
Abstract
Acutely following focal cerebral ischemia disruption of the microvessel blood–brain barrier allows transit of plasma proteins into the neuropil as edema formation that coincides with loss of microvessel endothelial β1-integrins. We extend previous findings to show that interference with endothelial β1-integrin–matrix adhesion by the monoclonal IgM Ha2/5 increases the permeability of primary cerebral microvascular endothelial cell monolayers through reorganization of claudin-5, occludin, and zonula occludens-1 (ZO-1) from inter-endothelial borders. Interference with β1-integrin–matrix adhesion initiates F-actin conformational changes that coincide with claudin-5 redistribution. β1-integrin–matrix interference simultaneously increases phosphorylation of myosin light chain (MLC), while inhibition of MLC kinase (MLCK) and Rho kinase (ROCK) abolishes the Ha2/5-dependent increased endothelial permeability by 6 h after β1-integrin–matrix interference. These observations are supported by concordant observations in the cortex of a high-quality murine conditional β1-integrin deletion construct. Together they support the hypothesis that detachment of β1-integrins from abluminal matrix ligands increases vascular endothelial permeability through reorganization of tight junction (TJ) proteins via altered F-actin conformation, and indicate that the β1-integrin–MLC signaling pathway is engaged when β1-integrin detachment occurs. These findings provide a novel approach to the research and treatment of cerebral disorders where the breakdown of the blood–brain barrier accounts for their progression and complication.
Keywords
Introduction
The blood–brain barrier is, physically and metabolically, an indispensable component of the neurovascular unit, contributing to the accurate function of the neurons and their supporting cells in the central nervous system (CNS). The permeability barrier consists of endothelial cells, the end-feet of astrocytes, extracellular matrix (ECM) proteins of the basal lamina, pericytes, and other cellular elements of cerebral microvessels. It has recently been demonstrated that adherence of the luminal endothelial cells and the abluminal astrocyte end-feet to the interposed basal lamina matrix (the “vertical barrier”) and the inter-endothelial adherens and tight junction (TJ) apparatus of the endothelium (the “horizontal barrier”) may function interdependently.1,2 These structures exclude or limit plasma proteins and circulating cells from the neuropil, and strictly regulate trans-endothelial transport of water, solutes, and nutrients. However, there is little known about how endothelial cell–matrix adherence could regulate inter-endothelial TJ fidelity and vascular permeability in the CNS.
Of the TJ proteins, claudin-5 is exclusively located at the junctional contacts of endothelial cells, but not at those of epithelial cells (e.g. astrocytes) in the CNS. 3 Claudin-5 protein expression levels correlate with endothelial permeability both in vivo and in vitro.4,5 Meanwhile, occludin has also been shown to be strongly and continuously expressed at the interfaces of brain endothelial cells with each other, and feature in the robustness of the paracellular barrier. 6 Those observations suggest that claudin-5 and occludin specifically contribute to the regulation of endothelial barrier permeability in cerebral microvessels. 7 And, zonula occludens-1 (ZO-1) directly interacts with F-actin and also contributes to management of the TJ complex through interactions with claudin-5 and JAM-A.8–10
During focal ischemia, disruption of the endothelial portion of the blood–brain barrier results in the extravasation of plasma proteins and accompanies the transit of circulating cells into the parenchyma. These events coincide with acute graded loss of β1-integrin immunoreactivity by the microvessel endothelium and αβ-dystroglycan by astrocyte end-feet that occur within 2 h following middle cerebral artery (MCA) occlusion in the non-human primate.2,11–13 Failure of normal water transit and exposure to plasma and circulating cells contribute to edema formation and parenchymal damage. Following focal cerebral ischemia, and reperfusion, in small animal models, loss of TJ proteins has been attributed to the appearance of several matrix metalloproteinases (MMPs), including MMP-2, MMP-9, or MMP-12.14–16 However, the mechanisms underlying these correlations and their signaling pathways have not been described.
In addition to the significant reduction in β1-integrin from cerebral microvessel endothelial cells that occurs during MCA occlusion in the non-human primate, 11 the losses of collagen IV, laminin, cellular fibronectin, and perlecan from the microvessel basal lamina also correlate temporally with increased vascular permeability and hemorrhagic infarction in this model.17–19 Endothelial cell β1-integrins adhere to collagen IV, laminin, and perlecan.20–22 Recently, Osada et al. 2 demonstrated that interference with the β1-integrin–collagen IV interaction by the selective anti-β1-integrin IgM Ha2/5 increased the permeability of cerebral endothelial cells in vitro and in vivo, in association with decreased expression of claudin-5. However, the impact of β1-integrin–matrix interference on the TJ apparatus, mechanism(s) of intracellular β1-integrin signaling to TJ protein expression, and the permeability changes in the cerebral microvasculature have not been rigorously examined.
The cytoplasmic domain of β1-integrin interacts with the 59 kDa integrin-linked kinase (ILK), which itself features in a number of signaling pathways that involve the cytoskeleton. 23 Because the TJ proteins are linked to the F-actin cytoskeleton, alterations in TJ organization might be expected with changes in F-actin polymerization. 24 Myosin light chain (MLC) is a downstream element in β1-integrin signaling pathways associated with the arrangement of cytoskeleton and TJ proteins in endothelial cells. 25
On the basis of reports that phosphorylated MLC (pMLC) can induce actin-myosin rearrangements that could lead to TJ disintegration and increased permeability,25,26 we hypothesized that disturbing the interaction of cerebral microvessel endothelial cell β1-integrins with their matrix ligands could increase endothelial permeability via down-regulation or reorganization of inter-endothelial TJ proteins (i) by increasing MLC phosphorylation, and (ii) altering F-actin conformation, (iii) independent of proteolysis.
Here, we demonstrate that disrupting β1-integrin–matrix interactions increases the permeability of confluent primary cerebral microvessel endothelium coincident with alterations in the TJ proteins claudin-5, occludin, and ZO-1 within the inter-endothelial borders. This is accompanied by MLC phosphorylation and alterations in F-actin structure coincident with increased permeability. These immunological and genetic studies significantly increase our understanding of microvessel barrier events that result from decreased β1-integrin expression during experimental focal ischemia.
Materials and methods
Animal subjects
All animal protocols and experiments were reviewed and approved by the University of Washington Institutional Animal Care and Use Committee. These studies were performed following institutionally approved procedures complying with the National Institute of Health’s Guide for the Care and Use of Laboratory Animals. They conform to the deliberations of STAIR and the ARRIVE Guidelines.27,28 All mice were on a C57Bl/6 background. All mice were maintained in a secured pathogen-free facility in a closed breeding colony before and during the studies.
Reagents
For functional blockade of all β1-integrins, a well-characterized purified hamster IgM monoclonal antibody (MoAb) against the mouse β1-integrin subunit was employed (clone Ha2/5, at 10 µg/mL; BD Pharmingen), as described previously (see Supplemental Materials). 2 This MoAb binds to β1-integrins in either active or inactive conformation, and interferes with β1-integrin matrix ligand adhesion. 29 The matched isotype IgM antibody (BD Pharmingen) and vehicle were used as controls.
For immunofluorescence histochemistry rabbit polyclonal antibodies against murine claudin-5 (nr 34-1600), occludin (nr 71-1500), or ZO-1 (nr 61-7300) (all Invitrogen) were used as primary immunoprobes. These were detected with Dylight 488-conjugated goat anti-rabbit IgG. For immunohistochemistry, β1-integrin was detected with an unconjugated rat anti-mouse MoAb (clone MB1.2, Millipore), which was detected with a biotin-sp-conjugated Affinipure donkey anti-rat IgG (H + L, Jackson Immunoresearch Laboratories). Extravasated IgG was detected with a biotin-sp-conjugated Affinipure donkey anti-mouse IgG (H + L, Jackson Immunoresearch Laboratories). Microglia were detected with the rabbit anti-mouse Iba1 biotin-conjugated polyclonal antibody (Wako Pure Chemical Industries, Ltd., Osaka).
For serial two-color flow cytometry studies interrogations were performed for β1-integrin with a phycoerythrin (PE)-conjugated hamster anti-mouse CD29 MoAb (nr 102208, Biolegend), for F-actin with an Alexa Fluor R488-conjugated mouse anti-F-actin MoAb (clone NH3; nr NB100-6479AF488, Novusbio), for claudin-5 with an unconjugated rabbit anti-claudin-5 polyclonal antibody (nr 34-1600, Invitrogen), and for ZO-1 with an unconjugated rabbit anti-ZO-1 polyclonal antibody (nr 61-7300, Invitrogen). For the latter two primary antibodies, the peridinin chlorophyl (PerCP)-conjugated AffiniPure F(ab′)2 fragment goat anti-rabbit IgG (H + L, Jackson Immunoresearch Laboratories) was employed as the secondary antibody. For three-color interrogations, the primary antibodies to β1-integrins, F-actin, and claudin-5 indicated above were employed.
Collagen I, collagen IV, and laminin (all Sigma-Aldrich) were used as growth substrates for the primary murine cerebral microvascular endothelial cells.
Primary cerebral endothelial cell culture
Primary cerebral microvessel endothelial cells from two- to three-month-old C57BL/6 mice (Jackson Laboratory, Bar Harbor, ME), in first or second passage (P1 or P2), were prepared as previously described (please see Supplemental Materials).12,30 Assessed by live immunostaining for the endothelial cell specific proteins CD31, platelet endothelial cell adhesion molecule-1 (PECAM-1), and by von Willebrand factor antigen, the confluent endothelial cell cultures were >99% pure. 2
Preparation of β1-integrin conditional knockout constructs
The Cre recombinase is under the control of the Mx1 promoter, which was induced to high levels of transcription (MxCre+). The Tie2Cre (Tek-Cre) transgene has the mouse endothelial-specific receptor tyrosine kinase (Tek or Tie2) promoter directing expression of Cre recombinase. The integrin deficient mouse models used, the MxCre+;β1-integrin flox/flox (β1 f/f), Tie2Cre+;α4-integrin flox/flox (α4 f/f), and the MxCre+;α4 f/f,β1 f/f, were each constructed in our laboratory as previously described. 31 The MxCre+;α4 f/f,β1 f/f mice (Dko) were generated by breeding α4 f/f mice to MxCre+β1 f/f mice. The β1-integrin deletion was induced by six intraperitoneal injections of poly-riboinosinic acid/polyribocytidylic acid (I:C) (Sigma-Aldrich), and mice were used after three weeks and before four weeks after induction.
The control animals for these studies were littermate β1-integrin flox/flox (β1 f/f) constructs that were Cre−, but induced with poly-I:C (these are termed β1+/+). Wild-type animals were C57Bl/6 that received no intervention.
TJ expression at the inter-endothelial junctions in primary endothelial cells
The impact of functional interference with established endothelial cell β1-integrin–matrix adhesion by Ha2/5 on the expression of TJ proteins at the inter-endothelial junctions (circumferential expression) was determined with the aid of quantitative video-imaging microscopy (please see Supplemental Materials). 2 Each glass slide-mounted insert was evaluated by capturing ten 1.80 mm2 non-contiguous regions of interest randomly chosen across the insert. Full-field photographic images were obtained, coded for inter-endothelial expression of TJ proteins detected by specific immunoprobes, and actively traced with resident video-imaging software, rendering the distances in µm. Cell numbers were identified by 4′,6-diamidino-2-phenylindole (DAPI) stain, so that the length of the intercellular circumferential TJ expression per cell was measured as (total TJ distance in µm)/(number of DAPI nuclei) for each image. This is termed “expression (µm/cell).” Measurements of the linear TJ expressions in the cell circumference were made by an observer blinded to the coded assignments.
Immunohistochemistry/Immunofluorescence
Antigens of interest in cerebral microvessels were identified on 10 µm frozen sections as previously described.2,19 Both single label and dual label studies were performed. Where two MoAbs were tested against each other the immunohistochemical process preceded the immunofluorescent process. For quantitation of specific epitopes (e.g. β1-integrin or IgG), video-imaging microscopy was performed as described in the Supplemental Materials to generate full color (RGB) full-field (1.80 mm2) non-contiguous images randomly chosen. These were converted to binary (0 versus 256, RGB color code) images that were then scanned and computed for the surface area of expression of the “immunoreactivity (pixels)” of the epitope using ImageJ software.
Phalloidin and F-actin in primary endothelial cells
In a time course study primary murine microvessel endothelial cells on inserts were exposed to vehicle, isotype IgM, or Ha2/5 for up to 24 h. After processing (please see Supplemental Materials), 200 µL of 1X Phalloidin iFluor 488 (ab176753, Abcam) solution was added into each insert, and further stained with fresh DAPI solution. The processed inserts were washed, mounted, and sealed with Vectashield (Vector Laboratories), and imaged under a modified Zeiss S100 Invert fluorescence microscope, equipped with AxioVision and KS 400 software (Zeiss, Oberkochen, Germany).
Western immunoblots
Immunoblots of endothelial cell lysates and secreted materials were performed as previously reported (please see Supplemental Materials). 32 The culture media was replaced with fresh endothelial cell growth medium (ECGM) containing Ha2/5 or isotype MoAb one day after the seeding on 24-well collagen IV-coated inserts (3.5 × 104 cells/insert). Proteins (2 µg/lane) were applied to 12% Tris-glycine gels, resolved, and transferred to polyvinylidene fluoride (PVDF) membranes that were probed for pMLC, MLC, and β-actin using resident techniques. Signals were detected with Pierce™ ECL Western Blotting Substrate (Thermo Scientific, USA) or Amersham ECL Prime Western Blotting Detection Reagent (GE Healthcare, UK) on X-ray film (LabScientific, Inc). Individual band optical densities were quantified using ImageJ software.
siRNA transfection
For the transfection experiments 3 × 104 primary cerebral microvascular endothelial cells were seeded in ECGM on 24-well inserts. The confluent endothelial cells were incubated for 6 h with ILK siRNA (sc-35667, Santa Cruz) or control siRNA (sc-37007, Santa Cruz), according to the manufacturer’s instructions (please see Supplemental Materials).
Cell viability assays
Cell survival in culture was determined by lactate dehydrogenase (LDH) release into the culture supernatant via kit assay according to the manufacturer’s instructions (Roche Diagnostics GmbH, Penzberg Basel, Switzerland). This is a reasonable surrogate for other markers of cell survival. 2
Permeability measurements and inhibitor studies
The barrier properties of confluent primary murine endothelial cells were assessed based on the partition of 40 kDa-fluorescein isothiocyanate (FITC)-dextrans (Sigma-Aldrich) (please see Supplemental Materials).2,33 Primary cerebral endothelial cells were seeded on 24-well collagen IV-coated inserts (3.5 × 104/insert) that were used for permeability after reaching complete confluence (after >2 days in culture). To the upper and the lower compartments, 95 µL and 400 µL of ECGM were added, respectively. Ha2/5 or isotype antibodies were added to the compartment below the inserts at 10 µg/mL. The apparent permeability coefficient (Papp, cm/s) of each group was calculated using the equation: Papp = (dM/dt)/(A × C), where dM/dt is the cumulative measured fluorescence intensity in the lower chamber per unit time (RFU/s) corrected for dilution due to sampling, A is the surface area of the insert membrane (0.33 cm2), and C is the initial concentration (RFU/mL) in the upper chamber.33,34
For the inhibitor studies, MLC kinase (MLCK) inhibitor peptide 18 (IP-18, R&D Systems), or Y-27632 (nr 688000, Calbiochem), or the combination of both (final concentrations of 10 µM and 2 µM, respectively) were added to the upper compartment and the lower compartment.
Two- and three-color flow cytometry
For two- and three-color flow cytometry primary endothelial cells were seeded on six-well inserts (7.5 × 104 cells/insert) pre-coated with collagen IV (10 µg/mL in phosphate-buffered saline (PBS)) (please see Supplemental Materials). Fresh ECGM containing Ha2/5 or isotype antibody was introduced to the cells after 72 h in culture. The endothelial cells were further incubated for 18 h at 37℃. At the end of the incubation, isolated cells were labeled with the PE-conjugated anti-mouse β1-integrin MoAb (CD29, nr 102208; Biolegend), then fixed and permeabilized in Cytofix/Cytoperm (BD Pharmingen). These processed cells were then labeled with the rabbit polyclonal antibody against claudin-5 and/or the anti-F-actin Alexa 488-conjugated MoAb diluted with Perm/Wash buffer (BD Pharmingen). The cells were washed twice again and then labeled with the PerCP-conjugated anti-rabbit IgG secondary antibody for 1 h on ice, washed twice, and re-suspended in 2% paraformaldehyde (PFA)–PBS. The PE-, FITC-, and PerCP-fluorescence intensities of labeled cells and fragments (“events”) were analyzed on a Becton Dickinson FACScan machine (San Diego, CA). All studies required capture of 10,000 events. Where appropriate, for each experiment, the mean fluorescence intensity (MFI) was calculated using FlowJo software (Tree Star, Inc, Ashland, OR).
Gelatin zymography
Sensitive quantitative gelatin zymography detected (pro-)MMP-related activities as previously described (Supplemental Materials).35,36 Quantitative band optical densitometry of the scanned gels was performed using NIH ImageJ software (NIH, Bethesda, MD).
Statistical analyses
All data are expressed as mean ± standard deviation of replicate experiments (with parallel duplicate or triplicate measures) on separate days. The number of measurements is shown in each figure. Detailed statistical methods are described in the Statistical Appendix of the Supplemental Materials, and routinely involved general linear models (GLM) or analysis of variance (ANOVA). All analyses were undertaken either in R v.3.2 (R Core Team (2015); R: A language and environment for statistical computing, R Foundation for Statistical Computing, Vienna, Austria) or SPSS v.22 (IBM Corporation, 2013). The statistician and co-authors were blinded to intervention assignment. Significance was set conventionally at p ≤ 0.05.
Results
Interference with endothelial cell β1-integrin–collagen IV adhesion, and permeability
Incubation of the murine primary cerebral microvessel endothelial cells in confluent monolayer on collagen IV with Ha2/5 for 24 h under normoxia significantly increased the permeability to 40 kDa-FITC-dextran compared to vehicle control or isotype IgM (Figure 1(a)). The flux (Papp) under Ha2/5 was significantly greater than with the isotype antibody (PappHa2/5 = 12.30 ± 2.09 × 10−7 cm/s versus PappisoIgM = 6.20 ± 1.56 × 10−7 cm/s; p = 0.0029; n = 6 each). No change in cell viability was observed (data not shown). The increase in Papp with functional blockade of β1-integrin–collagen IV adhesion confirms the observations of Osada et al.,
2
and set the conditions for the β1-integrin signaling studies in confluent cerebral microvessel endothelial cells below.
Changes in permeability (Papp) and tight junction (TJ) components of confluent primary cerebral microvessel endothelial cells at 24 h after β1-integrin–matrix interference. (a) Papp for 40 kDa-FITC-dextran significantly increased after Ha2/5 intervention compared to the vehicle control and isotype IgM (*p = 0.0029; n = 6 each). Papp under Ha2/5 was significantly higher than either control or isotype, which in turn did not differ (α = 0.05 for all pairwise multiple comparisons). (b) Circumferential expression of TJ proteins at the inter-endothelial interface. Ha2/5 (white bars) simultaneously produced significant reductions in the circumferential expression (µm/cell) of the three TJ proteins by 24 h compared to the isotype IgM antibody (black bars) when the endothelium was grown on the different matrix substrates (p < 1 × 10−33; n = 9 each). While significant differences among the three TJ proteins claudin-5, occludin, and ZO-1 were seen (p < 1 × 10−46; n = 9 each), there were no differences among the three matrix substrates (p = 0.44). (c) Profile plot (interaction plot) comparing the marginal means of TJ protein expression levels from panel B. The expression levels are significantly lower with Ha2/5 compared to isotype IgM. Claudin-5, occludin, and ZO-1 also significantly differed from one another with expression levels increasing from occludin (smallest) to ZO-1 to claudin-5 (largest) (at α = 0.05). Also, a significant interaction between the Ha2/5 and isotype IgM interventions and the three TJ proteins occludin, ZO-1, and claudin-5 was seen (p < 0.006). (d) Immunoblots of TJ protein expression (insets) from confluent primary endothelial cells following exposure to control, isotype IgM, or Ha2/5 interventions, normalized for control and β-actin (not shown). There were no significant differences in protein expression of claudin-5 or occludin, following exposure to vehicle control, isotype IgM, or Ha2/5 interventions. Expression of ZO-1 was significantly decreased relative to control following exposure to IgM or Ha2/5 (*p = 0.0012). Total protein contents of each culture did not differ (data not shown). Bars represent means of n = 3 observations. FITC: fluorescein isothiocyanate; ZO-1: zonula occludens-1.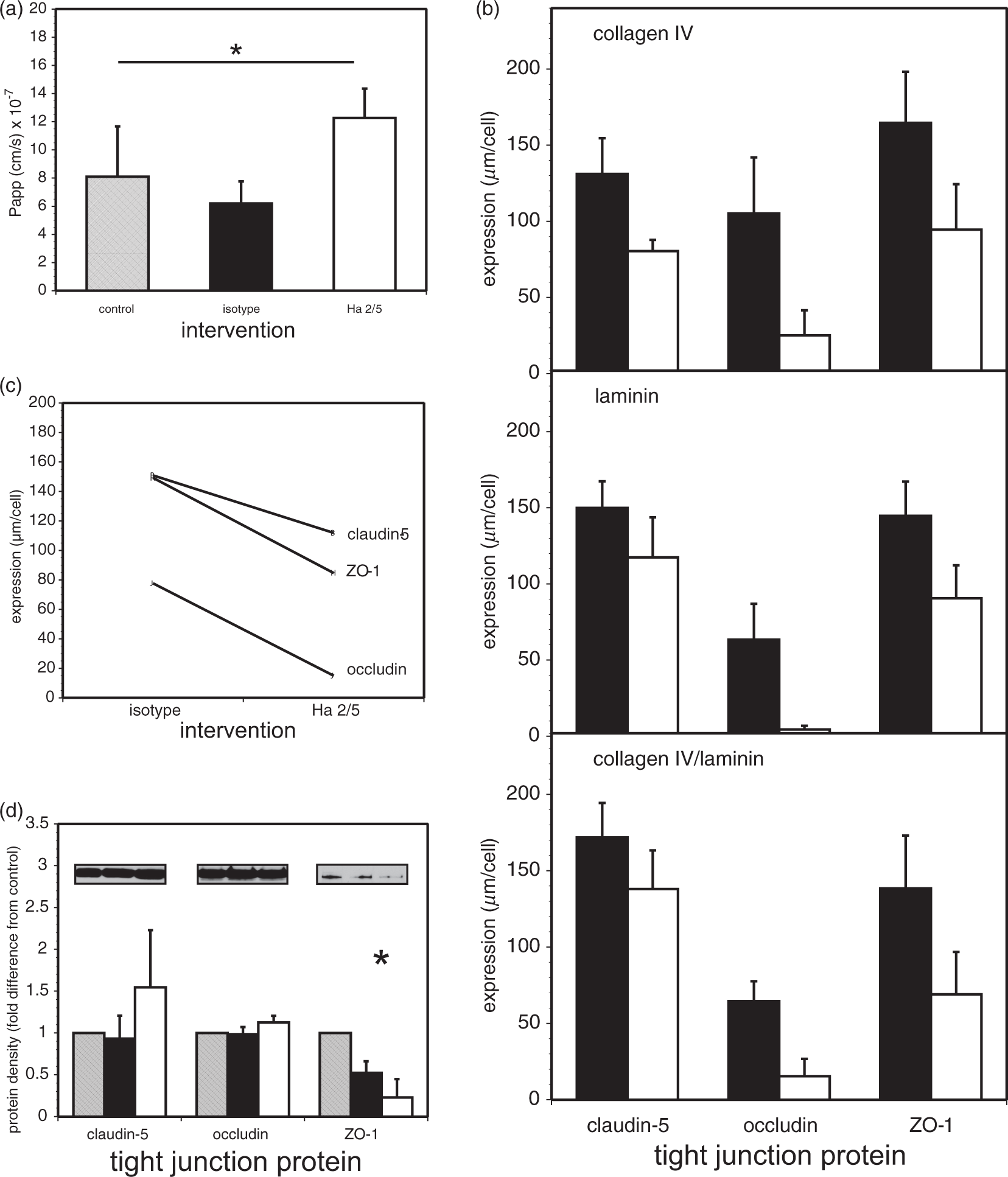
Ha2/5 decreases the expression of the TJ proteins claudin-5, occludin, and ZO-1
Interference with established β1-integrin–matrix adhesion with Ha2/5 for 18 h highly significantly altered the “circumferential” intercellular expressions of claudin-5, occludin, and ZO-1 in confluent microvessel endothelial cells in concert with the increased Papp at 24 h (F1,162 = 242.76, p < 1 × 10−33; n = 9 each) (Figure 1(b)). Significant differences among the TJ protein expressions were observed (F2,162 = 218.64, p < 1 × 10−46). However, the matrix substrate, whether collagen IV, laminin, or a 1:1 collagen IV/laminin mixture was not an important variable in the overall pattern of inter-endothelial TJ protein expression (F2,162 = 0.82, p = 0.44). Claudin-5 expression decreased further when cells were grown on collagen IV relative to the other substrates, as shown previously (Figure 1(b)). 2 Furthermore, no differences in cell morphology or number were detected (see Supplemental Materials), as reported previously. The interaction plot (Figure 1(c)) indicates that the impact of β1-integrin–matrix interference on inter-endothelial TJ expression differed across the three TJ proteins. The three profile lines would be parallel in the absence of interaction. However, while immunoblot studies demonstrated no change in claudin-5 or occludin expression, the expression of ZO-1 was significantly decreased (Figure 1(d)).
Interference with β1-integrin–collagen IV interactions increases MLC phosphorylation
In preliminary experiments a time-dependence of MLC signaling was observed (data not shown). In confluent murine primary cerebral endothelial cells grown on collagen IV, pMLC increased at 6 h after Ha2/5 exposure relative to isotype IgM (see Figure 2(a), upper panel), but had returned to baseline by 24 h (p = 0.5887) (data not shown).
Inhibition of established endothelial cell β1-integrin–collagen IV interactions induce MLC phosphorylation. (a) Effect of Rho kinase (ROCK) and MLC kinase (MLCK) inhibitors on endothelial cell pMLC and MLC expression during Ha2/5 exposure. Logged optical density ratios of pMLC/β-actin, MLC/β-actin, and pMLC/MLC differed significantly across the four conditions isotype IgM, Ha2/5, Ha2/5 + Y-27632, and Ha2/5 + IP-I8 (F3,20 = 5.18, p = 0.0083; F3,20 = 4.59, p = 0.0133; and F3,20 = 4.61, p = 0.0132, respectively). The ROCK inhibitor Y-27632 had a significant effect on the pMLC/MLC ratios generated by Ha2/5 as demonstrated by a post hoc Bonferroni comparison of the logged optical density ratios (* at α = 0.05). Immunoblot examples are shown in insets. pM: phosphoMLC; M: MLC; arrow: β-actin. Samples were taken from n = 6 separate cultures each. (b) The increased permeability of the endothelial cell monolayers to 40 kDa-dextran at 6 h caused by Ha2/5 was abrogated when inhibitor Y-27632 and IP-I8 were together with Ha2/5. From a post hoc Bonferroni comparison of the Papp by condition, both isotype IgM (black bar) and isotype IgM + Y-27632 + IP-I8 differed significantly from both Ha2/5 (white bar) and Ha2/5 + Y-27632. Ha2/5 differed significantly from Ha2/5 + Y-27632 + IP-I8 (at α = 0.05). Samples were taken from n = 14 (isotype IgM), 6 (isotype IgM + Y-27632 + IP-I8), 14 (Ha2/5), 8 (Ha2/5 + Y-27632), 8 (Ha2/5 + IP-I8), and 6 (Ha2/5 + Y-27632 + IP-I8) cultures. There was no effect on Papp at 24 h (p = 0.9012). (c) ILK suppression altered endothelial cell β1-integrin, F-actin, and claudin-5 expression (MFI of each, in three-color flow cytometry) by 24 h after transfection. From a general linear model analysis, the MFIs differed significantly by intervention (control (shaded bars), control-siRNA (black bars), or ILK-siRNA (white bars): F2,63 = 20.12, p < 1 × 10−6), and by epitope (β1-integrin, F-actin, or claudin-5: F2,63 = 281.85, p < 1 × 10−10). From post hoc overall comparisons (at α = 0.05), the three interventions were significantly different from one another, and the three epitopes were significantly different from one another. Each bar represents the mean of observations from n = 8 separate cultures. ILK: integrin-linked kinase; MFI: mean fluorescence intensity; MLC: myosin light chain.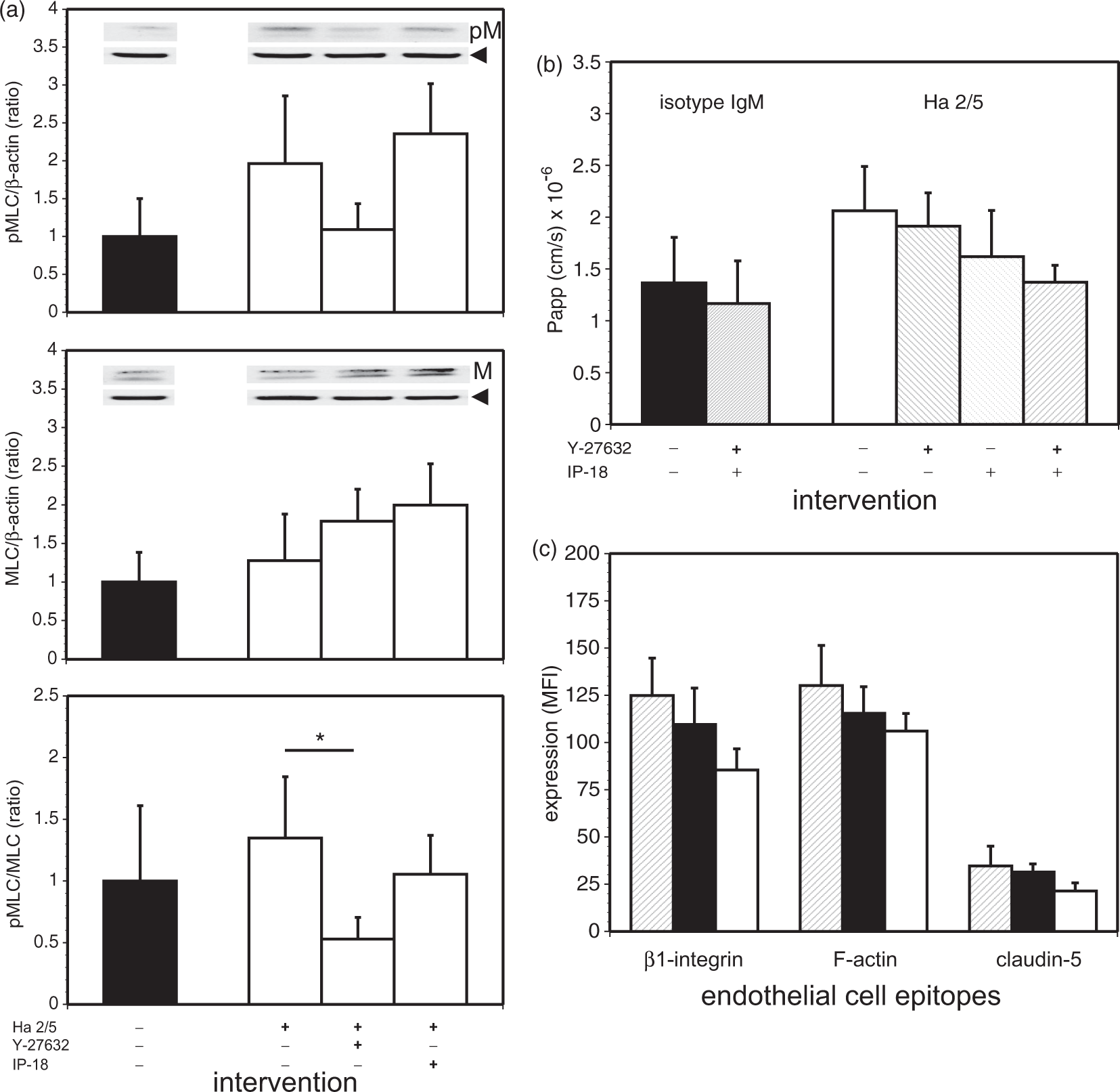
MLCK and ROCK inhibition suppresses Ha2/5-induced increased permeability
Because pMLC levels are generally regulated by MLCK or Rho kinase (ROCK),25,37,38 we interrogated the signaling cascades underlying the increase in MLC phosphorylation, and applied specific inhibitors against MLCK or ROCK. With exposure of confluent primary microvessel endothelial cells to isotype IgM or Ha2/5 a differential effect of the ROCK and MLCK inhibitors on pMLC and MLC was observed. Logged band optical density ratios for immunoblots of pMLC/β-actin, MLC/β-actin, and pMLC/MLC levels differed significantly across the four conditions isotype IgM, Ha2/5, Ha2/5 + Y-27632 (2 µM), and Ha2/5 + IP-18 (10 µM) (F3,20 = 5.18; p = 0.0083; F3,20 = 4.59; p = 0.0133; and F3,20 = 4.61; p = 0.0132, respectively; n = 6 each) (Figure 2(a)) (Supplemental Materials). The ROCK inhibitor Y-27632 had a significant effect on the pMLC/MLC ratios generated by Ha2/5 as demonstrated post hoc (p ≤ 0.05).
To determine the effects of ROCK and MLCK activity on monolayer permeability caused by interference with endothelial cell β1-integrin–collagen IV interactions in vitro, the effects of Y-27632 and IP-18 on Papp were observed after isotype IgM or Ha2/5 exposure (Figure 2(b)) (Supplemental Materials). The combination of IP-18 (10 µM) and Y-27632 (2 µM) offset the Ha2/5-induced increase in permeability of confluent endothelial cells to 40 kDa-FITC-dextrans at 6 h when applied together with Ha2/5 (p < 0.05) (Figure 2(b)), but had no effect on permeability at 24 h (p = 0.9012) (data not shown). From a post hoc Bonferroni comparison of the Papp by condition (see Statistical Appendix), both isotype IgM and isotype IgM + Y-27632 + IP-18 differed significantly from both Ha2/5 and Ha2/5 + Y-27632 (at α = 0.05). Hence, MLCK and ROCK are, in part, associated with increased permeability relatively early after interference with the established endothelial cell β1-integrin–collagen IV adhesion. These data also indicate that the increased permeability initiated by β1-integrin–matrix interference could be reversible until 6 h at least.
ILK suppression
As ILK signaling can also be modulated by β1-integrin–ligand binding, we interrogated the impact of ILK knockdown on permeability and on β1-integrin, F-actin, and claudin-5 expression using three-color flow cytometry. Using suitable reagents (Invitrogen), ILK knockdown significantly decreased β1-integrin expression by 24 h after transfection of confluent primary cerebral microvessel endothelial cells, that was accompanied by a significant decrease in claudin-5 expression, and a small significant change in F-actin (Figure 2(c)).
F-actin immunoreactivity
The structural consequences of interrupting β1-integrin–collagen IV adhesion on F-actin were further examined by flow cytometry. In three-color flow cytometry studies, a significant reduction in β1-integrin after Ha2/5 was accompanied by a variable decrease in claudin-5 in the same microvessel endothelial cells (Figure 3), whereas F-actin immunoreactivity was relatively unchanged (Figure 3(a) and (b)). This suggests that no major loss of F-actin occurred within established endothelial cell monolayers with the interruption of β1-integrin–collagen IV adhesion under these conditions (Figure 3(c)). However, limited change in F-actin immunoreactivity could still allow alterations in F-actin structural conformation.
F-actin and claudin-5 expression following β1-integrin–matrix interference in the same primary cerebral endothelial cells from confluent cultures using three-color flow cytometry. (a) Sample distributions of β1-integrin, F-actin, and claudin-5 expression in confluent primary cerebral endothelial cells (from the top) that were subject to isotype IgM (blue) or interference with β1-integrin–collagen IV adhesion by Ha2/5 (red). Note the bimodal distribution of each epitope, and the loss of β1-integrin (quantified in panel C) and changes in claudin-5 in the high expression subpopulation (seen in panel B, bottom pair). (b) Changes in the distributions of endothelial cell β1-integrin, F-actin, and claudin-5 shown in panel A from isotype IgM to interference in β1-integrin–collagen IV adhesion by Ha2/5. Note the altered distribution of claudin-5 with loss of β1-integrin, but the relatively unchanged distribution of F-actin vs. claudin-5 (middle pair). (c) The mean fluorescence intensities (MFI) of β1-integrin, F-actin, and claudin-5 described following treatment with isotype IgM (black bars) or Ha2/5 (white bars) antibodies. The MFIs differed significantly by intervention (p < 1 × 10−7) and by epitope (p < 1 × 10−10). The expression of β1-integrin was significantly reduced under Ha2/5 compared to isotype (*α = 0.05 for all comparisons), although there was no significant difference in either F-actin or claudin-5 expression. Each bar represents the mean of observations from n = 7 separate cultures.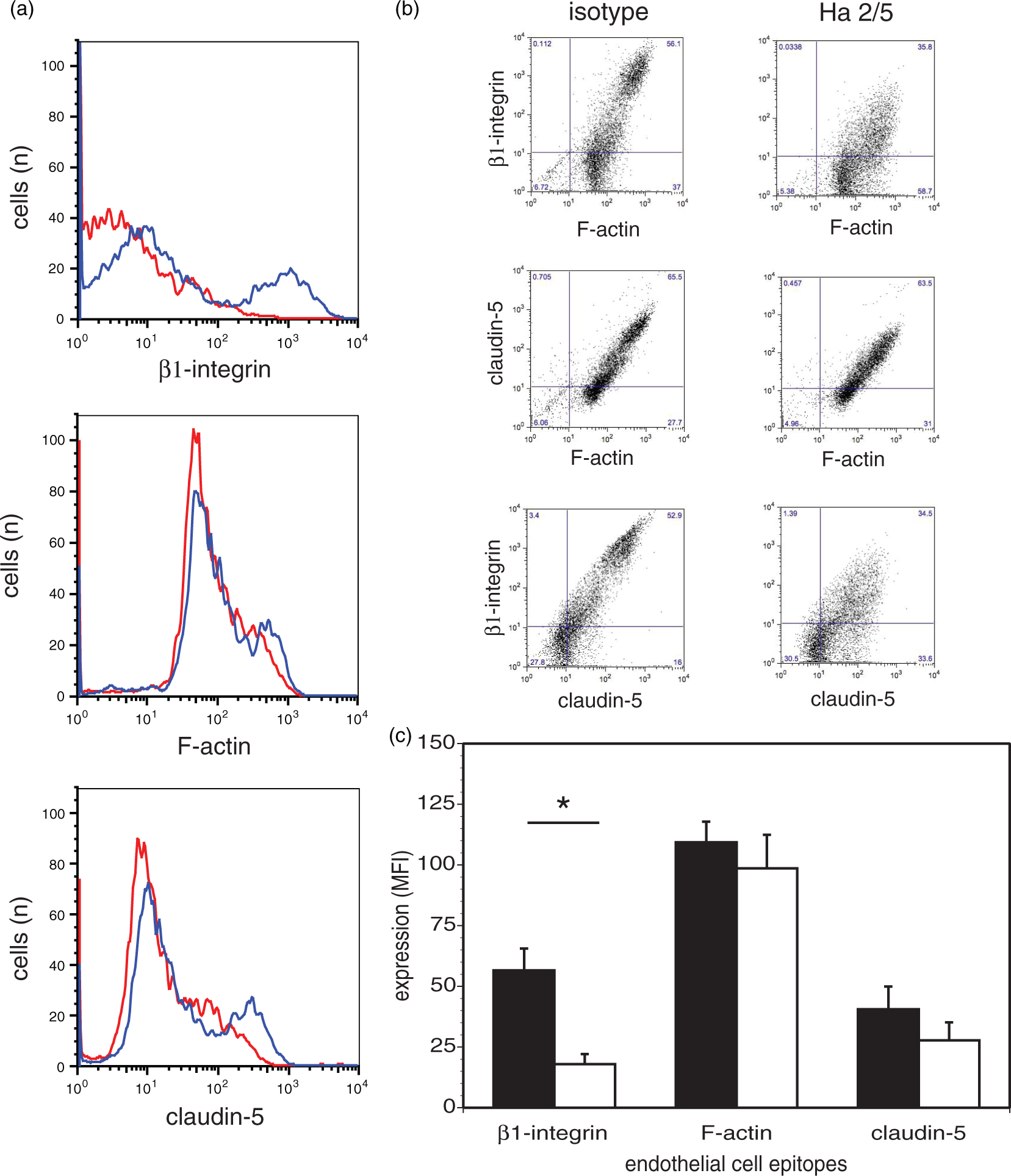
F-actin conformation
A time-dependent change in F-actin conformation in primary endothelial cells at confluence, displayed by a significant loss in total phalloidin fluorescence (Figure 4(a)), was seen at 24 h after initiation of β1-integrin–collagen IV interference (F2,9 = 20.24, p < 0.0005; n = 4 per intervention), corresponding to the significant increase in permeability (Papp) (Figure 1). Prior to this time total phalloidin fluorescence varied with intervention (Figure 4(b)). Up to 24 h of Ha2/5 exposure there was no difference in cell morphology or change in survival compared with the IgM or vehicle control (data not shown). So, although there was no major change in F-actin content after interference with β1-integrin–collagen IV adhesion or effect on cell viability, a significant change in F-actin conformation was observed (Figure 4(c)).
39
F-actin conformation changes following β1-integrin–matrix interference in confluent primary cerebral endothelial cells. (a) Left panel: fluorescent images of F-actin fibers (phalloidin) in confluent primary cerebral microvessel endothelial cells treated with vehicle control, isotype IgM, and β1-integrin-specific Ha2/5 antibodies from four separate animals (numbered) at 24 h after intervention. Montage of representative microscopic fields is from representative cultures derived from each animal subject. Right panel: montage of the binary images derived from the representative microscopic fields (fluorescent images of phalloidin) shown in the left panel. The binary images are generated as described in the “Materials and Methods” section and detailed in the Supplementary Materials. Magnification bar = 50 µm. (b) Time course of whole field phalloidin (F-actin) fluorescence intensity from cultures after intervention and conversion to binary data described in A above. The whole field fluorescence differed significantly by intervention (control, isotype IgM, or Ha2/5; p = 0.0002) and by time (3, 6, 18, or 24 h after intervention; p = 0.002). Each data point represents the mean of n = 4 observations. C: control; iso: isotype IgM. (c) Phalloidin (F-actin) fluorescence at 24 h differed significantly across the control, isotype IgM, and Ha2/5 interventions (*p < 0.0005; n = 4 per intervention). A post hoc Bonferroni all-pairwise comparisons procedure found that whole field fluorescence did not differ significantly between the control and isotype interventions, but both of these interventions had significantly greater intensities than Ha2/5 (at α = 0.05). C: control; iso: isotype IgM.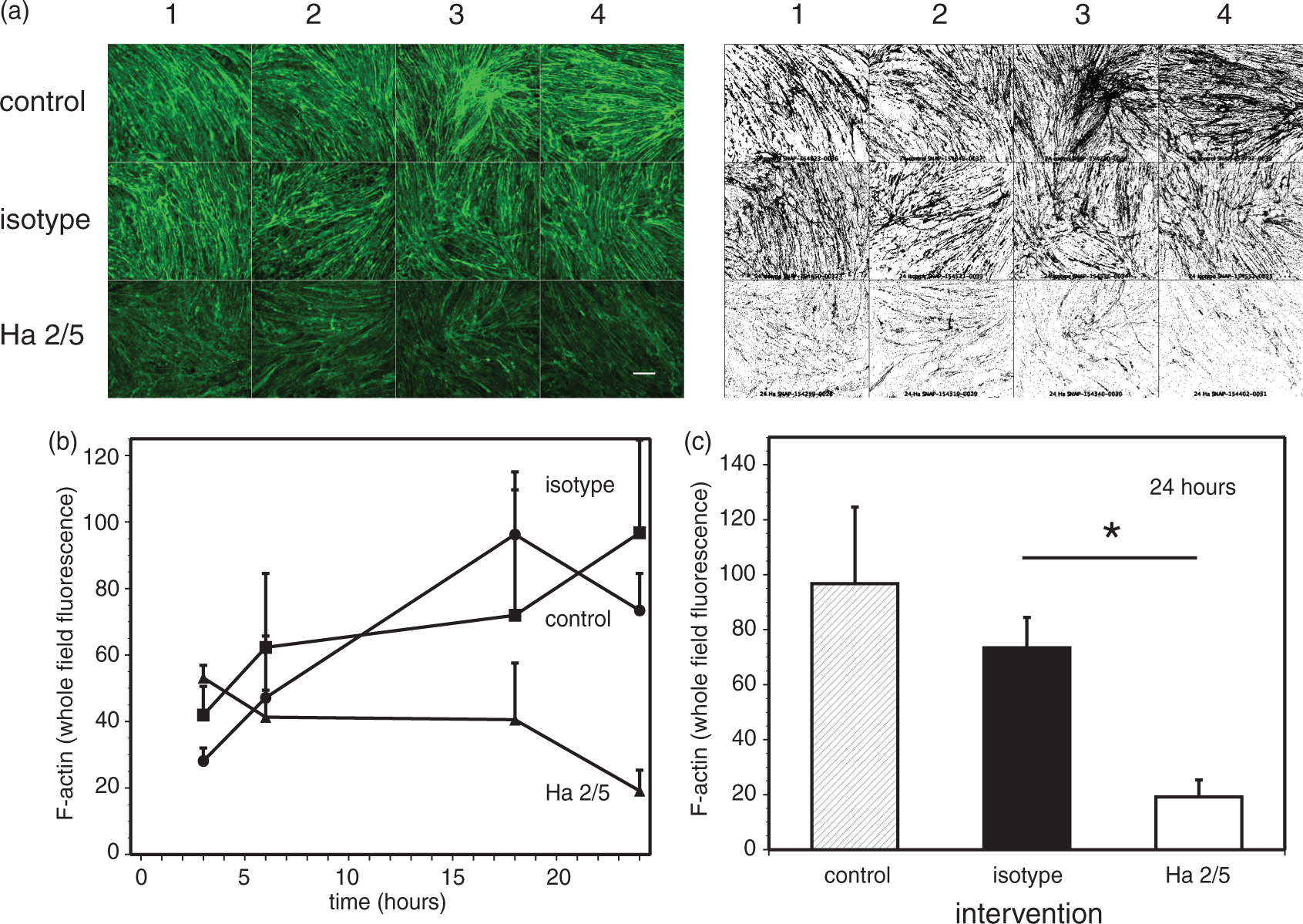
Inside-out signaling, permeability, and the matrix
The relationship of microvessel permeability to β1-integrin expression was then examined in vivo in adult whole animal β1-integrin conditional knockout (adult β1iKO) mice and their matched β1+/+ littermates.
40
The β1+/+ mice displayed β1-integrin immunoreactivity homogeneously throughout all cerebral vessel diameters, identical to wild-type. In the induced adult β1iKO animals cerebral microvessels displayed detectably less β1-integrin antigen than in the matched β1+/+ cerebral cortex (Figure 5(a)), indicating a preferential loss of β1-integrin immunoreactivity among microvessels in the adult β1iKO cortices. The β1-integrin loss varied by region.
Alterations in cerebral microvessel endothelial cell β1-integrin expression in perfused cerebral tissues from adult murine β1-integrin conditional knockout preparations. (a) β1-integrin expression in two pairs each (left and right panels) of conditional knockout mice (β1iKO), after induction of the knockout with poly-I:C, compared with matched β1+/+ litter-mate mice (that were Cre− β1-integrin flox/flox (β1 f/f) constructs, but induced with poly-I:C). The β1+/+ mice displayed β1-integrin immunoreactivity homogeneously throughout all cerebral vessel diameters, while the β1iKO mice demonstrated decreased microvessel β1-integrin expression through a range of microvessel diameters with regional heterogeneity. Left and right panels: β1-integrin immunoreactivity demonstrated 3-amino-9-ethylcarbazole (AEC) substrate (bluegray). Right panels: collagen IV immunoreactivity demonstrated with SG horse radish peroxidase substrate (red). Arrowheads indicate microvessels that display primarily collagen IV after conditional β1-integrin knockout in both sets. Note the greater intensity of the β1-integrin and the combined β1-integrin/collagen IV immunoreactivity in the respective β1+/+ images. Magnification bars = 100 µm. (b) Two-color flow cytometry studies demonstrate two populations of CD31+ cerebral microvessel endothelial cells in β1iKO subjects compared with a single population from the β1+/+ litter-mates. Overall, the mean fluorescence intensity (MFI) for β1-integrin expression was significantly decreased in the β1iKO endothelial cells compared to the β1+/+ samples (p = 0.05, single-tailed, n = 3). (c) Decreases in the microvessel basal lamina matrix content of collagen IV (see panel (a)) and laminin were observed in cerebral tissues from β1iKO subjects (white bars) compared to β1+/+ litter-mates (black bars) (**p < 0.005). The expressions were measured by computerized assisted video-imaging microscopy and rendering of the images in binary format by image processing software (see “Materials and Methods” section and Supplementary Materials). n = 5 each. (d) Increased permeability was observed in β1-integrin-deficient perfused cerebral tissues from β1iKO subjects (white bars) compared to β1+/+ litter-mates (black bars) in two separate subject pairs. Respective β1-integrin and IgG immunoreactivities were quantified as the pixel number of the binary images (0 vs. 256, a measure of surface area) within matched paired fixed microscopic fields (see “Materials and Methods” section). n = 8 paired fields each per subject. (*p < 0.05; **p < 0.005; ***p < 0.0001.).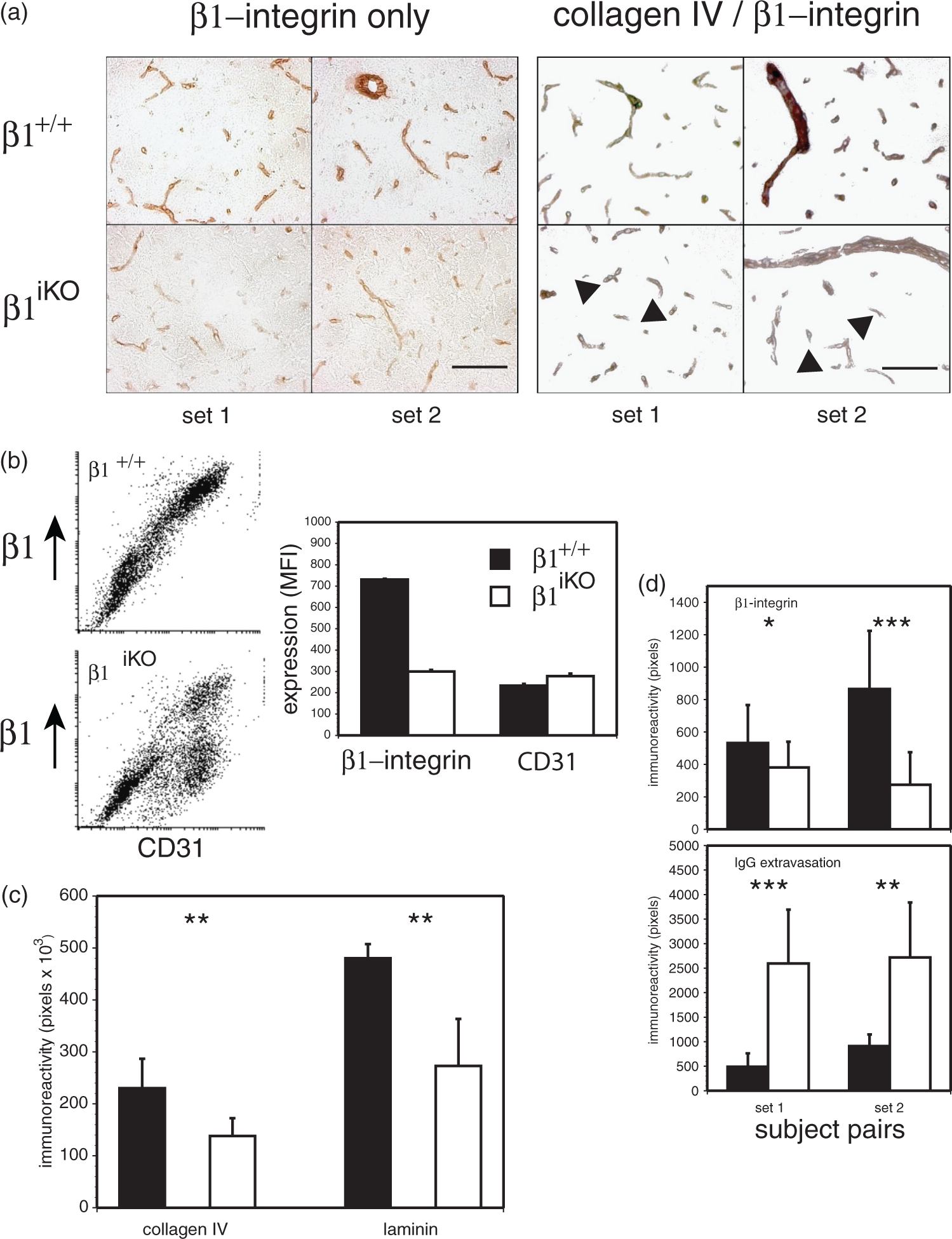
Furthermore, at least two detectable populations of endothelial cells from the β1iKO cerebral tissues reflected normal and ∼50% β1-integrin expression, consistent with variability in cerebral microvessel endothelial cell β1-integrin expression depending upon the degree of β1-integrin knockout and culture conditions (Figure 5(b)). Other populations that would have been CD31+/+ β1−/− were not detected. Overall, β1-integrin expression for the β1iKO endothelial cells was significantly less than the β1+/+ cells (p = 0.05, n = 3) (Figure 5(b)). Although hematopoietic ablation of β1-integrin is >90%, in non-hematopoietic cells it is variable and tissue-dependent.40,41 The 50% ablation estimate detected by flow-cytometry was measured following endothelial cell expansion in vitro and underestimates the true proportion of ablation, as wild-type endothelial cells proliferate while the β1-integrin deficient endothelial cells are slower to proliferate or do not at all (data not shown).
Importantly, in situ β1-integrin immunoreactivity in cortical microvessels of β1+/+ subjects roughly matched that of collagen IV by immunohistochemistry. However, the antigen content of both collagen IV and laminin (β1-integrin ligands) in the basal lamina of β1iKO subjects was significantly lower than in the matched β1+/+ cortex (Figure 5(a) and (c)). These findings are consistent with interdependence of endothelial cell β1-integrin expression and matrix protein composition of the cerebral microvessel basal lamina, and suggest the ability of microvessel basal lamina to change when endothelial cell β1-integrin decreases in the adult brain.
Direct observation of brains removed from unperfused animals suggested that cerebral volumes of adult β1iKO subjects were larger than their β1+/+ littermates. In separate perfused brain specimens, significantly increased and variable tissue IgG extravasation was observed in the β1iKO subjects compared to the β1+/+ littermates that appeared to reflect the decrease in microvessel β1-integrin expression (Figure 5(d)). Also confirmed was the variability in microvessel β1-integrin expression among and within the β1iKO subjects.
In separate experiments, the double α4β1-integrin deletion (Dko) construct was employed for preparation of cerebral microvessel endothelial cells for permeability studies as a prelude to perform Papp determinations with cultured β1iKO endothelial cells. Endothelial cells cultured from α4β1-integrin deletion mice displayed no difference in Papp for 40 kDa-dextrans compared to wild-type cells (3.30 ± 0.36 vs. 3.90 ± 1.21 × 10−6 cm/s, respectively, n = 3; p = 0.4569). Those studies imply that endothelial cells with low β1-integrin densities were supplanted by cells with the permeability characteristics of wild-type cells (see Figure 5(b)).
Regarding inflammation, histochemical stains (Wright-Giemsa) failed to show any polymorphonuclear (PMN) leukocytes within the cortex of either adult β1iKO subjects or their β1+/+ littermates. Activated microglia were not identified in the regions with or without IgG extravasation of the cerebral tissues of either β1-integrin gene mutation construct studied (data not shown).
Gelatinases and β1-integrin–matrix interference
In a number of accounts increased cerebrovascular permeability during focal ischemia has been associated with MMP-9 generation.42,43 Interference with endothelial cell β1-integrin–collagen IV adhesion in confluent monolayers by Ha2/5 had no impact on endothelial cell (pro-)MMP-2 expression or secretion, and did not promote (pro-)MMP-9 generation within the known period of β1-integrin-mediated signaling, by 6 h (Figure 6) or 24 h (data not shown). (Pro-)MMP-9 is not generated by primary murine microvessel endothelium in vitro or in vivo.
36
Gelatinase generation and secretion following β1-integrin–matrix interference in confluent primary cerebral endothelial cells. (a) Sample zymogram of (pro-)MMP-2 and (pro-)MMP-9 secreted from confluent primary murine cerebral microvascular endothelial cells exposed for 6 h to the interventions: vehicle control (lane 2), isotype IgM (lane 3), and Ha2/5 (lane 4), against human recombinant (pro-)MMP-2 (1 ng, lane 1a) and (pro-)MMP-9 (0.5 ng, lane 1b), which serve as markers. (b) Relative secretion of (pro-)MMP-2 following exposure to vehicle control (lane 2), isotype IgM (lane 3), and Ha2/5 (lane 4). No differences among these interventions were observed (p = 0.96). Samples n = 6 separate cultures each. (c) Relative secretion of (pro-)MMP-9 following exposure to vehicle control (lane 2), isotype IgM (lane 3), and Ha2/5 (lane 4) from the same samples. No differences among these interventions were observed (p = 0.09). Samples n = 6 separate cultures each. The three panels depict data from the same experiments. MMP: metalloproteinase.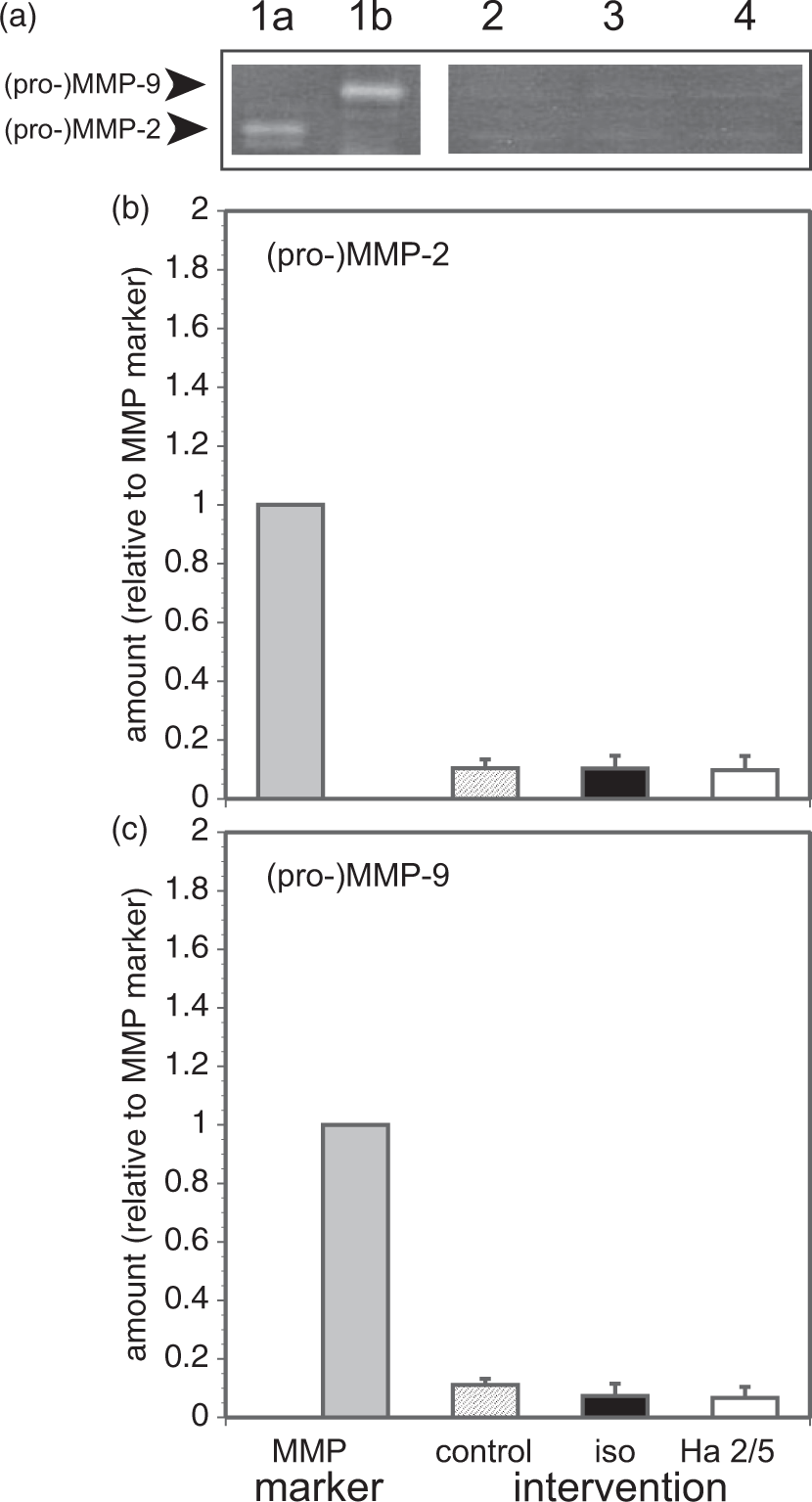
Discussion
Inter-endothelial cohesion within the cerebral microvessel permeability barrier requires intact adhesion of endothelial cells to the underlying basal lamina. For this, β1-integrin matrix adhesion receptors are predominantly expressed on the abluminal surface of endothelial cells.44,45 The acute loss of β1-integrin expression from cerebral microvessels following MCA occlusion in the primate coincides with permeability barrier failure, leakage of plasma proteins and fluid into the neuropil, and the evolution of edema. Osada et al. demonstrated that interruption of established endothelial cell β1-integrin–matrix interactions alone increased monolayer permeability in vitro and in vivo in mice, and was associated with altered claudin-5 expression. 2 The aim of this follow-on study was to (i) extend the observations of Osada et al. to the major TJ proteins, (ii) confirm the role of β1-integrins in barrier fidelity, (iii) investigate whether β1-integrin expression can affect matrix ligand expression, (iv) determine intracellular signals that connect β1-integrins to TJ protein changes in cerebral microvessels, and (v) identify mechanisms relevant to the change in permeability. We show here that specific interference with β1-integrin–matrix interactions, by competition with an anti-mouse β1-integrin-specific IgM MoAb, alters the organization of inter-endothelial TJ complexes in cerebral microvessels, and the fidelity of the permeability barrier via “outside-in” signaling, with cytoskeletal (F-actin) changes.
Integrin–matrix interactions
β1-integrins are fundamental structural components required for the maintenance of vascular endothelial cell integrity and viability. 46 Curtis et al. showed that incubation of calf pulmonary artery endothelial cells with polyclonal antibodies to integrin α5β1 elicited a time-dependent increment in permeability to albumin. 47 However, the impact on TJ protein expression was not investigated. Here, we confirm that inter-endothelial claudin-5 expression significantly decreases when β1-integrin–collagen IV interactions are interrupted, and extend the observations to the loss of ZO-1 and of occludin at the inter-endothelial interface. These changes matched parallel observations with identical material under conditions in which permeability was increased, and cell viability preserved. We suggest that all three TJ proteins are redistributed when β1-integrins are disengaged (Figure 1(d)) (Supplemental Material). In vivo conditional loss of β1-integrin subunits in adult mice caused a heterogeneous increase in vascular permeability to IgG that varied with β1-integrin expression regionally and among animals (Figure 5). These findings indicate that the configuration of the three major TJ proteins is in part controlled by “outside-in” signaling and that permeability barrier properties can be determined by endothelial cell β1-integrin–matrix adherence.
Relevant to these interactions is the composition of the matrix. In the CNS, endothelial cell β1-integrins interact with collagen IV, laminin, and perlecan,20,22 while the presence of β1-integrins and perlecan determine the organization of laminin within the cell surface. 21 In β1-integrin conditional knockout animals, loss of microvessel β1-integrin expression in the adult brain accompanied concomitant loss in basal lamina collagen IV and laminin content. The degree to which matrix composition can modulate β1-integrin expression and the permeability properties of the confluent endothelial cell monolayers is unknown. We propose that endothelial β1-integrin expression, permeability, and matrix ligand expression are linked, in addition to regional specificity of microvessel basal lamina matrix protein content. 48 This is under study.
Modulation of matrix–adhesion receptor interactions
Certain MMPs, and other matrix proteinases have been correlated with increased cerebral vascular permeability under conditions of focal ischemia, or have been correlated with basal lamina loss in several systems.2,35,42,43 Activated “MMPs” have also been reported to alter or degrade TJ proteins directly, and to cleave claudin-5 and occludin, under specific conditions in the brain.14,15,49–51 However, that inter-endothelial TJ proteins could be degraded by MMP-9 acutely has not so far been shown. The studies here demonstrate that TJ protein expression can be significantly altered independent of (pro-)MMP-2 or -9 generation and secretion by the endothelium, which do not change, and suggest alternative mechanisms via interference with endothelial cell β1-integrin–matrix adhesion.
Signaling
Extracellular environmental cues launch “outside-in” signals into the cell through integrins, and integrins interact with specific tyrosine kinases to regulate cell proliferation, differentiation, and survival. 52 The link between the phosphorylation state of MLC and inter-endothelial cohesion has been thoroughly investigated. The status of MLC phosphorylation depends on the balance of the activity of MLC kinases and MLC phosphatase. Rho-associated protein kinase (ROCK) and MLC kinase (MLCK) phosphorylate MLC, 53 and Rho-kinase inhibits MLC phosphatase, both of which can increase pMLC.37,38 Here, we demonstrate that interference with β1-integrin–collagen IV adhesion in confluent endothelial monolayers facilitates (i) MLC phosphorylation, (ii) the reorganization of the three components of the inter-endothelial TJ complex, claudin-5, occludin, and ZO-1, (iii) F-actin conformational change, and (iv) increased permeability, together.
Here, combined MLCK and ROCK inhibition together with β1-integrin blockade (Ha2/5) prevented the increased permeability changes at 6 h, but not at 24 h in vitro. Recently, Shen et al. demonstrated that MLC phosphorylation is itself sufficient for the changes in occludin and ZO-1 redistribution and increased permeability of epithelial cells in culture. 53 Separately, antibody (JB1A) blockade of β1-integrins attenuated shear stress-enhanced MLC phosphorylation in bovine aortic endothelial cells in vitro. 54 Tyagi et al., however, reported that treatment of primary rat cardiac microvascular endothelial cells with function-blocking MoAbs to α5-integrin (5H10-27) and β1-integrin (Ha2/5) together did not change the permeability to albumin, although a subtle and insignificant increase in the permeability was detected. 55 That outcome, while appearing inconsistent with our data linking β1-integrin adhesion, pMLC generation, and permeability, suggest that α-subunit specificities are relevant, or that culture conditions could be at play (see below). Nonetheless, the impact of β1-integrin–matrix interference here is consistent with the recognized roles(s) of MLC as a downstream effector of increased permeability by a number of signaling pathways. 56 More broadly, modulation of MLC phosphorylation and preservation of the binding of endothelial cell β1-integrins to microvessel matrix could provide new candidates for therapeutic approaches to preserve the endothelial permeability barrier disrupted by CNS diseases.
The central role of active β1-integrin-dependent signaling in microvessel endothelium and its requirement for barrier maintenance is fundamental, and may not be evident by deletion of a single integrin α-subunit because of compensatory effects through other β1-integrin α-partners in cerebral vascular settings. 57 Therefore, these studies are also required for identification of the relevant functional integrin α-subunit partners that maintain the barrier in confluent endothelium.
Timing of signal and subsequent changes
Preliminary time course experiments demonstrated a maximum MLC generation by 6 h after Ha2/5 exposure. Signal changes evident at 6 h post-stimulus (Ha2/5) translated into significant F-actin conformational alterations (Figure 4) and irreversible permeability changes by 24 h in this murine in vitro system. The permeability changes were reversible by 6 h. Three relevant points are that (i) this in vitro system is isolated from the normal vessel environment for the purpose of defining relationships between β1-integrin signaling and permeability, (ii) it cannot be ruled out that the competition for adhesion by Ha2/5 could also activate non-physiologic processes, and (iii) modulation of pMLC production by other signaling pathways initiated by β1-integrin is also possible, and requires exploration.
In further support of a role for β1-integrin in TJ protein expression, ILK knockdown studies indicated a significant decrease in β1-integrin expression and changes in claudin-5 expression with small changes in F-actin levels within the same cells (Figure 2(c)). ILK is essential for endothelial cell survival, vascular development, and cell integrin–matrix interactions in mice. 58 Yet, there is little information about the role(s) of ILK in TJ protein expression or permeability in the CNS.
Preliminary condition-setting experiments demonstrated that the effects of Ha2/5 depend on the endothelial cell density on inserts and the timing of the cell harvest (data not shown). It is possible that the influences of β1-integrin–matrix blockade on endothelial cell signaling and permeability depend also on the arterial, venous, or capillary origin of the mixed endothelial cells, the external stimulus applied, the timing when the signals are investigated, and the growth factors contained in the endothelial growth medium used for the cell cultures and assays. Overall, these experiments indicate that the MLC signal transduction pathways link alterations in endothelial β1-integrin–matrix interactions, TJ protein function, and permeability in murine cerebral microvessel endothelial cells.
Focal adhesion complexes and F-actin–TJ interactions in endothelium
β1-integrins participate in focal adhesion complexes (FACs). The interactions of the intracellular structural elements that secure the TJ proteins and the signaling processes involved in their positioning are not completely understood. Polymerized F-actin participates in integrin-based-FACs, whose characteristics can reflect mechanical forces in addition to adhesion complex composition. 59 One mechanism for increased permeability could involve F-actin disassembly causing instability of ZO-1 which links F-actin to claudin-5. 60 Among known triggers for changes in F-actin are stimuli for cellular migration and flow, which also involve cell adhesion. In confluent endothelial cells significant conformational changes in F-actin were apparent by 24 h following β1-integrin adhesion blockade in a time course study (see Figure 4), that paralleled large changes in the β1-integrin subunits with only modest changes in F-actin and claudin-5 content (see Figure 3). There was no change in viability or morphology ((see Supplemental Materials) and del Zoppo et al. 36 ). The studies here support the hypothesis that alterations in endothelial cell β1-integrin-mediated matrix adhesion can subtly affect F-actin structure, and by extension variably disrupt the endothelial barrier.
Relationship to oxygen-glucose deprivation (OGD)/focal ischemia
This approach to interfering with endothelial β1-integrin–matrix adhesion (i) is taken as a potential mimic of acute loss of endothelial cell β1-integrins that have been observed in the non-human primate following MCA occlusion, 11 and (ii) is a necessary control to examine the mechanisms of endothelial β1-integrin changes during and following experimental ischemia in vitro. MCA occlusion in the non-human primate is accompanied by the concurrent acute appearance of (i) edema and (ii) matrix protease generation, with (iii) loss of matrix in the microvessel basal lamina in the ischemic core, and (iv) variable hemorrhagic transformation. It has been postulated that specific matrix proteases (e.g. cathepsin-L, certain MMPs) released under ischemic conditions could account for the degradation of microvessel matrix. 61 This could in turn cause disordered matrix adhesion by several mechanisms, including loss of β1-integrins, that could conceivably result in MLC phosphorylation within the endothelium. Events or interventions that decrease endothelial cell MLC phosphorylation could maintain TJ expression. Further work on these aspects is underway.
Another consequence of these findings relates to endothelial cell morphology in the setting of focal ischemia. A number of reports have indicated that during MCA occlusion in animal models swelling of the vascular endothelium can occur, while other reports distinctly demonstrate that the endothelial thickness appears normal within the ischemic territories. 62 The experiments here suggest that partial depolymerization, due to interference with β1-integrin–matrix adhesion, might cause F-actin untethering and, hence, cellular shape change, subject to the composition and osmolality of the perfusion fluids used for sample acquisition.
Limitations
In addition to specific issues already mentioned, other limitations might constrain the interpretation of our results. The primary P1/P2 microvessel endothelial cultures here derive from a mixture of arterial, venous, and capillary endothelial cells and have heterogeneous endothelial phenotypes and perhaps cell adhesion properties. Co-culture of endothelial cells with astrocytes increases inter-endothelial TJ expression and barrier function, 63 and cerebral microvessel endothelial cells express specific proteins only in co-culture with astrocytes. 64 Because αβ-dystroglycan-dependent signaling is associated with astrocyte–matrix adhesion, 33 and degradation of the matrix can cause functional disruption of the barrier it will be important to investigate the effect of astrocytes and their activation state on the matrix–endothelial β1-integrin–pMLC axis for TJ maintenance and blood–brain barrier fidelity. Moreover, other cells comprising the neurovascular unit, not involved in this study, including pericytes and neuronal input, are known to contribute to barrier integrity.
Implications
These observations support the novel thesis that adherence of microvessel endothelium β1-integrins to the subjacent basal lamina is essential to maintain intact the blood–brain barrier by the proper positioning of the TJ proteins, via β1-integrin-dependent MLC signaling and F-actin conformation. Intact β1-integrin adhesion would explain the normal stability of the microvessel permeability barrier. Implied by these observations is that any process(es) that interrupt β1-integrin–matrix interactions might alter TJ configurations and thereby increase permeability. Removal of β1-integrins acutely, or sustained or permanent interference with endothelial β1-integrin function could have an irreversible impact on the permeability barrier, as observed acutely during focal ischemia.2,11 Other early in vivo events during focal ischemia, including edema, matrix protease generation, and innate inflammation may prevent restitution of β1-integrins. However, in this simple in vitro model the changes in permeability appear independent of protease-mediated TJ loss as no generation of MMP-9 (or MMP-2) is observed. Therefore, increased endothelial permeability does not require matrix, TJ, or adhesion receptor proteolysis; although, it does not preclude the presence of proteases under specific conditions.
Another implication is that any disorder or process of the astrocyte end-feet or neuropil that interferes with endothelial cell β1-integrin expression and/or alters FAC structure would be likely to alter the permeability barrier by altering inter-endothelial cell TJ expression. For instance, disorders affecting the brain parenchyma could alter permeability through this avenue. Structural and functional disruption of the microvessel permeability barrier is associated with a variety of neural diseases, 65 including cerebral small vessel disease, 66 Alzheimer’s disease, 67 and multiple sclerosis. 68 Ischemic injury to the neuropil (which is more sensitive to ischemia than the cerebrovascular endothelium) may be a proximate cause of the loss of β1-integrins from the endothelium observed in vivo. 11 A corollary is that alterations in basal lamina matrix composition and/or structure, with loss or alteration in β1-integrin ligands, might lead to altered β1-integrin expression by the endothelium, thereby also causing increased permeability via “outside-in” signaling. Studies are underway to explore this.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: research grants NS 053716 and NS 038710 from the National Institutes of Health (GJdZ); the Astellas Foundation for Research on Metabolic Disorders and the Mochida Memorial Foundation (MK). The laboratory also received funds from Mr and Mrs A Gonsalves whom it wishes to thank.
Acknowledgments
We thank Ms GI Berg for her expert assistance, and Ms AL van Westrienen for organization.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
The studies were conceived and planned by Drs del Zoppo, Spatz, and Papayannopoulou in conjunction with Drs Izawa and Koziol. The experiments were performed by Drs Izawa, Gu, Osada, Kanazawa, and Hawkins, and the β1-integrin mouse constructs were prepared in the laboratory of Dr Papayannopoulou. The authors were variously blinded to the intervention assignments or to the study results of other members of the group. Dr Koziol performed the statistical analyses for all experimental studies, and prepared the Statistical Appendix (see Supplemental Materials). He received coded data sets. All authors participated in the preparation of the manuscript, reviewed the data and their analyses, and reviewed and confirmed all components of the materials here.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.