
Abstract
Despite the potential obstacle represented by the blood–brain barrier for extravasating malignant cells, metastases are more frequent than primary tumors in the central nervous system. Not only tightly interconnected endothelial cells can hinder metastasis formation, other cells of the brain microenvironment (like astrocytes and microglia) can also be very hostile, destroying the large majority of metastatic cells. However, malignant cells that are able to overcome these harmful mechanisms may benefit from the shielding and even support provided by cerebral endothelial cells, astrocytes and microglia, rendering the brain a sanctuary site against anti-tumor strategies. Thus, cells of the neurovascular unit have a Janus-faced attitude towards brain metastatic cells, being both destructive and protective. In this review, we present the main mechanisms of brain metastasis formation, including those involved in extravasation through the brain vasculature and survival in the cerebral environment.
Introduction
Brain metastases are life-threatening pathologies with limited therapeutic options, representing a major cause of death. 1 Although endothelial cells of brain capillaries are tightly interconnected, therefore difficult to penetrate, metastases occur 10 times more frequently than primary brain tumors in adults and have a prevalence of 8.3–14.3/100,000 persons. 2 The number of diagnosed brain metastases is constantly increasing partly because of the improved diagnostic techniques and partly due to better therapeutic possibilities targeting primary tumors and non-cerebral metastases, prolonging the life of patients, thus allowing tumor cells to disseminate into and proliferate in the brain.
Although several different cancer cell types can colonize the brain (renal, colorectal, ovarian, prostate, etc.), tumors originating from lung cancer, breast cancer and melanoma are the most common, representing 67–80% of metastases of the central nervous system (CNS). 2 Lung cancer accounts for 39–56% of brain metastases; non-small cell lung cancer (NSCLC), especially adenocarcinoma being the most frequent source of metastatic brain disease. 2 In addition, the brain is a common secondary tumor site for small cell lung cancer (SCLC). 3 The second most frequent cause of CNS metastases is breast cancer (representing 13–30% of the cases) 2 ; brain metastases occurring more frequently in triple negative (i.e. negative for estrogen receptors, progesterone receptor and Her2) and Her2 overexpressing mammary tumors. 4 Although much less prevalent than lung cancer or breast cancer, melanoma (responsible for 6–11% of brain metastases) 2 has the highest risk to spread into the CNS among all cancer types. 5 According to autopsy reports, approximately 75% of patients dying of melanoma have brain metastatic lesions. 6 Patients with BRAF or NRAS mutations are more likely to have CNS involvement 7 ; however, direct correlation between BRAF mutations and development of brain metastatic lesions is a question of debate. 8 Brain involvement – and generally metastasis formation – is an early event in melanoma and lung cancer and typically occurs late in breast cancer.9,10
The most frequent intracranial metastatic site is the brain parenchyma (cerebrum, cerebellum and brainstem), most commonly the cerebral gray matter–white matter border; however, the dura, the leptomeninges, the pituitary, the pineal gland, the choroid plexus and the ventricles can also be affected. 11 Brain metastases often occur in conjunction with extracranial metastases, of which lung metastases are the most frequent. Brain metastatic lesions are either single or multiple, the prevalence of these latter increasing from 39% in the 1980s to 71% between 2005 and 2009. 12 Brain secondary tumors present the tendency of having sharp borders; although infiltrative growth patterns have also been described with a variable prevalence (0–64%).13–16 The surrounding brain parenchyma is often edematous. The main symptoms are non-specific, like headache, vomiting, nausea, hemiparesis, visual changes and seizures.
Despite significant therapeutic advances in non-cerebral malignancies, management of brain metastases is still a significant challenge. Besides palliative treatments, surgery and radiotherapy (whole-brain radiotherapy and stereotactic radiosurgery) remain the first therapeutic choices. 17 In addition, chemotherapy, immune therapy and targeted therapy can be applied.18–20 Unfortunately, uptake of systemic agents is highly limited by the blood–brain barrier (BBB) 21 and brain metastases have an extremely poor prognosis. Therefore, development of new preventive and therapeutic strategies is urgently needed. This, on the other hand, depends on the expansion of our knowledge on the biology of brain metastasis formation.
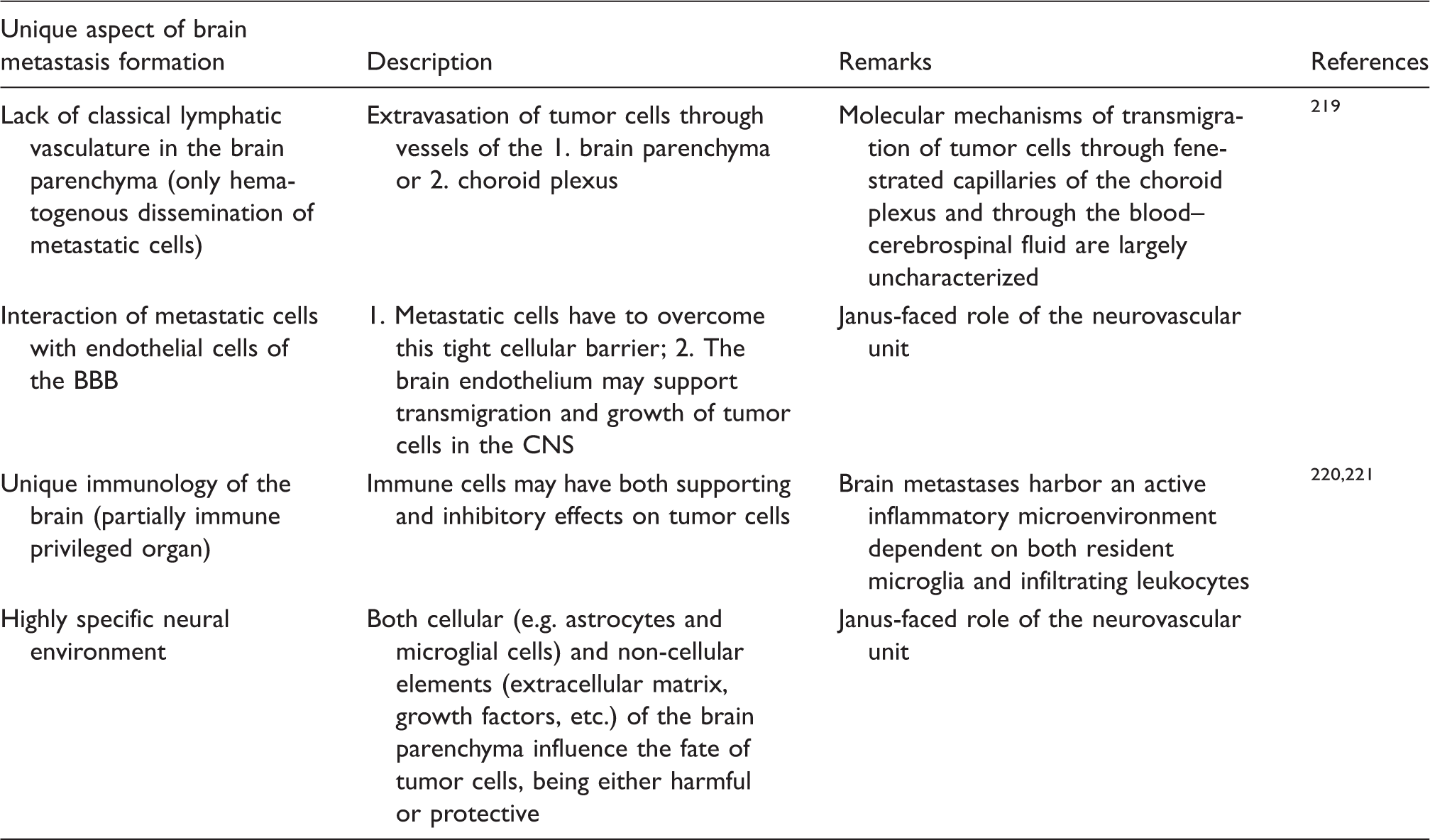
Unique aspects of brain metastasis development
Unique aspects of brain metastasis formation.
Model systems used for studying brain metastasis formation
The number of studies focusing on mechanisms of brain metastasis formation has been constantly increasing in the last few years. Main methodological approaches addressing this problem include in vitro studies, mouse models and analyses of clinical samples. Although translational relevance of data obtained in cell cultures is limited, in vitro models 26 can primarily differentiate between mechanisms involved in extravasation of tumor cells through the BBB and those responsible for survival and proliferation in the brain environment. In vivo models recapitulate the complexity of the human disease; however, they have limitations as well. Xenograft models (i.e. human tumor cells injected into immunocompromised mice) exclude the involvement of the full immune response, which might have a crucial importance. 27 Clinical relevance of results obtained in allograft (syngeneic) mouse tumor systems, on the other hand, is limited by interspecies differences between human and mouse. This might especially be important in melanoma, since mice rarely develop this disease. 28 Several results obtained in mouse models (mainly changes in the expression of certain proteins) were partly confirmed in patient samples. In addition, matched pairs of primary and metastatic tumor tissues were used to distinguish among genetic and epigenetic alterations driving formation of the primary tumor, those involved in general metastatic capacity of tumor cells and those required for the tropism of metastatic cells specifically to the CNS.
In the present paper, we comprehensively discuss mechanisms involved in brain metastasis formation, taking into account the relevance of different model systems in each aspect of the disease.
Mechanisms of extravasation through the BBB
Selective molecular characteristics of brain-seeking tumor cells
Structure of the microvasculature of the host tissue may have decisive roles in metastatic infiltration. In this respect, sinusoids or fenestrated capillaries might be more permissive than endothelial cells of the BBB interconnected by continuous tight junctions (TJs), 29 supported by pericytes and astrocytic endfeet. Therefore, metastatic cells might need to acquire specific characteristics to extravasate into the brain. However, several molecular aberrations found to be associated with brain metastasis formation can also mediate tumor spread to other metastatic locations, which is consistent with clinical data indicating that patients with brain involvement frequently have extracerebral metastases as well. For example, basal-like (mainly triple negative), less-differentiated and claudin-low (typically negative for claudin-3, -4 and -7) 30 breast cancer cells were found to exhibit a high probability to metastasize to the brain and lung, probably because these cells may initiate the metastatic cascade. 31 Similarly, cyclooxygenase COX-2 – an inducible cyclooxygenase – and the epidermal growth factor receptor (EGFR) ligand HB-EGF (heparin-binding EGF-like growth factor) mediate breast cancer metastatic infiltration of both the brain and the lungs. 32 COX-2 is also responsible for driving breast cancer cells from the parenchyma into the cerebrospinal fluid; these cells being able to move further to the systemic circulation to potentiate metastatic recurrence. 33
Molecular characteristics of tumor cells involved in their transmigration through the BBB.
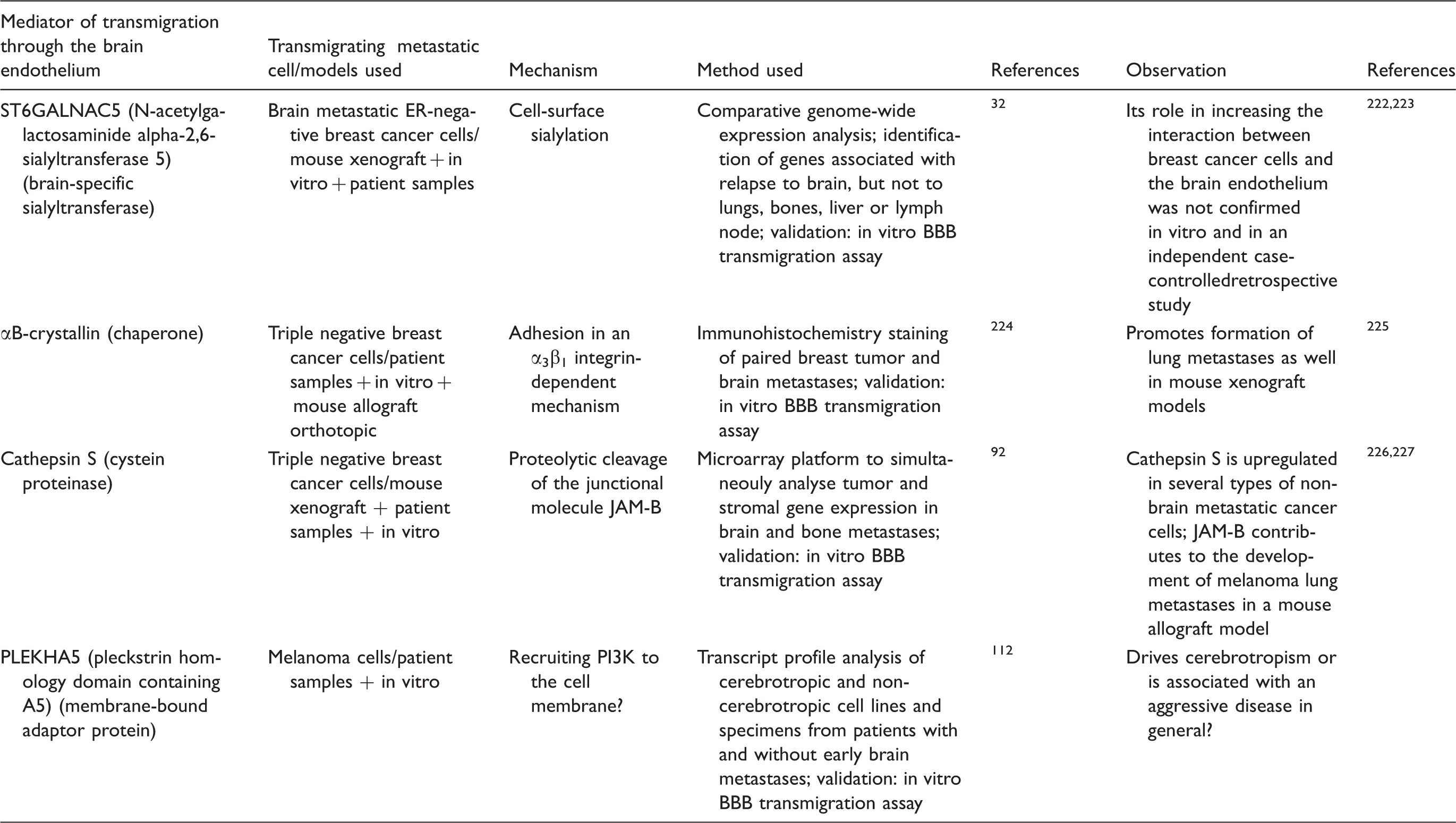
BBB: blood–brain barrier.
Molecular characteristics which differentiate tumor cells metastasizing to the brain parenchyma and those with high affinity to the meninges.
More and more data indicate that organotropism of tumor cells is primarily determined by released extracellular vesicles (EVs, mainly exosomes), which are taken up by organ-specific cells. By transforming resident cells, exosomes are able to prepare the pre-metastatic niche, facilitating metastasis formation of tumor cells. 34 Exosomes are equipped with adhesion molecules addressing them to specific organs; e.g. integrin expression profile of exosomes correlates with their tissue organotropism. 35 Exosomes carry several bioactive compounds, including microRNAs (miRs), protecting them from degradation and delivering them to distant sites.
Several recent studies have explored the role of miRs in mediating metastatic diseases. Brain metastases of breast cancer show reduced levels of miR-509 compared to primary tumors. Importantly, miR-509 suppresses transendothelial migration of tumor cells by blocking RhoC-induced matrix metalloproteinase-9 (MMP-9) expression and prevents TNF (tumor necrosis factor)-α-induced BBB opening. Therefore, downregulation of miR-509 might have a substantial role in the formation of brain metastases of breast cancer. 36 In contrast to miR-509, miR-181c promotes brain metastasis formation of breast cancer cells. Brain-seeking breast cancer cells release EVs containing miR-181c, which downregulates 3-phosphoinositide-dependent protein kinase-1 (PDPK1) resulting in cofilin dephosphorylation, modulation of actin dynamics and consequent breakdown of the BBB. 37 Moreover, breast cancer cells secreting EVs with high levels of miR-105, acquire greater metastatic potential through destroying endothelial barriers in the lungs and in the brain. 38 In addition, breast cancer stem-like cells (CSCs), which are highly metastatic to the brain, were shown to have reduced levels of miR-7, resulting in increased expression of Kruppel-like factor 4 (KLF4). Low miR-7 and high KLF4 expression was found in brain metastases of breast cancer and correlates with the ability of CSCs to migrate through the brain endothelium. 39 Downregulation of miR-7 and miR-509 and upregulation of miR-105 and miR-181c in brain metastatic breast cancer cells have been confirmed in in vitro BBB models and mouse xenografts.
In NSCLC brain metastases and those primary lesions which give rise to brain metastases miR-378 is overexpressed, contributing to the cerebrotropism of NSCLC cells. 40 In addition, combination of miR-328 and miR-330-3p was found to differentiate NSCLC patients positive and negative for brain metastasis. 41 MiRs have been exploited as prognostic and diagnostic biomarkers in melanoma as well. In a recent study, a molecular signature of four miRs (miR-150-5p, miR-15b-5p, miR-16-5p and miR-374b-3p) was found to greatly improve prognostic accuracy of brain metastasis development in primary melanoma. Among these, expression of miR-150-5p is likely to derive from infiltrating leukocytes and not from the tumor cells themselves, indicating the importance of immune response in controlling the disease. 42 Interestingly, two of these miRs (miR-150 and miR-15b) were identified as serum biomarkers for recurrence in melanoma.43,44
As a conclusion, several molecular markers determining transmigration of metastatic cells selectively through the BBB have been identified; however, many of these turned out to be responsible for metastasis formation in general (Table 2). Certain exosomal proteins and miRs are the most promising candidates to predict cerebrotropism of metastatic cells; however, further studies are needed to validate the data obtained so far.
Steps of extravasation of metastatic tumor cells through the BBB
Extravasation of malignant cells through the BBB consists of arrest in the brain vasculature, followed by adhesion to the luminal surface of CECs and finally transmigration through the endothelium (Figure 1). Recently, incorporation into the endothelial monolayer has been described as an intermediate step between adhesion and transmigration.
45
Extravasation of tumor cells through the BBB. Successful metastasis formation is dependent on arrest of tumor cells in the microvessels, followed by the adhesion and transmigration step. Extravasating tumor cells survive for days in the capillary lumen before transmigration is completed. During this process, tumor cells activate the Rac and PI3K signaling pathways and release TGF-β and proteases. CECs may also enhance transendothelial migration of metastatic cells through activation of COX-2 and secretion of MMP-2. Reactive astrocytes and microglia are recruited at initial steps of extravasation. Astrocytes may secrete cytokines, chemokines and proteases to enhance transendothelial migration of tumor cells. Microglia may also enhance invasion of the brain serving as transporters for malignant cells.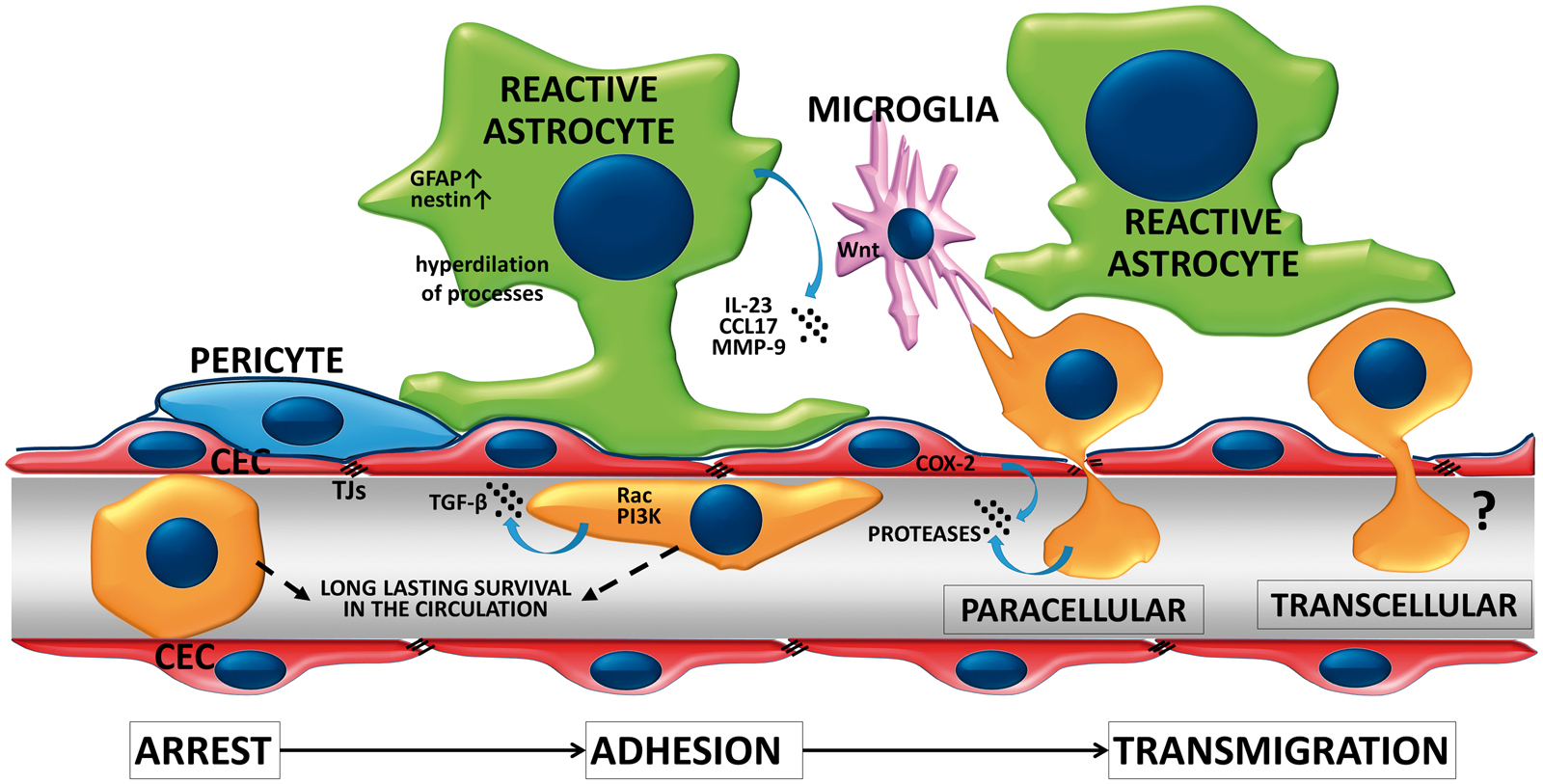
Arrest of tumor cells in the brain vasculature – which usually takes place at vessel branching points in capillaries and venules 58 – might depend on size restriction and complex hemodynamic conditions. In vitro, tumor cells preferentially tether to adhesive hot spots. 46 This initial step of extravasation implies build-up of tether/adhesion forces between tumor cells and the endothelium, which are determined by surface molecules (glycocalyx, adhesion molecules) and regulatory signaling pathways. Using single cell force spectroscopy, the total adhesion strength between melanoma cells and the brain endothelium was observed to be of approximately few hundred pN and composed of elementary units of roughly 20 pN in size. 47 The number of elementary events and the total tether/adhesion force was found to grow in the presence of ROCK (Rho-kinase) inhibitors. 48 Increase in the adhesion force between melanoma and brain endothelial cells in response to ROCK inhibition probably depends on flattening of melanoma cells (i.e. increase in the adhesive surface) and possibly on selectins, on the molecular level. Mechanical and molecular properties of the endothelial glycocalyx probably also strongly modulate the adhesion force; however, this aspect of metastasis formation has only been studied in non-cerebral endothelial cells. 49 Nevertheless, adherent NSCLC and breast cancer cells degrade the glycocalyx of CECs to expose adhesion molecules.50,51
Mechanisms of adhesion of tumor cells to microvascular endothelial cells are presumably partly similar to that of leukocytes52,53; however, much less known. Cancer cell extravasation depends on adhesion molecules expressed in tumor cells and on the luminal surface of CECs. As shown in mouse xenograft and allograft models, ALCAM (activated leukocyte cell adhesion molecule)/ALCAM and VCAM (vascular cell adhesion molecule)-1/VLA-4 (very late antigen-4, α4β1 integrin) interactions play a major role in breast cancer seeding to the brain. 54 Furthermore, both in vivo and in vitro models indicate that the ability of melanoma cells to cross the BBB depends on their high expression of melanotransferrin 55 or low expression of claudin-1. 56 In addition, according to data obtained in preclinical models, breast cancer and melanoma cells can also utilize gap junction proteins (connexin-43 and connexin-26, respectively) to initiate extravasation into the brain. 57
The time of extravasation – starting form arrest of the metastatic cell in the brain vasculature of mice until completion of transendothelial migration – is much longer in the brain than in other organs. The difference is given by the time spent inside the vessel lumen and not the transmigration itself. Therefore, successful brain metastasis formation depends on the ability of arrested tumor cells to survive for long time (at least two to three days) intravascularly,58,59 which seems to be a critical step only in the brain. Some surviving cells may start proliferating in the vascular lumen, serving as a sustained release source of tumor cells, 60 while others proceed to transmigration.
The transmigration step is completed within a few hours; however, aggressive melanoma cells can migrate through the brain endothelium in already 20 min, at least in vitro. 61 Theoretically, cells can either migrate through the endothelium by using the paracellular pathway (through interendothelial junctions) or the transcellular pathway (through the cytoplasm of endothelial cells) (Figure 1). Transcellular migration is well characterized for leukocytes,62,63 while in case of tumor cells, the majority of data refer to the paracellular pathway.
Melanoma cells are able to degrade TJ proteins and to disintegrate the junctional complex of CECs, which is indicative of paracellular transmigration. 64 Similarly, SCLC cells can also disrupt TJs of CECs 65 through release of placental growth factor which activates Rho and ERK (extracellular signal-regulated kinase) signaling. 66 In mammary tumor cells, β4 integrin expression mediates VEGF secretion, which enhances adhesion to the intercellular junctions instead of cell bodies 67 and promotes transmigration through altering brain endothelial integrity. 68 Besides in vitro data, in vivo results also suggest that VEGF contributes to brain metastasis formation of breast cancer cells 69 ; however, in these latter studies, the angiogenic effect of VEGF cannot be separated from its possible direct role on extravasation. Breast cancer cells can also induce TJ opening through secretion of the neuropeptide substance P, which activates CECs to secrete TNF-α and angiopoietin-2 (Ang-2), resulting in redistribution of TJ proteins. 70 Brain endothelial Ang-2 secretion can also be induced in response to the VEGF released by breast cancer cells, and both VEGF and Ang-2 contribute to increase in BBB permeability. 71 Therefore, breast cancer cells might open the TJs of CECs to extravasate paracellularly. However, recent in vitro data indicate that melanoma cells are more effective in breaking down the paracellular barrier than breast cancer cells, 61 while breast cancer cells might possibly be more effective in the transcellular type of migration. Nevertheless, transcellular migration of tumor cells has only been described for intravasating breast cancer cells,72,73 and not during extravasation into the brain. Therefore, the possibility of transcellular migration of tumor cells through the BBB needs further investigations.
Transendothelial migration of tumor cells may be facilitated by other cell types of the NVU. Reactive astrocytes – having increased expression of intermediate filament proteins – were described in close proximity to cancer cells already before extravasation. 58 Astrocytes may facilitate melanoma cell transendothelial migration through secretion of MMP-9, 58 the CCR4 ligand CCL1774 and IL-23 which upregulates MMP-2 in melanoma cells. 75 In addition, microglia – activated by metastatic breast cancer cells – enhance invasion of breast cancer cells through activation of JNK (c-Jun N-terminal kinase) in tumor cells. Microglia were observed to actively prepare the way for breast cancer cells to invade and colonize the brain tissue in a Wnt-dependent way. 76
Among non-cerebral cells, cancer-associated fibroblasts (CAFs) were shown to mediate BBB disruption and transmigration of breast cancer cells in in vitro models. 77 Indeed, circulating CAFs can be detected in the blood of metastatic breast cancer patients 78 ; however, their role in brain metastasis formation is largely uncharacterized. Similarly, the role of other circulating cell types, like neutrophils, macrophages and platelets might also be relevant, as shown in non-cerebral metastases.79–81
Proteolytic mechanisms involved in the transmigration of tumor cells through the BBB
Proteolytic enzymes secreted by both tumor cells and host cells might play key role in several steps of brain metastasis formation, including extravasation through the brain endothelium.
The best-studied proteases in the pathogenesis of cancer and metastasis formation are matrix metalloproteinases (MMPs). They are Ca2+-dependent Zn2+-endopeptidases which participate in many physiological and pathological processes in the brain and at the BBB, 82 including metastasis formation.83,84 Accordingly, increased serum MMP-9, but not MMP-2 lytic activities could be detected in patients with brain metastasis compared to healthy controls. 85 In addition, higher expression and activity of MMP-1 and MMP-2 were detected in brain metastasis variants of melanoma 86 and higher expression of MMP-1 and MMP-9 was found in brain-seeking breast cancer cells in comparison to bone-seeking and parental cells. 87 MMPs have the ability to degrade components of the endothelial glycocalyx 88 ; however, proteolytic degradation of the glycocalyx of CECs by tumor cells during brain metastasis formation has not been adequately addressed so far.50,51 The brain endothelial junctional complex is also a target of MMPs, and high expression of MMP-1 in brain metastatic breast cancer cells contributes to the degradation of key TJ proteins and opening of the BBB. 89
Proteolytic mechanisms might also be involved in opening of brain endothelial junctions induced by melanoma-released S100A4, 90 a Ca2+-binding protein dysregulated in many human cancers. 91 The mechanism of S100A4-induced VE (vascular endothelial)-cadherin downregulation may be chelation of Ca2+ or stimulation of MMP production in endothelial cells.
Besides MMPs, other types of proteases are also involved in brain metastasis formation, including the cysteine proteinase cathepsin S expressed by brain metastatic breast cancer cells 92 and serine proteases involved in transmigration of melanoma cells through the BBB. Melanoma cells express plasminogen activators (PAs), which catalyze proteolytic conversion of the inactive plasminogen to the active serine protease plasmin, which was found to mediate extravasation of melanoma cells into the brain. 93 In vitro, gelatinolytic serine proteases – including the membrane-bound seprase – were shown to be involved in the transmigration of melanoma cells through the BBB. 64 Seprase (fibroblast activation protein-α/FAP-α) expression correlates with the invasive phenotype of melanoma and carcinoma cells 94 and is transcriptionally upregulated in invasive melanoma cells via the canonical TGF (transforming growth factor)-β signaling pathway. 95
After transmigration through the brain endothelium, breast cancer and melanoma cells migrate along the external surface of brain vessels to distant sites, but remain perivascular.60,96 Proliferation of metastatic cells in the brain starts along the vessels, the basement membrane acting as an active substrate for tumor cell growth. Interestingly, tumor cell clones with highest propensity to colonize the brain express high levels of heparanase,97,98 an endoglycosidase which cleaves heparan sulfate to remodel the extracellular matrix, releasing growth factors, chemokines, angiogenic factors and bioactive heparan sulfate fragments. 99 Heparanase expression is inversely regulated by miR-1258, levels of which are reduced in highly brain metastatic breast cancer cells. 100 Moreover, heparanase – which can be secreted not only by tumor cells, but astrocytes as well 101 – can activate EGFR signaling in brain metastatic breast cancer cell, which is a survival pathway for the tumor cells. 102 Besides heparanase, MMPs secreted by tumor and host cells can also modulate the extracellular matrix. 82 Brain metastases have gelatinase activity and express high levels of MMP-2, -3 and -9 proteins. 103 MMPs, besides degrading the extracellular matrix, may have other pro-metastatic effects, e.g. MMP-1 secreted by brain metastatic breast cancer cells can activate latent TGF-α, a ligand for EGFR. 104
On the whole, proteases play an important role in the transmigration of tumor cells through the BBB, by degrading components of the capillary wall (TJs, basement membrane proteins and probably the glycocalyx). Therefore, it is not surprizing, that the protease-dependent mesenchymal type of movement (Figure 2) is primordial in the transmigration of tumor cells through the BBB.
48
Phenotypes of migrating tumor cells. During individual migration, tumor cells either acquire the amoeboid (leukocyte-like) or the mesenchymal (fibroblast-like) phenotype. Amoeboid cells have a rounded morphology and can change their shape to move through narrow gaps. The needed force is generated by the actin cytoskeleton, which is controlled by the small GTPase RhoA and its effector Rho-kinase (ROCK). On the other hand, the mesenchymal type of movement is proteolysis-dependent. Mesenchymal cells are elongated, having lamellipodia and filopodia produced under the control of the small GTPase Rac1. Cancer cells can shift from one migration type to the other depending on the environment they are moving in. During transmigration through the BBB, the mesenchymal phenotype seems to be more favorable.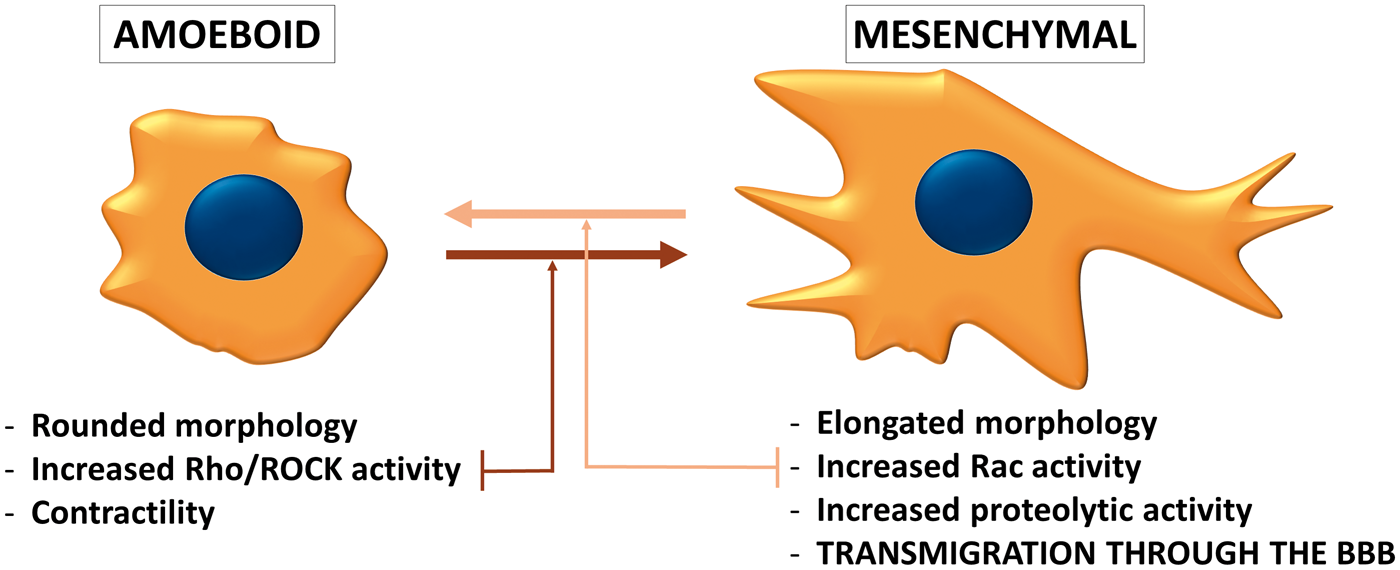
Signaling pathways activated in tumor cells during extravasation into the brain
Interaction of malignant cells with the cerebral endothelium involves complex signaling mechanisms, which are not well understood. The two best-characterized signaling pathways involved in transmigration of tumor cells through the brain endothelium are PI3K (phosphoinositide 3-kinase)/Akt and small GTPase (Rho and Rac) signaling.
PI3Ks are key regulators of growth and cancer development-related processes. Key elements of the pathway involved in tumorigenesis are: PIK3CA, encoding the class IA PI3K catalytic subunit p110α, the negative regulator PTEN (phosphatase and tensin homolog), Akt (PKB/protein kinase B) and the mTOR (mammalian target of rapamycin) complexes (mTORC1 and mTORC2). The oncogene PIK3CA and the tumor suppressor PTEN are frequent targets of somatic mutations in several cancer types 105 ; therefore, dysregulation of the PI3K/PTEN/Akt/mTOR pathway is clearly associated with the development of tumors, including breast cancer and melanoma.106,107
Development of brain metastases and penetration of malignant cells through the BBB have also been linked to alterations in PI3K signaling. PI3K-aberrant squamous cell lung cancers appear as an aggressive subset associated with brain metastases. 108 In metastatic melanoma, cerebrotropism also seems to highly depend on this pathway.109,110 Loss of PTEN – i.e. activation of the PI3K pathway – in melanoma cells is associated with significantly shorter time to brain, but not to liver, lung or bone metastasis formation. 111 Furthermore, PLEKHA5 – which has been associated with transmigration of melanoma cells through the BBB – has a phosphoinositide-binding specificity; therefore, it was suggested that its crosstalk with the PI3K/Akt pathway might be responsible for the guidance of the cerebrotropic phenotype in melanoma cells. 112 In addition, inhibition of PI3K was shown to significantly reduce the number of transmigrating breast cancer and melanoma cells in in vitro BBB models. 61
The PI3K pathway was shown to regulate Rac signaling in breast cancer cells.113,114 Rac1 is a member of the Rho family of GTPases playing important role in actin dynamics. Rac activation is involved in the acquisition of the mesenchymal (fibroblast-like) phenotype of tumor cells, characterized by elongated morphology and extracellular proteolysis.115–117 On the other hand, the amoeboid (leukocyte-like) type of tumor cell migration is characterized by rounded morphology, increased acto-myosin contractility and extensive RhoA signaling. Cancer cells can switch between these two phenotypes (Figure 2) depending on environmental conditions. 118 It has been proposed that during transmigration of tumor cells through the BBB, the mesenchymal migration is preferred to the amoeboid one. Inhibition of Rho/ROCK signaling (i.e. triggering of the mesenchymal phenotype) induces a significant increase in the number of melanoma cells migrating through CECs and promotes formation of parenchymal brain metastases. 48 On the other hand, inhibition of Rac impedes transmigration of breast cancer cells and melanoma cells through cultured CECs. 61
In addition to PI3K and Rac signaling, activation of Src kinases can also be critical in extravasation of Her2-positive and triple negative breast cancer cells into the brain parenchyma. Src-activated cells are able to more efficiently disrupt TJ integrity of CECs to facilitate brain metastasis formation. 119
Therefore, activation of PI3K, Rac and Src signaling pathways in tumor cells coming in contact with the cerebral endothelium promotes their extravasation through the BBB. However, activation of these pathways is not restricted to the development of brain metastases.
Changes in the brain endothelium during extravasation of tumor cells
Interaction of tumor cells and CECs is bidirectional and these latter also suffer changes during extravasation of malignant cells. During transendothelial migration, metastatic cells damage the integrity of the endothelium. Vessel wall destruction is probably not very extensive; however, fibrin deposition and platelet aggregation were observed in vivo, 58 while apoptosis could only be detected in vitro. 64 Signaling pathways activated in the cerebral endothelium during tumor cell transmigration were studied in in vitro models. The Rho/ROCK pathway was shown to be involved in the transendothelial migration of SCLC cells. 65 This is in contrast with Rho/ROCK activation in tumor cells, which impedes extravasation through the BBB. 48 As a consequence of Rho/ROCK activation in CECs, actin reorganization occurs through phosphorylation of myosin light chain and cofilin, and cytoskeletal changes proved to be responsible for TJ reorganization. 65
In addition, TGF-β-dependent endothelial-mesenchymal transition (EndMT) has been recently described as a potential mechanism involved in brain metastasis formation of melanoma and breast cancer cells.120,121 In response to TGF-β released by cancer cells, CECs lose their endothelial markers and junctions, gain expression of fibroblast-specific and mesenchymal proteins and differentiate into α-smooth muscle actin-positive myofibroblasts. This process was demonstrated to play an important role in metastatic transendothelial migration in vitro. Since EndMT development is a long-lasting process, one can speculate whether the long time spent by tumor cells inside the blood vessels of the brain might be needed for “mesenchymal transformation” of the underlying endothelium. Importantly, TGF-β secreted by melanoma cells not only acts on brain endothelial cells, but also in an autocrine manner to regulate seprase activity and invasive capacity of melanoma cells. 95
In addition to being a barrier against tumor cells, CECs can also facilitate their transmigration. According to an in vitro study, this phenomenon is dependent on activation of COX-2 in brain endothelial cells, which in turn upregulates expression and activates MMP-2 in response to interaction with breast cancer cells. 122
An interesting question is the role of environmental factors in brain metastasis development. Pollutants like polychlorinated biphenyls can upregulate adhesion molecules and alter expression of brain endothelial TJ proteins to promote brain metastasis formation in in vivo models.123,124 On the other hand, physical exercise might help in the maintenance of BBB integrity thereby protecting the brain during metastatic progression. 125
The brain metastatic environment
Early interaction of extravasated tumor cells with the brain vasculature
The majority of extravasated brain metastatic cells die in the brain tissue and only a small proportion proliferates to form micro- and macrometastases. 59 Survival of metastatic cells depends on their interaction with cellular and non-cellular elements of the highly unique microenvironment of the CNS. After transmigration through brain microvessels, brain metastatic lung cancer, breast cancer and melanoma cells remain in contact with the extraluminal surface of capillaries, attaching to them in a pericyte-like position.59,126 Angiotropism (attachment to vascular abluminal surfaces) of melanoma cells has recently been shown to correlate with the expression of serpin B2, leading to migration and spreading of tumor cells along the abluminal vascular surfaces of microvessels, called pericytic mimicry. 127
Nevertheless, loss of perivascular contact leads to cancer cell death. Even if directly injected into the brain, tumor cells preferentially attach to vessel walls 126 and incorporate endothelial cells and pericytes, detaching astrocytic endfeet. 128 Interaction of tumor cells with endothelial cells may be mediated by integrins – e.g. LFA-1 (lymphocyte function-associated antigen-1, integrin αLβ2) expressed by breast and lung cancer cells 129 – and gap junctions – i.e. connexin-43- and connexin-26-based gap junctions used by breast cancer and melanoma cells, respectively. 57 These interactions provide growth signals and protection to malignant cells.
Besides gap junction communication and endothelin signaling, 130 another line of CEC-dependent chemoprotection is provided by efflux transporters. 131 Among these, ABCB1 (P-glycoprotein/P-gp, MDR1/multidrug resistance protein 1) and ABCG2 (BCRP/breast cancer resistance protein) are probably the most important. 132 They are expressed in both endothelial and tumor cells; therefore, convey double resistance to anti-cancer drugs in brain metastases. 133
Extravasated tumor cells not only associate with CECs, but have strong connections with the vascular basement membrane in vivo. In this process, L1 cell adhesion molecule (L1CAM) was found to be crucial. 134 Moreover, integrin α3β1 was shown to mediate adhesion of NSCLC cells to laminin in the extracellular matrix. 135 Expression of α3β1 integrin is dependent on ADAM-9 (a disintegrin and metalloprotease 9) overexpression, characteristic to highly brain-metastatic NSCLC cells. 136
Much less is known about the direct interaction of pericytes and tumor cells. Our current knowledge about the role of pericytes in brain metastasis formation is restricted to vascularization mechanisms (see later). However, multipotent stem cell potential of pericytes 137 renders them a tumor-promoting character, as shown in primary brain tumors. 138 In vitro, pericytes isolated from normal fetal brain enhance proliferation and migration of triple negative breast cancer cells. 139 Neoplastic pericytes may derive from and can also generate CSCs to promote tumor development and can also fuse with glioblastoma cells.140–142 However, these mechanisms still need to be verified in secondary tumors of the CNS.
Surviving tumor cells attached to the extraluminal surface of the vessels either start to proliferate or remain dormant and become active later (Figure 3). The perivascular location is a niche favorable for cells with stem cell-like characteristics, including neural and glial stem cells and dormant cancer cells.
143
Dormant cells persist as single cells for several weeks or even years without proliferating or regressing, slowly moving in the perivascular niche.59,144 Dormant tumor cells are reversibly growth arrested and are resistant to therapy; therefore, they can be responsible for the clinical phenomenon of latent disease, leading to relapses or late manifestations.
145
Several endothelial factors are involved in mediating quiescence, survival and drug resistance of dormant cancer cells in different tissue-specific perivascular niches,
145
including thrombospondin-1 localized to the basement membrane of resting (i.e. not sprouting) microvessels.
146
Fate of extravasated tumor cells in the brain microenvironment. After migration through the vessel wall, large part of metastatic cells dies in the brain microenvironment. Astrocyte-released PA is involved in killing serpin-negative tumor cells. Surviving cells either remain dormant, closely attached to the vessel wall or start proliferating in response to signals arising from the brain microenvironment. Strong reactive astrocytosis persists during growth of the metastatic lesion. Astrocytes support proliferation of malignant cells through release of soluble factors and exososmes and formation of heterocellular gap junctions. Astrocyte-dependent Notch signaling is involved in the maintenance of the CSC phenotype in brain metastatic breast cancer cells. Response of microglial cells to cancer cells is heterogeneous. Increased secretion of cathepsin S and release of chemokines from microglia might promote viability and migration of tumor cells in the brain. GJ: gap junction.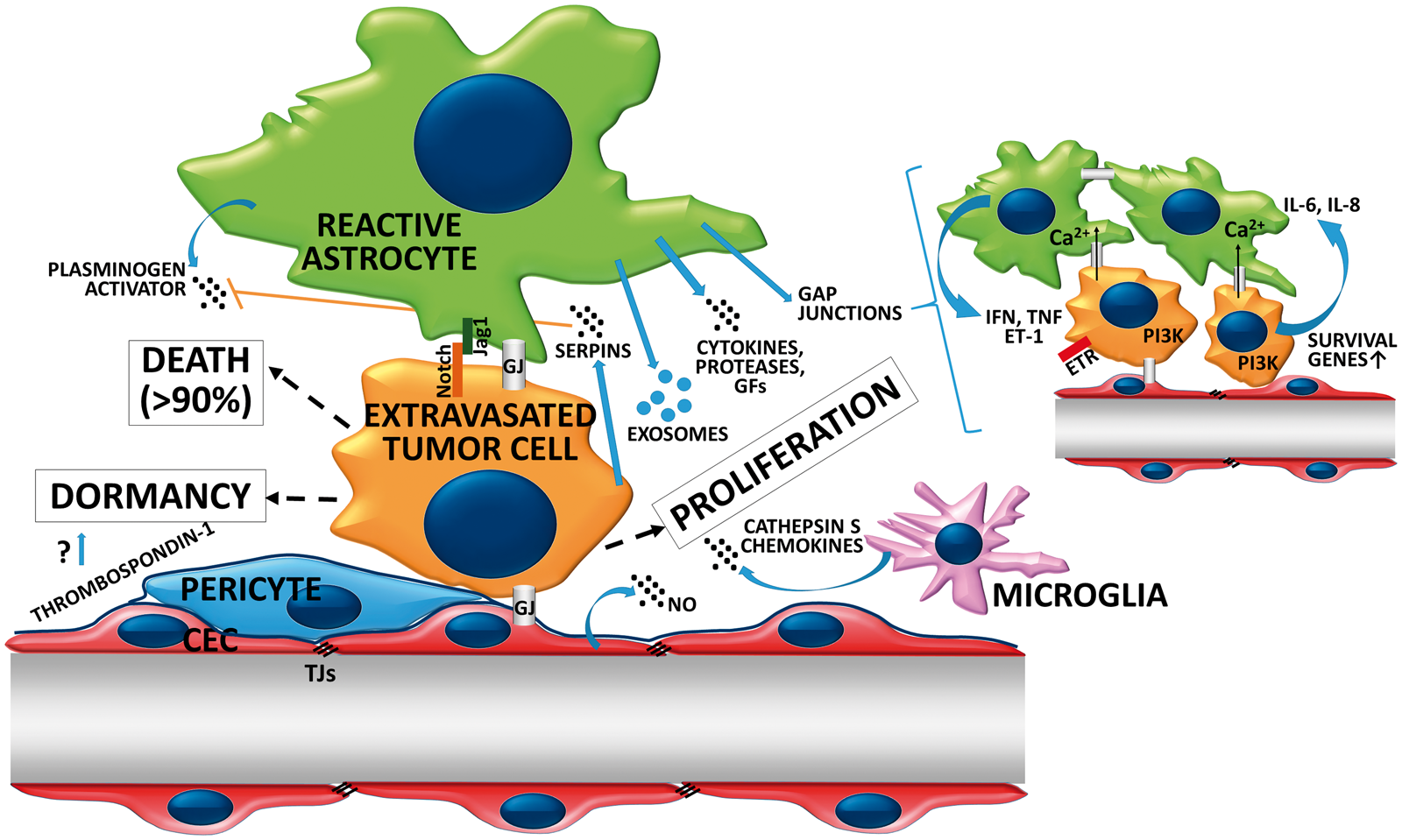
In conclusion, survival of metastatic cells in the brain highly depends on their ability to remain attached to the outer surface of the vessel wall. Interaction with the basement membrane seems to be critical in the early post-extravasation period.
Interaction of brain-homed tumor cells with cells of the NVU
Besides vascular cells, other cells of the CNS are also important in determining the fate of tumor cells. 147 According to recent results obtained in mouse models of brain metastasis, LFA-1 (integrin αLβ2) seems to be critical in the interaction of breast and lung cancer cells with cells of the NVU. It mediates tumor cell attachment to astrocytes, microglial cells, neurons and endothelial cells, upregulating COX-2 expression and VEGF secretion in astrocytes and consequently NO release from CECs, which enhances tumor growth. 129
Metastatic tumor cells in the brain grow in close contact not only with vessels, but with glial cells as well. On the other hand, tumor lesions are generally separated from neurons by edema. Islands of reactive astrocytes and microglial cells are localized in the interior of most of the tumors and stimulate anchorage independent growth of brain metastatic breast cancer cells. 148
Factors released by astrocytes during crosstalk with metastatic tumor cells.
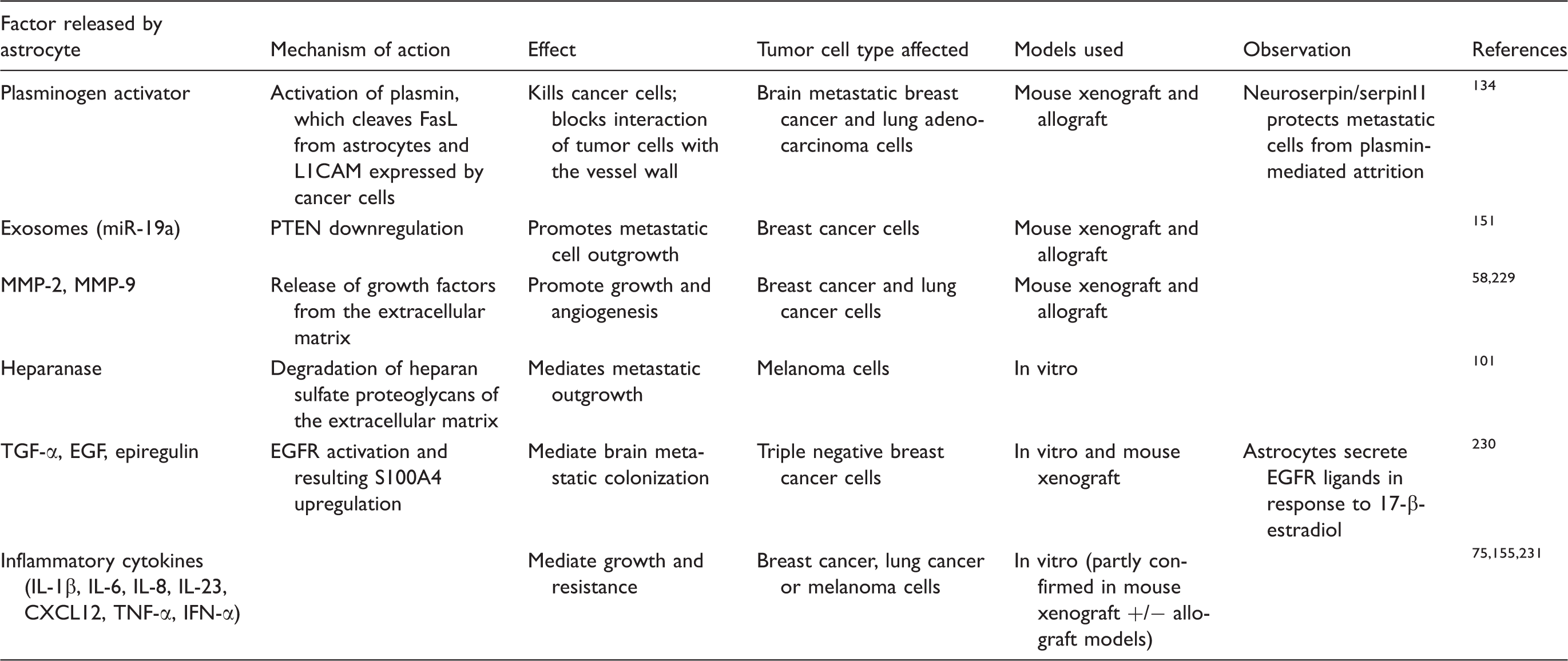
Factors released by metastatic cells during crosstalk with astrocytes.

CSC: cancer stem-like cells.
In addition, astrocytes can establish direct contacts with metastatic cells through JAG-Notch interaction and gap junctions. JAG1 can be upregulated in astrocytes in response to IL-1β secreted from brain metastatic breast CSCs. In turn, the JAG1-Notch-signaling pathway promotes self-renewal of CSCs. 152 Notably, activation of the Notch signaling pathway has been shown to induce migratory and invasive properties 153 and to maintain the CSC phenotype of brain metastatic breast cancer cells. 154
Gap junctions between astrocytes and brain metastatic cells comprise protocadherin-7- and connexin-43-dependent interactions and may result in tumor growth, 155 and resistance to chemotherapy by sequestering excess of Ca2+ from the cytoplasm of tumor cells. 156 It is noteworthy that gap junction communication and network formation promote survival of primary brain tumor (i.e. astrocytoma) cells as well. 157 Heterocellular gap junctions between astrocytes and metastatic cells can also lead to activation of PI3K signaling, upregulation of survival genes and secretion of inflammatory cytokines through endothelin (ET) signaling.130,158 Not surprising, endothelin receptor B (ETBR) was found to facilitate growth of melanoma cells within the CNS. 159
Moreover, astrocytes may elicit metastatic cell-type specific effects as well. Expression of reelin, an extracellular neuronal protein, is induced by astrocytes only in Her2-positive, but not in triple negative breast cancer cells, leading to increased proliferation of Her2-positive cells in the brain environment. 160
Besides astrogliosis, microgliosis also starts already before extravasation. Tumor-associated microglia/macrophages are predominantly resident microglia, but can also be monocytes/macrophages entering the brain from the bone marrow. 161 Interestingly, microglial reaction is heterogeneous throughout metastatic growth. Some extravasated cells or lesions recruit large amounts of activated and reactive microglia, while others can be completely free of microglial cells. 58
Microglial cells may also exhibit Janus faces towards metastatic cells by having dual (tumor destructive and supportive) role in brain tumor progression. 162 Factors secreted by cultured microglia may inhibit proliferation of lung cancer cells. 163 Nevertheless, surviving and proliferating tumor cells take benefit of a favorable and more permissive brain microenvironment created by microglial cells (Figure 3). Microglia can communicate with tumor cells through release of cytokines, chemokines, growth factors, proteases and exosomes; however, these mechanisms have mainly been studied in primary brain tumors. 164 Among proteases, microglia/macrophage-derived cathepsin S was shown to play significant role in the development of breast cancer brain lesions. Only depletion of both tumor-derived and stromal cathepsin S could reduce formation of experimental brain metastases. 92 Although not studied specifically in the brain, this cysteine endopeptidase might modify the extracellular matrix and stimulate angiogenesis. 165
Chemokine signaling is also bidirectional and is involved in the modulation of both tumor cells and microglia. High CCL2 expression in brain metastases of breast cancer was shown to recruit CCR2-positive microglia/macrophages. 151 In addition, the CCR4 ligands CCL22 and CCL17 are secreted by brain stromal cells, including microglia. Microglia-derived soluble factors upregulate expression of CCR4 in brain metastatic melanoma cells, promoting viability and even migration of tumor cells through the brain endothelium. 74 Breast cancer cells metastasized to the brain parenchyma may enhance activation of microglia/macrophages towards the M2 state, with reduced expression of MHC (major histocompatibility complex) class II, CD11c, iNOS (inducible nitric oxide synthase) and arginase-1 and higher expression of CD206 (mannose receptor). 161 In vitro no upregulation of M2-specific cytokines was observed in microglial cells cocultured with breast cancer cells. 76 Therefore, cytokine profile of activated microglia of brain metastatic lesions needs to be clarified.
Although the BBB provides a partial immune-privilege to the CNS, infiltrating leukocytes might also influence metastatic cells in the brain. Regulatory T cells actively infiltrate melanoma and NSCLC brain metastases 166 and possess immunosuppressive activity, contributing to tumor immune evasion. By inhibiting Stat3 activity in regulatory T cells, the antitumor activity of CD3-positive/CD8-negative/CD25-negative T cells can be restored. 167
Taken together, cells of the NVU have a dual role in the crosstalk with brain metastatic cells, being both offensive and protective. Communication between tumor cells and cells of the CNS is bidirectional, and fate of tumor cells (death, dormancy or proliferation) depends on their response to signals coming from the brain microenvironment.
Characteristics of brain-homed tumor cells
Environmental adaptation of tumor cells requires transient activation of genes associated with homeostasis and stress, followed by activation of genes involved in more advanced tissue-specific functions. 168
In order to identify characteristics determining adaptation of tumor cells to the brain, matched primary and brain metastatic tumors were compared. These studies revealed several copy number variations (CNVs), single nucleotide polymorphisms (SNPs) and differentially expressed genes. Genes found to be amplified in brain metastatic lung adenocarcinoma tumors are involved in migration and organ development, while those having a lower copy number in the secondary tumor negatively affect cell proliferation and adhesion. 169 Aberrations in brain metastatic breast tumor specimens may affect genes related to stem cell pluripotency and genes frequently amplified in primary breast cancers, like PIK3CA. Comparison of matched pairs of primary and brain metastatic breast cancer samples identified both similar and divergent CNVs. 170 Some studies did not find significant differences in the mutation profiles, 171 but identified several differentially expressed genes (CXCL12, MMP-2, MMP-11, VCAM-1 and MME/membrane metalloendopeptidase) between primary breast cancers and breast cancer brain metastases. 172 Altered expression of cell cycle regulatory proteins (particularly, upregulation of p27kip1 and cyclin D1), growth factors and hormone receptors 173 and overexpression of DNA double-strand break repair genes 174 have been described in brain metastases compared to matched breast primary cancers. Breast cancer brain metastases may also harbor mutations absent in the primary tumor. 175 Interestingly, driver alterations are homogeneous within multiple regions of the same lesion and among distinct brain lesions in the same patient. These mutations target a few signaling molecules and pathways associated with brain metastases, including cyclin-dependent kinases, the PI3K/Akt/mTOR pathway, Her2, EGFR, the MAPK (mitogen-activated protein kinase) pathway and others, predicting sensitivity to targeted therapies. 175
These signaling pathways are indeed aberrantly activated in brain tumors. The PI3K pathway – besides its role in extravasation of metastatic cells through the BBB, as described previously – is involved in the survival of tumor cells in the brain. In aggressive brain metastatic tumor cells, activating mutations of the PI3K/PTEN/Akt/mTOR pathway can either develop in the primary tumor or in the brain.108,175 In line with the role of the PI3K pathway in the development of brain metastases, expression of mTORC1/2-related proteins was found to be increased in brain metastatic lesions compared with primary tumors in lung adenocarcinoma. 176 Moreover, alterations in the PI3K/Akt pathway, i.e. activating mutations in PIK3CA or inactivating mutations in PTEN, are partly responsible for the therapy resistance of BRAF-mutant melanoma brain metastases. 177 BRAF V600K mutations have the highest incidence in melanoma, 178 resulting in constitutive activation of the MAPK/ERK signaling pathway. This pathway is also overactivated by mutations in HRAS, KRAS and NRAS, which have been detected in brain metastatic tumors. 179 Taken together, activation of PI3K and MAPK pathways is not specific to brain metastases, but has important implications in the development and treatment of secondary tumors of the CNS.
The PI3K and MAPK pathways can be activated by members of the ErbB family – including EGFR (also known as ErbB1 or Her1/heregulin 1), Her2 (neu, ErbB2) and Her3 (ErbB3) – which are involved in brain metastasis formation of breast and lung tumors.180–182 These receptors regulate cell migration, invasion and proliferation responding to growth factors released in the brain. Interestingly, aberrant EGFR signaling may be directly and indirectly influenced by miR-145-5p downregulation, which was shown to contribute to the development of lung cancer brain metastases in humans and mice. 183 Moreover, as described in the previous chapters, the brain metastatic environment (e.g. astrocytes) can activate EGFR signaling through secretion and activation of its ligands.
The PI3K and the MAPK pathways are downstream effectors of neurotrophin receptors (NTRs) as well, ligands of which – neurotrophins (NTs) – are abundantly expressed in the brain. NTs (NGF/nerve growth factor, BDNF/brain-derived neurotrophic factor, NT-3 and NT-4/5) are growth factors promoting neuronal survival, differentiation and cell death. Melanoma cells express the low-affinity (p75NTR) and the high-affinity tyrosine kinase NTRs (TrkA, B and C) 184 which regulate proliferation, motility and invasive capacity of brain metastatic melanoma cells.184,185 Therefore, melanoma cells, which are neural crest-derived cells, can respond to NTs secreted in the central nervous tissue, and this mechanism has been considered decisive in homing of melanoma cells to the CNS. 186
Interestingly, NTR signaling may not be confined to brain metastasis development of melanoma. In breast cancer cells, TrkB can heterodimerize with Her2, resulting in a survival advantage in the brain. 187 Moreover, in brain metastatic breast cancer cells, NT-3 expression may also be increased, promoting mesenchymal-epithelial transition, upregulation of Her2, proliferation in the brain and reducing microglial activation. 188 Indeed, mesenchymal-epithelial transition and re-expression of E-cadherin were shown to be induced in the brain environment. 189
In addition to excessive activation of survival pathways, adaptation of energy metabolism to the brain microenvironment is also important for survival of tumor cells in the brain. This adaptation can comprise a switch to anaerobic glycolysis (i.e. conversion of glucose to lactate) which is known as the Warburg effect, 190 even in the presence of oxygen. To compensate for the inefficiency of this process, brain metastatic cells can express high levels of hexokinase 2. 191 In addition, breast cancer-secreted miR-122 reduces glucose consumption in brain astrocytes (and also lung fibroblasts) through downregulation of pyruvate kinase M2 and the glucose transporter GLUT1 (SLC2A1), leading to enhanced cancer cell proliferation probably partially mediated by increased glucose availability. 192 In addition to anaerobic glycolysis, brain tumors are able to oxidize glucose in the tricarboxylic acid cycle as well. Besides glucose, acetate can also be simultaneously oxidized, and conversion of acetate into acetyl-coenzyme A is dependent on the increased expression of ACSS2 (acyl-coenzyme A synthetase short-chain family member 2). 193
In addition to these rather general mechanisms, brain-specific metabolic changes might also exist. One such mechanism is the capacity to metabolize neurotransmitters. In this respect, brain metastatic breast cancer cells may take up and catabolize GABA (γ aminobutyric acid) into succinate, entering the tricarboxylic acid cycle. 194 On the other hand, glutamate seems not to be an energy source in breast cancer and melanoma brain metastases, 193 glutamate rather being involved in signaling processes in brain metastatic melanoma. 195 Acquisition of neuron-like characteristics (i.e. expression of neurotransmitter receptors) in breast cancer 194 and melanoma cells 195 might also be part of the adaptation scenario of tumor cells to the brain environment.
In conclusion, aberrant activation of signaling and metabolic pathways determines the ability of tumor cells to overcome the selective pressure of the brain environment. However, characteristics involved in survival of tumor cells in the brain tissue cannot always be unambiguously distinguished from features required for extravasation through the BBB or from those generally determining aggressiveness and metastatic capacity of tumor cells.
Vascularization and the blood–tumor barrier of metastatic brain tumors
Tumor cells that have survived and started to proliferate in the brain result in micrometastases and later in larger lesions. These tumor masses need proper vascularization.
As previously shown, metastatic cells start to proliferate in close proximity to pre-existing microvessels, which determines a special vascularization process, called vascular cooption. Cooption is characteristic to highly vascularized tissues, 196 such as the brain, and is a mechanism rendering tumors less likely to respond to anti-angiogenic therapy. 197 Indeed, this is the main vascularization mechanism in the brain for breast cancer and melanoma cells.59,126 On the molecular level, vascular cooption is dependent on L1CAM and neuroserpin. 134
Besides cooption, other mechanisms of tumor vessel formation might also be relevant in the brain. Accordingly, melanoma cells can secrete factors activating the MAPK and PI3K/Akt survival pathways, enhancing angiogenic properties of brain endothelial cells. 198 In addition, VEGF – which is a key factor inducing sprouting angiogenesis – can be secreted by brain metastatic tumor cells. VEGF release can be induced by several mechanisms. In breast cancer cells, activation of αvβ3 integrin promotes expression of VEGF in normoxic conditions inducing angiogenesis selectively in the brain. 199 In melanoma cells, activated Stat3 enhances brain metastasis formation through induction of angiogenesis mediated by VEGF, basic fibroblast growth factor (bFGF) and MMP-2. 83 However, melanoma cells tend to grow by vascular cooption in the brain despite high expression of VEGF. 200 VEGF secreted by melanoma cells can induce dilation of coopted brain vessels with subsequent permeability increase. This means that VEGF released by metastatic melanoma cells can modulate the pre-existent vasculature (i.e. own vessels of the brain coopted by the tumor); therefore, blood supply of VEGF-secreting brain metastatic melanoma does not necessarily depend on induction of sprouting angiogenesis. 201 Therefore, vessel cooption is unquestionably important in the development of the vasculature of breast cancer and melanoma brain metastases; however, angiogenic neovascularization might also contribute to the formation of tumor vessels. On the other hand, lung cancer cells may present early angiogenesis in the brain. 59 As a consequence, anti-VEGF treatment is more effective in inhibiting brain metastasis formation in NSCLC than in breast cancer patients 202 ; although combination of Her2 inhibitors with an anti-VEGF receptor-2 antibody may have a significant survival benefit compared to Her2 inhibition alone in cerebral metastases of Her2-amplified breast cancer. 203
The vasculature of the tumor evolved through vessel cooption, sprouting angiogenesis or possibly other mechanisms
204
forms a similar barrier to the BBB, the so-called BTB (Figure 4). In endothelial cells of the BTB, VEGF and CD31 expression increases, while ZO-1 and GLUT1 (SLC2A1) expression may decrease.149,205 Previous results suggested that the BTB is intact in small metastases and altered in larger lesions.
25
However, recent studies did not find correlation between BTB permeability and the size of the metastatic lesion in breast cancer brain metastasis models.131,206 Permeability of mammary brain metastases is highly heterogeneous, in contrast to the relatively homogeneously high permeability of gliomas.
207
In metastases, the BTB remains sufficiently intact to impair drug delivery in therapeutically relevant concentrations.
131
However, it is not clear whether these data derived from mouse models adequately recapitulate the human disease in terms of drug delivery to brain tumors.
208
The blood–tumor barrier. The vasculature of the growing tumor forms the BTB. Permeability of BTB in metastatic lesions is heterogeneous and usually sufficiently low to impair penetration of relevant amounts of therapeutic agents. Tumor cells secrete proteolytic enzymes to modulate the extracellular matrix. Astrocytes loose polarity and secrete proteases and VEGF, which increase permeability of the BTB. Expression of PDGFR-β is decreased in pericytes. In lesions with higher permeability, pericytes are desmin-positive and the amount laminin-α2 is decreased in the basement membrane.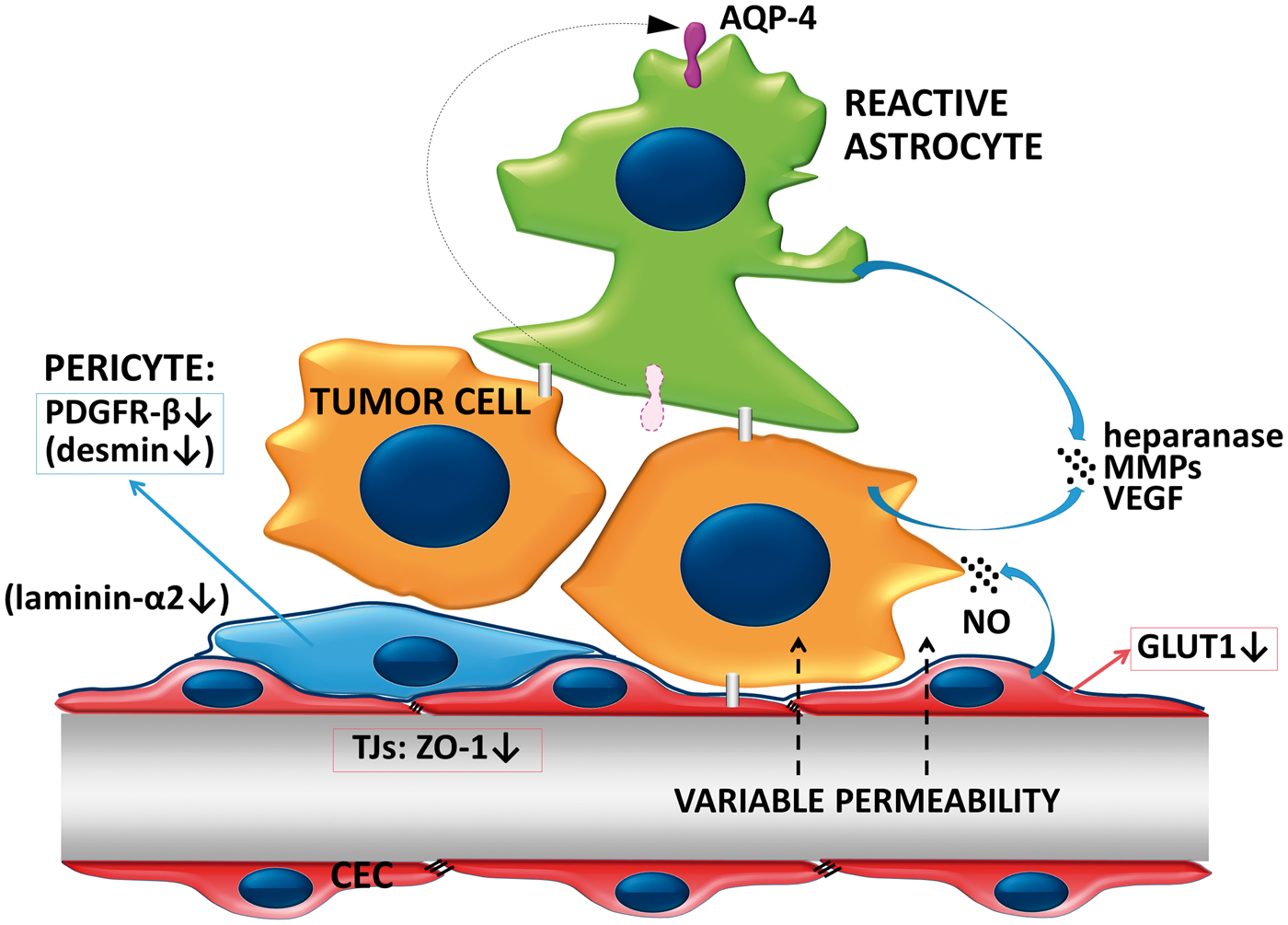
Recent data suggest that gadolinium-DTPA-impermeable cerebral breast metastases have significantly more proliferative nuclei compared to gadolinium-DTPA-permeable tumors in the mouse brain. 209 In contrast, another study showed that melanoma metastases having a permeable vasculature grow faster than those having an intact BTB. 144 This study also demonstrated that only brain-permeable PI3K inhibitors are active against metastatic lesions, and that targeting micrometastases and even dormant cells is more effective than treating large lesions. Interestingly, TNF or lymphotoxin may selectively permeabilize the BTB, as demonstrated in a mouse model of breast cancer brain metastasis. 210
Important factors in the induction of an increased BTB permeability are changes in pericytes. In mouse intracranial melanoma, NG2 ablation-induced pericyte deficiency (i.e. delayed pericyte maturation and reduced association with endothelial cells) decreases basal lamina deposition and vessel patency, increases vessel leakiness and results in reduced tumor progression and intratumoral hypoxia.211,212 In experimental animals, metastatic lesions show decreased expression of PDGFR (platelet-derived growth factor receptor)-β, a specific protein of pericytes. In metastatic tumors with higher permeability, vessels are surrounded by desmin-positive pericytes. Moreover, a tendency of reduced CD13 staining in pericytes was observed. 205 Increased BTB permeability also correlates with the decrease in the expression of the parenchymal basement membrane component laminin-α2 expressed by astrocytes and pericytes. 205 Absence of laminin-α2 leads to increased permeability of blood vessels, hypertrophy of astrocytic endfeet with lack of appropriately polarized aquaporin 4 (AQP4) channels. 213 Loss of polarization of astrocytic endfeet was also observed at the level of the BTB in breast cancer metastases; however, no significant differences were observed between highly permeable and poorly permeable lesions. 205 In conclusion, depending on changes in pericytes, permeability of BTB of metastatic lesions is heterogeneous, but low enough to impede drug delivery. Vasogenic edema increased interstitial fluid pressure and hypoxia also contributes to low drug penetration.214–216
Along with the growth of metastatic lesions, the enhanced permeability and retention (EPR) effect might enable nanoparticles and macromolecular drugs to enter the brain tumor. This unique feature of tumor vessels – i.e. highly increased permeability towards high molecular weight drugs and nanosystems compared to normal vessels – is dependent on structural abnormalities in the endothelium of tumor blood vessels. 217 However, the EPR effect is significantly weaker in the cerebral microenvironment than in the periphery, as shown in primary brain tumors. 218 Further studies will elucidate involvement of the EPR effect in treatment of cerebral metastases.
In conclusion, vascularization of the growing tumor mass has important therapeutic implications. Vascular cooption might render metastatic tumors of the brain resistant to anti-angiogenic therapy. Relatively preserved tightness of the BTB and expression of efflux transporters by both CECs and tumor cells highly limit the efficacy of systemic therapies.
Conclusions
The Janus-faced attitude of cells of the NVU towards metastatic cells.
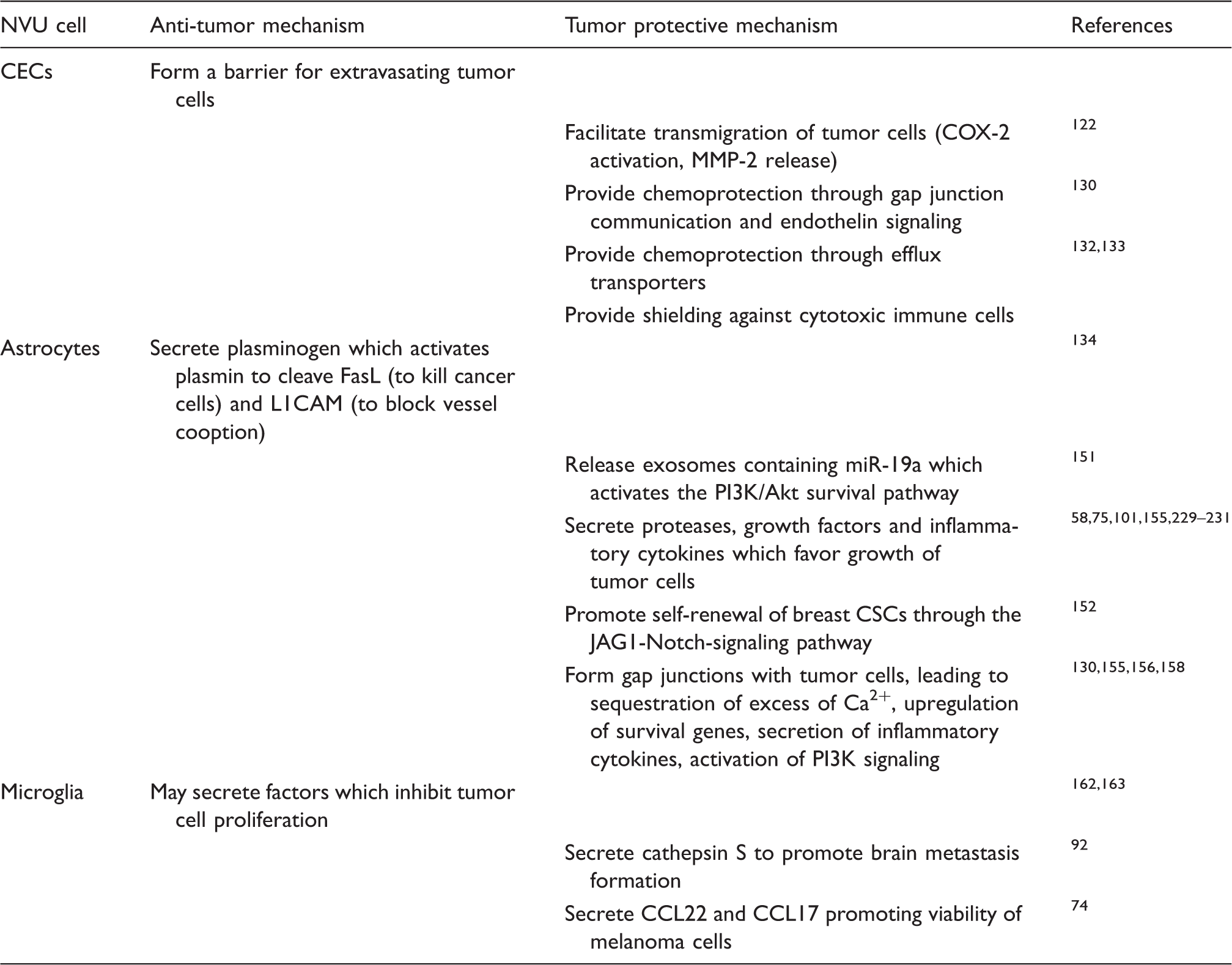
NVU: neurovascular unit; CEC: cerebral endothelial cell; CSC: cancer stem-like cell.
From clinical point of view, it would be important to identify selective and specific molecular markers in primary tumors or circulating cancer cells which determine formation of brain metastases. Exosomes and miRs are the most promising emerging biomarkers; however, further studies are needed to find and validate miR patterns determining cerebrotropism. Although several alterations are common in the primary and the brain metastatic tumor, molecular profile of the brain secondary tumors is different from primary and extracranial lesions. Therefore, identification of key mechanisms of adaptation of the tumor cells to the brain microenvironment is a prerequisite for the design of new therapeutic strategies.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Work of IAK is supported by the National Research, Development and Innovation Office (grant numbers: OTKA K-116158, GINOP-2.3.2-15-2016-0020, GINOP-2.3.2-15-2016-0030 and GINOP-2.3.2-15-2016-0034). IW is supported by NKFIH OTKA FK-124114, CF is supported by NKFIH OTKA PD-121130; AGV is supported by the NKFIH OTKA PD-115697 grant of the National Research, Development and Innovation Office. IW and AGV are supported by the János Bolyai Research Fellowship of the Hungarian Academy of Sciences (BO/00334/16/8 and BO/00598/14/8, respectively).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.