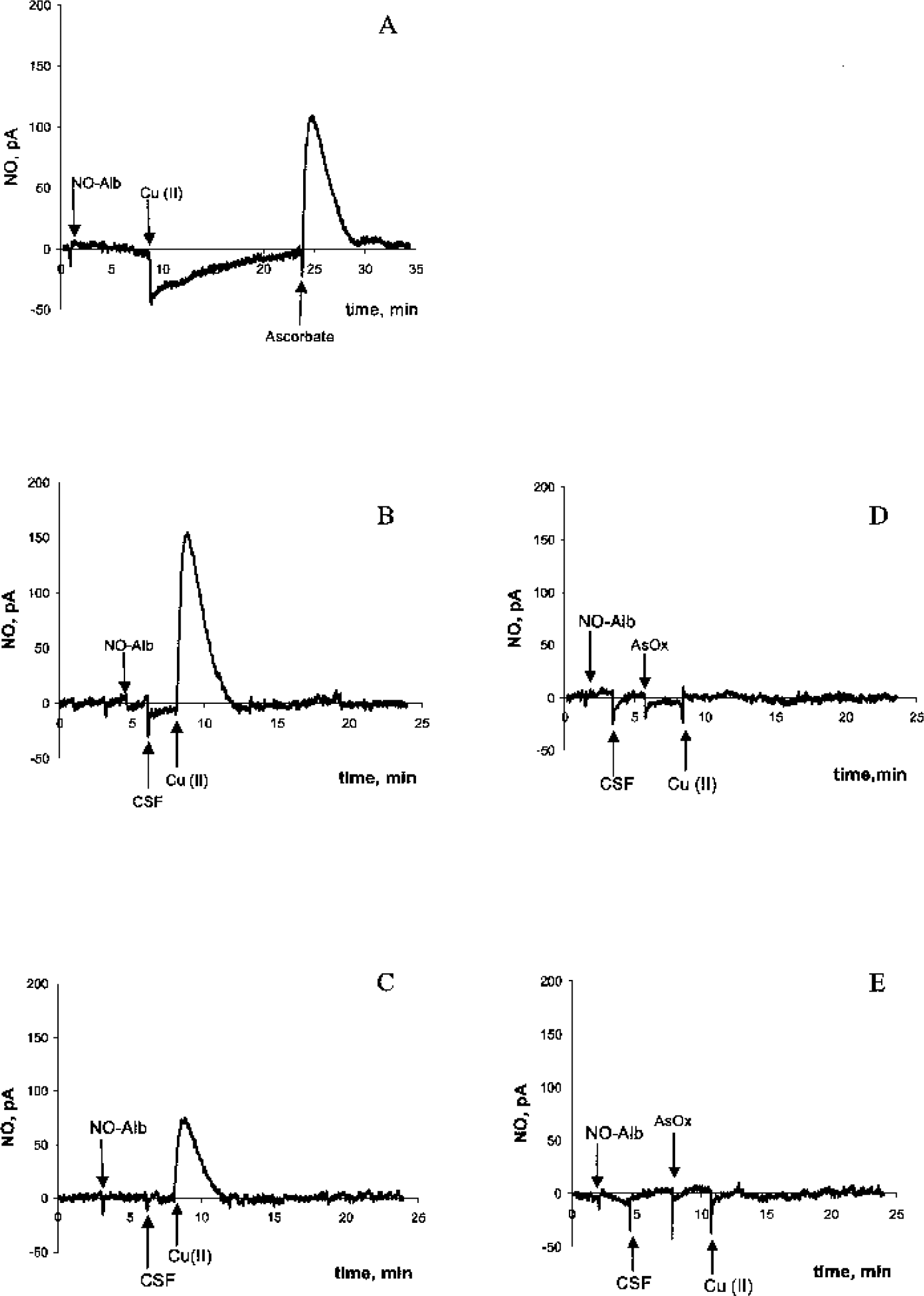
Typical examples of electrochemical detection of S-nitrosothiol (RSNO) decomposition in phosphate-buffered saline (PBS) showed that ascorbate was essential for nitric oxide (NO) release in the presence of low concentrations of copper
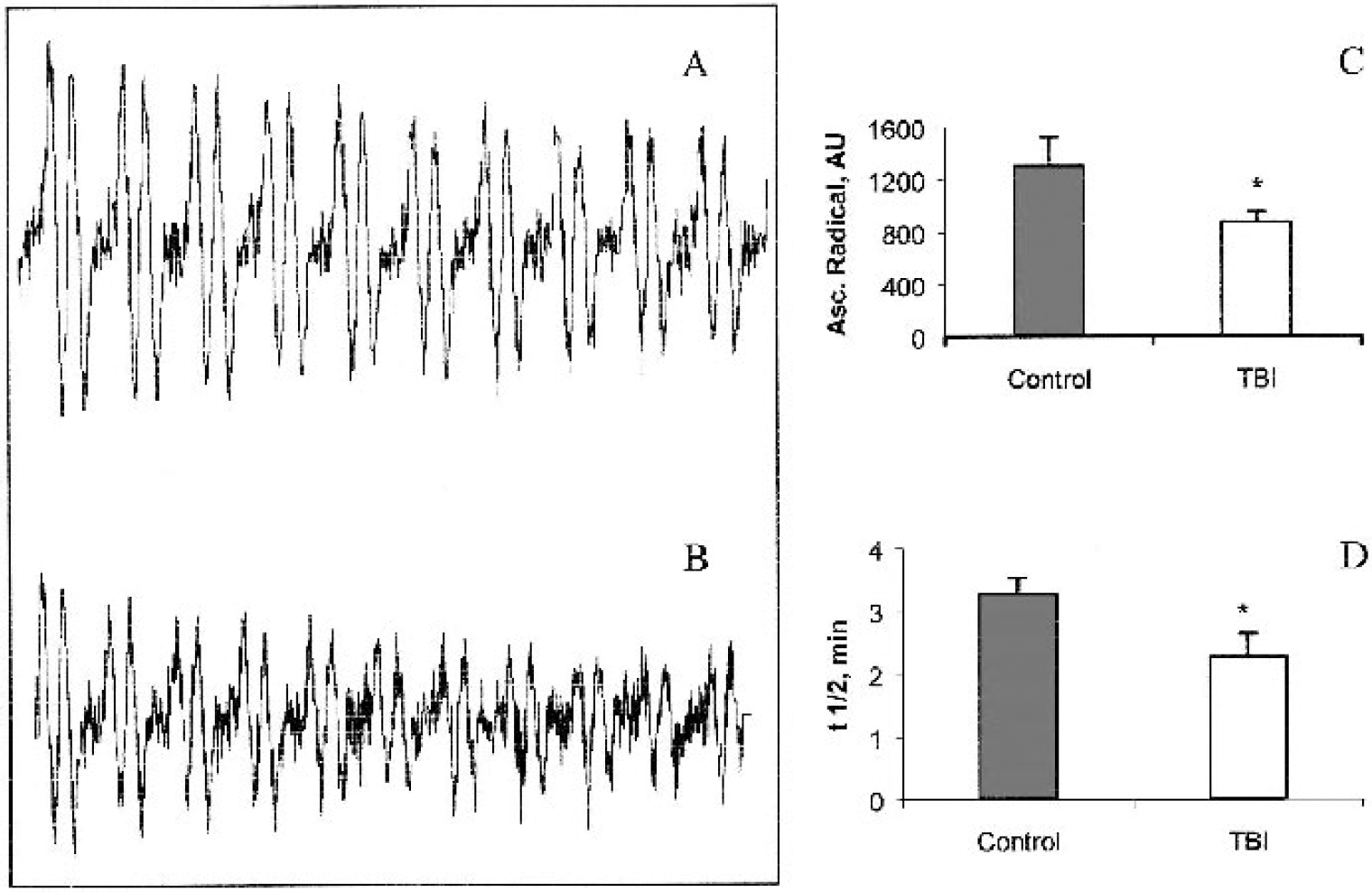
Demonstration of ascorbate oxidation during S-nitrosothiol (RSNO) decomposition. Ascorbate radicals are detected in controls
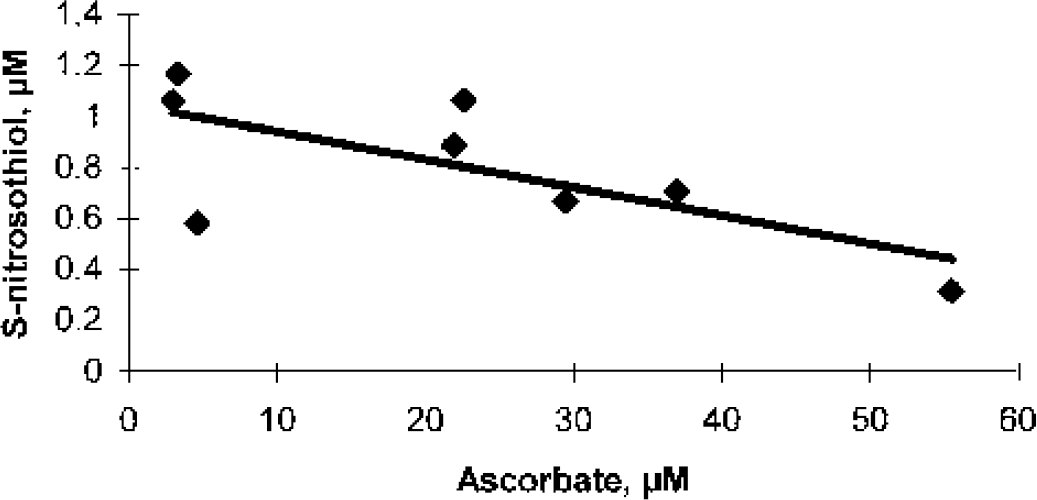
Cerebrospinal fluid ascorbate concentrations and S-nitrosothiol levels were linearly correlated in patients with traumatic brain injury (R2 = 0.495, P < 0.05).
Clinical correlations
Multivariate analysis of CSF RSNO concentration and physiologic variables at the time of sample collection revealed that ICP and CSF RSNO concentration were inversely associated (P < 0.05), suggesting the possibility that resolution or successful treatment of intracranial hypertension leads to increase in RSNO concentration. Barbiturate use for treatment of refractory intracranial hypertension was directly associated with CSF RSNO levels after TBI. Time after injury was directly associated with CSF RSNO concentration, suggesting gradual increase in RSNO levels after TBI. There was no association between mechanism of injury, age, outcome, and CSF RSNO concentrations (Table 2).
Multivariate effect of select variables on cerebrospinal fluid S-nitrosothiol levels
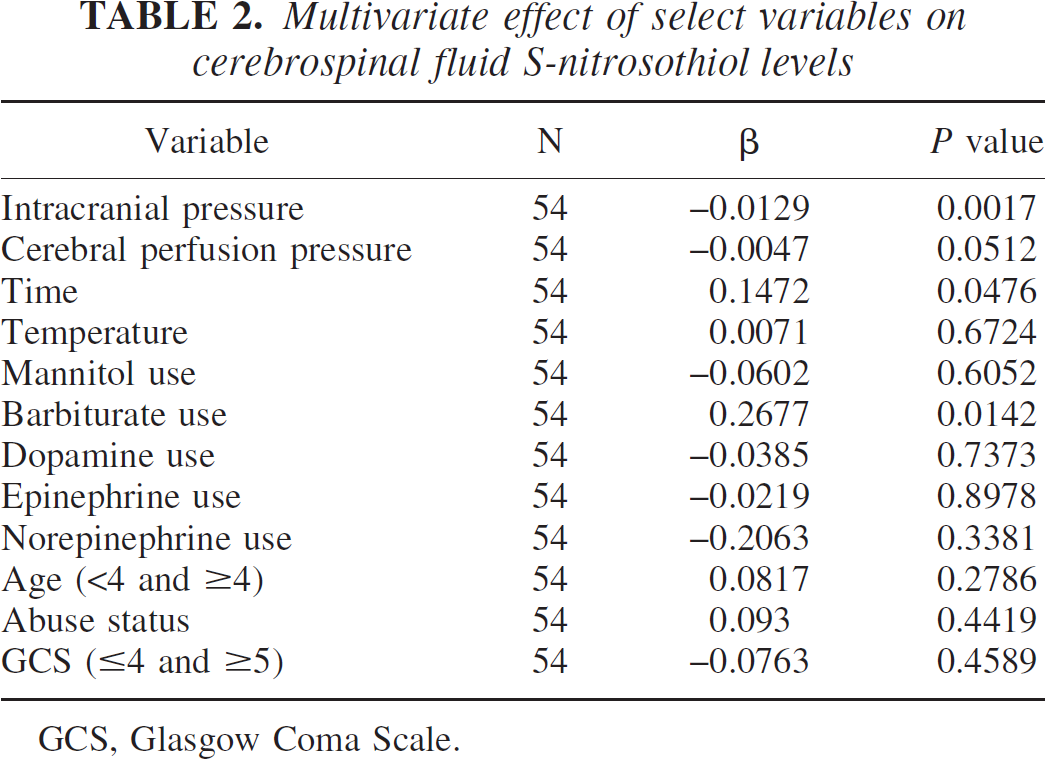
GCS, Glasgow Coma Scale.
DISCUSSION
Content of S-nitrosothiols in cerebrospinal fluid
We demonstrate, for the first time, the presence of RSNO and S-nitrosoalbumin in human CSF. Another RSNO, S-nitrosoglutathione, has been shown to be present in the rat cerebellum in 6 to 8 μmol/L concentrations by liquid chromatography-mass spectrometry (Kluge et al., 1997). We used a sensitive fluorescence assay for measurements of RSNO in CSF. The assay is based on a combination of a commonly used decomposition of RSNO by ultraviolet irradiation (Stamler et al., 1992; Marley et al., 2000), and subsequent capture of released NO by a fluoregenic reagent, DAF-2 (Kojima et al., 1998). 4,5-Diaminofluorescein was developed and introduced as a specific reagent for NO that does not react with NOx or peroxynitrite (Kojima et al., 1998). The sensitivity of our assay using DAF-2 to detect RSNO in CSF was 50 nmol/L.
Although the majority of the reported levels of RSNO in biologic fluids are in micromolar concentrations, their levels covering almost three orders of magnitude ranging from 20 nmol/L to 10 μmol/L have been reported (Marley et al., 2000; Stamler et al., 1992; Tyurin et al., 2001). To confirm the presence and micromoles per liter concentrations of RSNO in CSF measured by fluorescent assay, we quantified CSF RSNO levels by a method independent of photolysis, namely, a biotin switch assay, which showed CSF S-nitrosoalbumin concentration of 0.25 μmol/L (Fig. 5). To eliminate the possibility that nitrites and nitrates may be the source of DAF-positive response, we performed experiments using nitrites and nitrates in PBS. A fluorescence response with DAF-2 was generated only in nitrite and nitrate concentrations 20-fold higher than the levels reported for CSF after ultraviolet irradiation (V. A. Tyurin et al., unpublished observations, 2002).
We also report, for the first time, that CSF RSNO levels are ∼1.7-fold higher 3 days after TBI versus control. Physiologically relevant changes in endogenous tissue levels of RSNO are seen in other disease conditions such as pneumonia and asthma (Gaston et al., 1993). S-nitrosothiols have a wide range of biologic activities including vasodilation (Kowaluk and Fung, 1990), platelet inhibition (Simon et al., 1993), bronchodilation (Gaston et al., 1993), and antimicrobial effects (Morris and Hansen, 1981). The biologic effects of these compounds have been attributed to NO release with homolytic cleavage of S-N bond. Several studies, however, suggest that biologic activities of RSNO may not be solely caused by NO release. S-nitrosothiols can serve as intermediates in the activation of guanylate cyclase (Ignarro et al., 1981; Mellion et al., 1983; Keaney et al., 1993; Scharfstein et al., 1994). The heterolytic pathway of RSNO decomposition with resultant NO transfer between the RSNO and sulfhydryl groups of proteins may predominate in many biologic systems (Heuil et al., 2000).
Content and decomposition of S-nitrosoalbumin in cerebrospinal fluid
We also obtained albumin-free and albumin-enriched fractions of CSF and performed measurements of RSNO in these two fractions. We found that the albumin fraction contained 59% of total CSF RSNO in patients with TBI and 46% in control samples. About 72% of the increase in total RSNO levels after TBI was detected as S-nitrosoalbumin. Administration of albumin reduces histopathologic damage and improves neurologic outcome after experimental TBI (Belayev et al., 1999). Albumin-treated rats show less brain edema compared with vehicle-treated rats after transient cerebral ischemia (Belayev et al., 1998). In addition, administration of albumin normalizes metabolism-flow uncoupling that occurs after TBI (Ginsberg et al., 2001). Although additional clinical trials are needed, a randomized controlled trial showed decreased brain edema and improved neurologic outcome after cerebral contusion in patients treated with albumin (Tomita et al., 1994). Besides potential oncotic effects, albumin possesses numerous properties that may also contribute to its neuroprotective actions in CNS injury. It scavenges free radicals, binds free fatty acids and metal ions, and prevents related lipid peroxidation (Halliwell, 1988). The neuroprotective effects of albumin may also be due to its S-nitrosylation. S-nitrosoalbumin is the most abundant RSNO in human plasma (Stamler et al., 1992). S-nitrosoalbumin has endothelium-derived relaxing factor-like properties (Keaney et al., 1993) and inhibits platelet aggregation (Amano et al., 1994) and neutrophil adhesion to endothelium (Gluckman et al., 2000). Locally administered S-nitroso-bovine serum albumin bound preferentially to areas of vessel injury and inhibited intimal proliferation and platelet deposition after vascular arterial balloon injury in rabbits (Marks et al., 1995).
S-nitrosylation of albumin may play regulatory roles in the binding and transport of organic anions and heavy metals in circulation (Kashiba-Iwatsuki et al., 1997). Supporting this finding, S-nitrosylation of human serum albumin confers redox-cycling activity to its complex with copper (Y. Gryzunov et al., unpublished results, 2002). This in turn may result in oxidative stress and further depletion of reductants with subsequent decrease in decomposition of S-nitrosoalbumin. If the proposed pathway of events occurred in vivo, it could create a vicious circle with enhanced oxidative damage by redox active copper bound to nitrosylated albumin. Future studies addressing the putative damaging effects of S-nitrosoalbumin mishandling copper in experimental models of TBI are warranted.
Our S-nitrosoalbumin decomposition experiments clearly show that reducing properties of ascorbate and its one-electron oxidation, detectable as the formation of ascorbate radical, are essential to the decomposition of RSNO with resultant NO release in CSF. If RSNO were exerting their neuroprotective effects via NO release, the observed depletion of ascorbate would likely be detrimental in the setting of TBI. In this study, however, increased RSNO concentration was associated with low ICP and use of barbiturate therapy. Monitoring ICP and application of therapies (such as mannitol, barbiturate) targeting brain swelling are standard treatment modalities after TBI (Bullock and Povlishock, 1996). Cerebral swelling invariably develops and results from edema or increased cerebral blood volume, the first being the predominant mechanism of brain swelling, after TBI (Marmarou et al., 2000). Cerebral edema develops by one of three mechanisms: cellular swelling, blood-brain barrier (BBB) injury or osmolar swelling (Kochanek et al., 2001). Nitric oxide and peroxynitrite disrupt the BBB (Boje and Lakhman, 2000) leading to cerebral edema. In addition, scavenging of peroxynitrite has been reported to decrease both intracranial pressure and BBB leakage in experimental bacterial meningitis (Kastenbauer et al., 1999). Accordingly, increased RSNO—an NO sink—may attenuate cerebral edema, leading to decreased ICP after TBI. Additionally, increased RSNO levels may have direct neuroprotective effects after TBI by sustained nitrosylation of N-methyl-D-aspartate receptor (Lipton et al., 1993), thereby decreasing excitotoxicity or by nitrosylation of caspases (Mannick et al., 1999) leading to inhibition of apoptosis. Recently, accelerated RSNO breakdown has been implicated in pathophysiology of amyotrophic lateral sclerosis, supporting a beneficial role for RSNO in CNS (Johnson et al., 2001).
Clinical implications
Taken in light of the results of a number of prior studies by our group showing excitotoxicity (Ruppel et al., 2001), ischemia (Adelson et al., 1997), apoptosis (Clark et al., 1997), and oxidative stress (Bayir et al., 2002) after TBI in infants and children, our data support the following speculative synthesis. In the setting of excitotoxicity and ischemia early after TBI, the production of reactive oxygen species channel NO toward peroxynitrite as evidenced by nitrotyrosine positivity in contusions 24 hours after experimental TBI (Whalen et al., 1999). It is possible that RSNO increases as patients are recovering from excitotoxicity and intracranial hypertension. Induction of barbiturate coma for refractory intracranial hypertension acutely reduces brain interstitial glutamate levels, ischemia, and ICP (Goodman et al., 1996). Also consistent with this hypothesis, iNOS expression generally occurs in a delayed fashion in the contusions after experimental TBI (Clark et al., 1996b; Wada et al., 1998b), although studies in cold-induced injury have suggested more rapid iNOS expression (Stoffel et al., 2000). Similarly, peak nitrate and nitrite concentrations in human CSF are seen 30 to 42 hours after TBI (Clark et al., 1996a). Furthermore, iNOS appears to exhibit an important beneficial effect on long-term outcome in experimental TBI (Sinz et al., 1999). Specifically, iNOS knock-out mice exhibited extremely poor functional outcome versus wild type after TBI. The specific role RSNO play during recovery remains to be determined.
Limitations of study
Despite the small sample size and considerable variability in patient age, mechanism of injury, and treatment, statistical significance was achieved between injured and control subjects for all the measurements. In our study sample (n = 18 patients), 4 children died and 3 infants were victims of child abuse, limiting the analysis of the relation of RSNO levels to outcome and/or mechanism of injury. Nevertheless, this sample is very representative of our pediatric patient population with severe TBI. Further studies with a larger sample size including a comprehensive analysis of biochemical data as they relate to outcome, mortality, mechanism of injury (accidental trauma vs. child abuse), and age are needed. Assessment of the relation between RSNO and associated mechanisms of secondary damage such as apoptosis (Clark et al., 1997), excitotoxicity (Ruppel et al., 2001) and inflammation (Kochanek et al., 2000) could also be revealing. Traumatic brain injury is associated with BBB damage; there is a possibility that detection of RSNO in CSF is reflecting injury to the barrier with transfer of RSNO from blood to CSF. Experimental studies suggest that BBB disruption is maximal early after injury and then gradually recovers (Tanno et al., 1992). The RSNO levels demonstrated a biphasic time course, with peaks on day 1 and day 3 after TBI. The delayed peak in S-nitrosoalbumin on day 3 is unlikely to be explained by BBB disruption. In addition, the CSF albumin levels did not differ between patients with TBI and controls (0.37 ± 0.24 vs. 0.21 ± 0.07 μg/μL, P = 0.14, respectively), although they are dramatically higher in plasma (30–45 μg/μL). Taken together, these facts argue against substantial contribution of plasma RSNO to their CSF levels as a result of BBB disruption on day 3 after TBI. We recognize, however, that the contribution of BBB disruption to RSNO levels (particularly on day 1) cannot be completely ruled out by the results of this study.
CONCLUSIONS
S-nitrosothiols and S-nitrosoalbumin can be detected in human CSF and are increased after TBI versus controls. The progressive increase in RSNO in CSF may result from increased formation of peroxynitrite and increased production of NO by overexpression of iNOS or other NOS isoforms and/or decreased decomposition of RSNO because of depletion of reductants such as ascorbate. Increased RSNO in CSF is associated with low ICP and barbiturate use. Formation of RSNO, more specifically S-nitrosoalbumin, in CSF may attenuate nitrosative stress and serve as a reservoir facilitating transport of NO to sites distal to its production. Further studies are needed to evaluate the role of RSNO in CNS.