
Abstract
Increases in cortical cerebral blood flow are induced by stimulation of basal forebrain cholinergic neurons. This response is mediated in part by nitric oxide (NO) and reportedly involves both nicotinic and muscarinic receptors, some of which are possibly located in the vessel wall. In the present study, the vasomotor response(s) elicited by acetylcholine (ACh) on isolated and pressurized bovine and/or human intracortical penetrating arterioles were investigated, and pharmacological characterization of the receptor involved in this response was carried out. Acetylcholine (10−11 to 10−4 mol/L) dose dependently dilated bovine and human intracortical arterioles at spontaneous tone (respective pD2 values of 6.4 ± 0.3 and 7.2 ± 0.3 and EAmax of 65.0 ± 26.8 and 43.2 ± 30.1% of the maximal dilation obtained with papaverine) and bovine arterioles after preconstriction with serotonin (pD2 = 6.3 ± 0.1, EAmax = 80.0 ± 17.9% of induced tone). In contrast, nicotine (10−8 to 10−4 mol/L) failed to induce any vasomotor response in bovine vessels whether at spontaneous or at pharmacologically induced tone. Application of the nitric oxide synthase (NOS) inhibitor Nω-nitro-L-arginine (L-NNA; 10−5 mol/L) elicited a gradual constriction (∼20%) of the arterioles, indicating the presence of constitutive NO release in these vessels. Nω-Nitro-L-argigine (10−5 to 10−4 mol/L) also significantly blocked the dilation induced by ACh. The muscarinic ACh receptor (mAChR) antagonists pirenzepine, 4-DAMP, and AF-DX 384 dose dependently inhibited the dilatation induced by ACh (10−5 mol/L) with the following rank order of potency: 4-DAMP (pIC50 = 9.2 ± 0.3) ≫ pirenzepine (pIC50 = 6.7 ± 0.4) > AF-DX 384 (pIC50 = 5.9 ± 0.2). These results suggest that ACh can induce a potent, dose-dependent, and NO-mediated dilation of bovine and/or human intracortical arterioles via interaction with an mAChR that best corresponds to the M5 subtype.
The brain intraparenchymal microcirculation consists mainly of arterioles, small venules, and capillaries. These vessels are known to be innervated by fibers originating from brain intrinsic neurons (Reis and Iadecola, 1989; Lou et al., 1987), which can impart vasomotor as well as other functions via the release of neurotransmitters. For instance, intracortical and/or basocortical cholinergic nerve fibers have been shown to intimately associate with arterioles and capillaries in the rat (Arnerić et al., 1988; Vaucher and Hamel, 1995) and human (Mesulam et al., 1992; Tong and Hamel, 1999) cerebral cortex. These perivascular cholinergic nerve fibers may play a role in the control of local cerebral blood flow (CBF), as suggested by the cortical increases in CBF following stimulation of basal forebrain neurons (Biesold et al., 1989; Lacombe et al., 1989). Both nicotinic and muscarinic acetylcholine (ACh) receptors-some of which are possibly localized at the microvascular level-as well as nitric oxide (NO) have been implicated in these local changes in CBF (for review, see Sato and Sato, 1995).
Stimulation of muscarinic ACh receptors (mAChRs) in cerebrovascular tissue can induce either a vasodilation or a vasoconstriction depending on the species, the concentration of ACh, the presence of the endothelial cell layer, the subtypes of mAChRs present, and their localization in the vessel wall. For instance, activation of smooth muscle mAChRs with a pharmacological profile corresponding to the M1 subtype induces constriction of cerebral arteries (Dauphin et al., 1991). In contrast, endothelial mAChRs pharmacologically similar to the M3 mAChR subtype seem to mediate dilation of cerebral blood vessels (Dauphin and Hamel, 1990; Shimizu et al., 1993; Dauphin and MacKenzie, 1995). This endothelial-dependent dilation is mediated by NO or an NO derivative (for review, see Faraci and Brian, 1994). With respect to brain intraparenchymal circulation, ACh has been shown to dilate isolated rat cerebral arterioles (Dacey and Bassett, 1987) as well as microvessels in hippocampal slice preparations (Sagher et al., 1993). Except for the demonstration that this response was atropine sensitive, therein involving mAChRs (Dacey and Bassett, 1987), the mechanism(s) and the receptor subtype(s) implicated still remain to be established.
Radioligand binding studies have demonstrated the presence of both nicotinic (Kalaria et al., 1994) and muscarinic ACh receptors (Estrada et al., 1983; Linville and Hamel, 1995) in brain microvessels of several species, including humans. Additionally, we showed by radioligand binding studies, reverse transcriptase polymerase chain reaction, and second messenger assays that isolated human brain microvessels are endowed with heterogeneous mAChR subtypes (Linville and Hamel, 1995; Elhusseiny et al., 1999). Functional M2 and M5 mAChRs have been identified in human cerebromicrovascular endothelial cell cultures (Elhusseiny et al., 1999), allowing speculation that an endothelium-dependent ACh vasodilation could be mediated by either of these mAChRs.
In this study, we examined the effects of ACh and nicotine on isolated and pressurized bovine and/or human penetrating intracortical arterioles. The putative role of NO in mediating the spontaneous tone of these intracortical resistance microvessels and the ACh-induced dilation was assessed with the nitric oxide synthase (NOS) inhibitor Nω-nitro-L-arginine (L-NNA). In addition, the mAChR subtype mediating the ACh-induced dilation was characterized using mAChR antagonists with a selective profile of activity at the various mAChR subtypes, namely, 4-DAMP, pirenzepine, and AF-DX 384 (Dörje et al., 1991; Alexander and Peters, 1998; Caulfield and Birdsall, 1998). Parts of these results have appeared in abstract form (Elhusseiny and Hamel, 1998).
MATERIALS AND METHODS
Intracortical penetrating arterioles were isolated from bovine (frontoparietal cortex; Abattoir Forget and Abattoir Ecolait, Terrebonne, Québec, Canada) and human (biopsies of temporal and frontal cortex obtained at surgery for the treatment of epilepsy and with approval from the institutional research ethics committee) cerebral cortex following a similar procedure to that described by Dacey and Duling (1982). In brief, brain slices (1 to 2 mm thick) were cut parallel to the brain surface with the overlying pial layer still intact. The slices were pinned down in a petri dish in cold morpholinopropanesulfonic acid (MOPS) solution (4°C, pH 7.4 ± 0.1) with a basic composition (in mmol/L) of NaCl, 144; KCl, 3.0; CaCl2, 2.5; MgSO4, 1.5; glucose, 5; pyruvate, 2.0; ethylenediaminetetraacetic acid (EDTA), 0.02; MOPS, 2.0; and NaH2PO4, 1.2 (Dacey and Bassett, 1987). The pial membrane was reflected from the brain surface, exposing the underlying penetrating vessels, which were then isolated by gentle tugging. Upon removal, the vessels were carefully suctioned along with dissection fluid into a modified pasteur pipette to be transported into a cold holding chamber until later use for cannulation.
Vessels were maintained in vitro in an arteriograph chamber system (Living Systems Instrumentation, Burlington, VT, U.S.A.) modified for continuous on-line monitoring and maintenance of temperature (37 ± 1°C) and pH (7.4 ± 0.1) and filled with a debubbled MOPS/albumin (albumin 1%) buffer. Arterioles were placed in the chamber, cannulated, secured (with ultrafine string; Living Systems) at one end to a glass micropipette (20- to 30-μm diameter) filled with a MOPS/albumin solution, and attached to tubing leading to a pressure-servo micropump (Living Systems).
Albumin is required on the inside of the arterioles to stabilize and maintain the endothelial cell layer (Muller et al., 1993). A small amount of flow was applied to the pipette by the pressure-servo system to wash out any remains of blood or other substances from the vessel lumen. The distal end of the vessel was then sealed and secured to the opposing glass micropipette (∼40-μm diameter) with ultrafine string. Vessels were pressurized to 60 mm Hg (Dacey and Bassett, 1987), superfused (6 mL/min) with MOPS solution, and allowed to stabilize and acquire spontaneous tone (45 minutes to 1 hour) before any experimentation. Vessels were magnified (400- to 800-fold) on an inverted microscope (Leica, Montréal, QC, Canada) coupled to a video caliper (Imagen Instrumentation, Trenton, NJ, U.S.A.); on-line measurement of intraluminal diameters was performed with a closed circuit video system (National Electronics, Living Systems, Burlington, VT, U.S.A.). All compounds were added to the superfusing MOPS solution and thus were applied, at the desired concentrations, extraluminally for a period of 3 minutes. Smooth muscle reactivity was tested with 70 mmol/L K+ in MOPS.
Graded concentrations of ACh (10−11 to 10−4 mol/L) and nicotine (10−8 to 10−4 mol/L) were added sequentially to isolated bovine or human arterioles either at spontaneous tone or in bovine vessels after induction of a pharmacological tone (amounting to 76.0 ± 11.7% of the spontaneous tone) with serotonin (5-HT; 10−6 mol/L). The NOS inhibitor L-NNA (10−5 and/or 10−4 mol/L) was used (30 minutes preincubation) to study the role of NO on vessel tone and in the ACh-mediated dilation of bovine and human arterioles at spontaneous tone or after preconstriction of vessels with 10−6 mol/L 5-HT (in bovine vessels only). Papaverine (10−4 mol/L) was added at the end of each experiment to maximally dilate all vessels, including those incubated with L-NNA.
To characterize the subtype(s) of mAChRs involved in the ACh-induced dilation, inhibition curves with a fixed concentration of ACh (10−5 mol/L) were generated in the presence of increasing concentrations of selected mAChR antagonists applied to a single vessel segment. This method was chosen over the classic Schild analysis because the latter requires a series of repeated full dose-response curves, a criterion that could not be performed in a consistent manner in these delicate arterioles (unpublished observation). Bovine arterioles were thus successively preincubated (15 minutes) with different concentrations of the discriminative mAChR antagonists 4-DAMP (10−11 to 10−7 mol/L; RBI, Natick, MA, U.S.A.), pirenzepine (10−9 to 10−5 mol/L; RBI), and AF-DX 384 (10−8 to 10−5 mol/L; generously supplied by Dr. H. Doods, Boehringer-Ingelheim Pharma KG, Biberach, Germany), preconstricted with 5-HT (10−6 mol/L; Sigma, St. Louis, MO, U.S.A.), and then exposed to ACh (10−5 mol/L). A control for the reproducibility of the ACh-mediated response was performed in an identical manner as described above except that no mAChR antagonist was added.
Agonist dose-response curves were generated and the agonists maximal response (recorded EAmax) and potency (pD2 values or negative log of EC50) determined. The efficacy of the mAChR antagonists in blocking the ACh-mediated dilation was determined based on pIC50 values (negative logarithm of the molar antagonist concentration that induces 50% inhibition of the ACh response) from the antagonists concentration-response curves. The antagonists vascular potencies were compared by rank order of potency (Dörje et al., 1991; Alexander and Peters, 1998; Caulfield and Birdsall, 1998) and regression analyses to their published affinities at the cloned human mAChRs (Dörje et al., 1991).
Calculations and statistical analysis
All results are expressed as the mean ± SD. The significance of changes of diameter between control values at spontaneous tone and at different time points following application of L-NNA (Fig. 1) was determined by repeated measures analysis of variance followed by a Dunnett comparison test. The significance of the differences between the dose-response curves of ACh and ACh with L-NNA (10−5 and 10−4 mol/L; Fig. 2) was determined by one-way analysis of variance with a Bonferroni comparison between sets of data, whereas the difference between the pD2 values was analyzed by unpaired Student's t-test. The means of two measurements for ACh and ACh with L-NNA in preconstricted arterioles (Fig. 3) was determined by unpaired Student's t-test for independent observations. One-way analysis of variance followed by a Dunnett comparison test was used to analyze the inhibitory effects of muscarinic receptor antagonists on the ACh dilation (Fig. 4, top). In all cases, P < 0.05 was considered significant. All statistics and curve fittings (nonlinear regression analysis for sigmoidal dose-response curves) were performed with the software GraphPad Prism (GraphPad Software, San Diego, CA, U.S.A.).
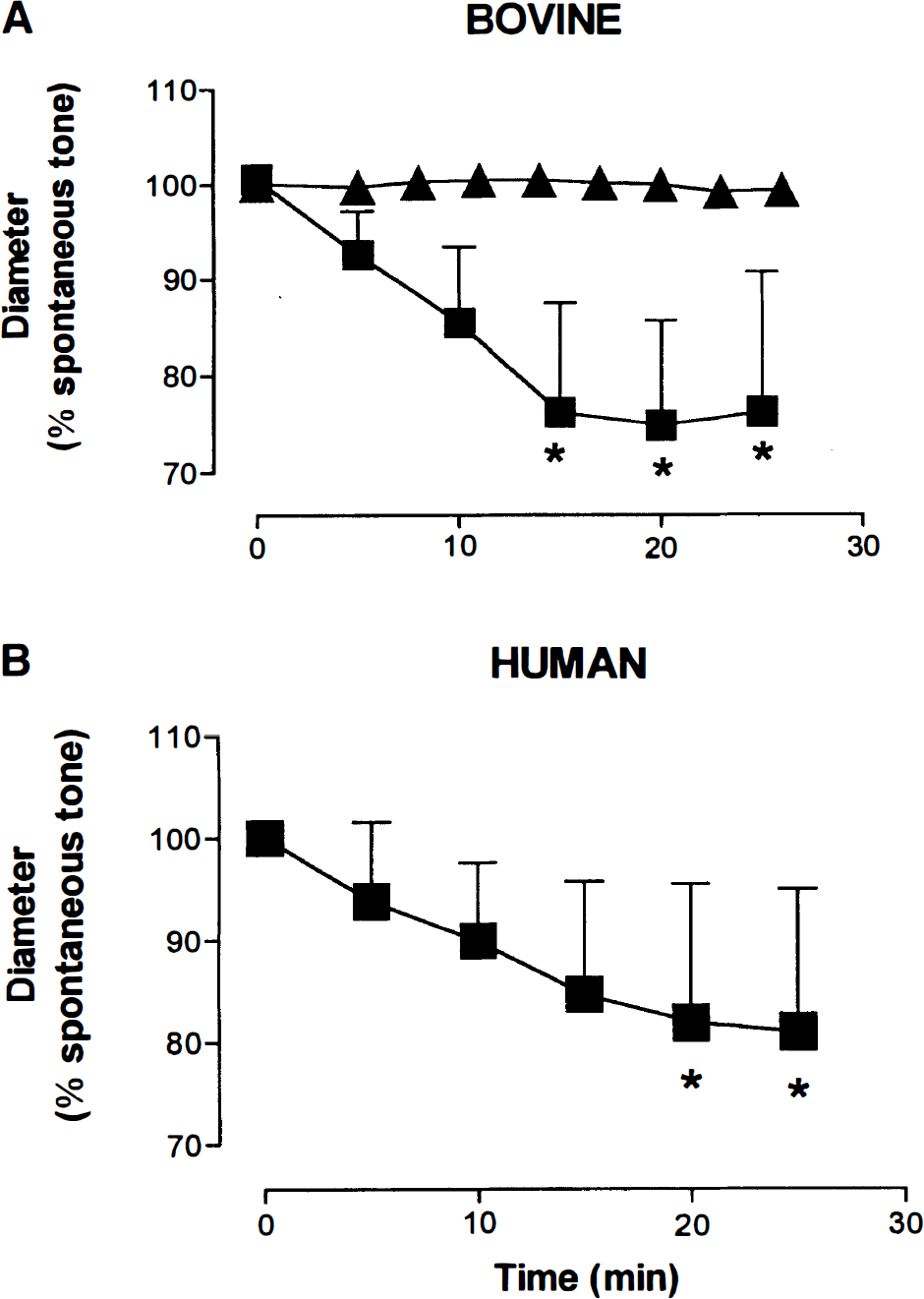
Representation of the progressive decrease over time in the diameter of bovine
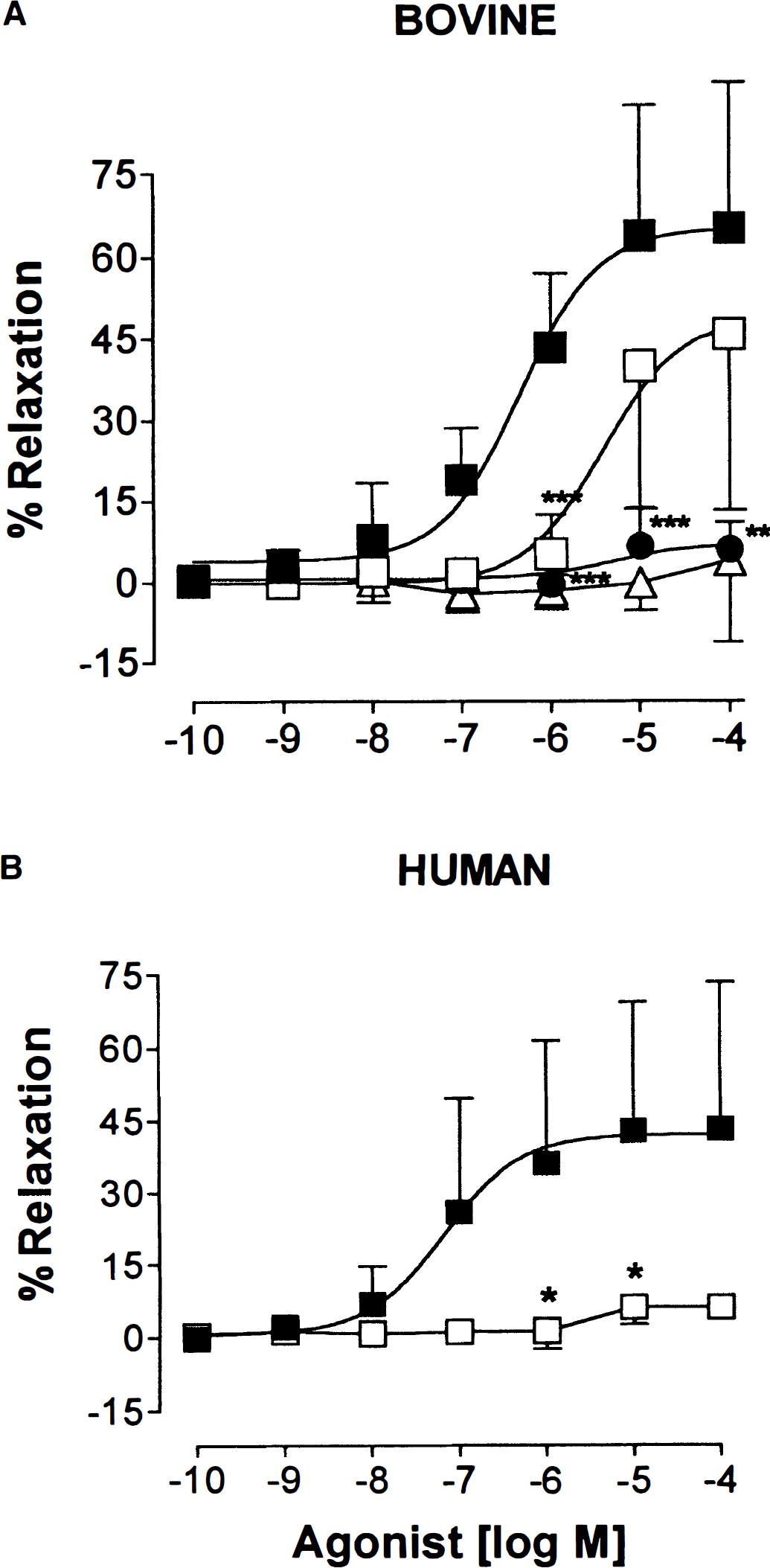
Concentration-dependent dilation elicited by acetylcholine in bovine
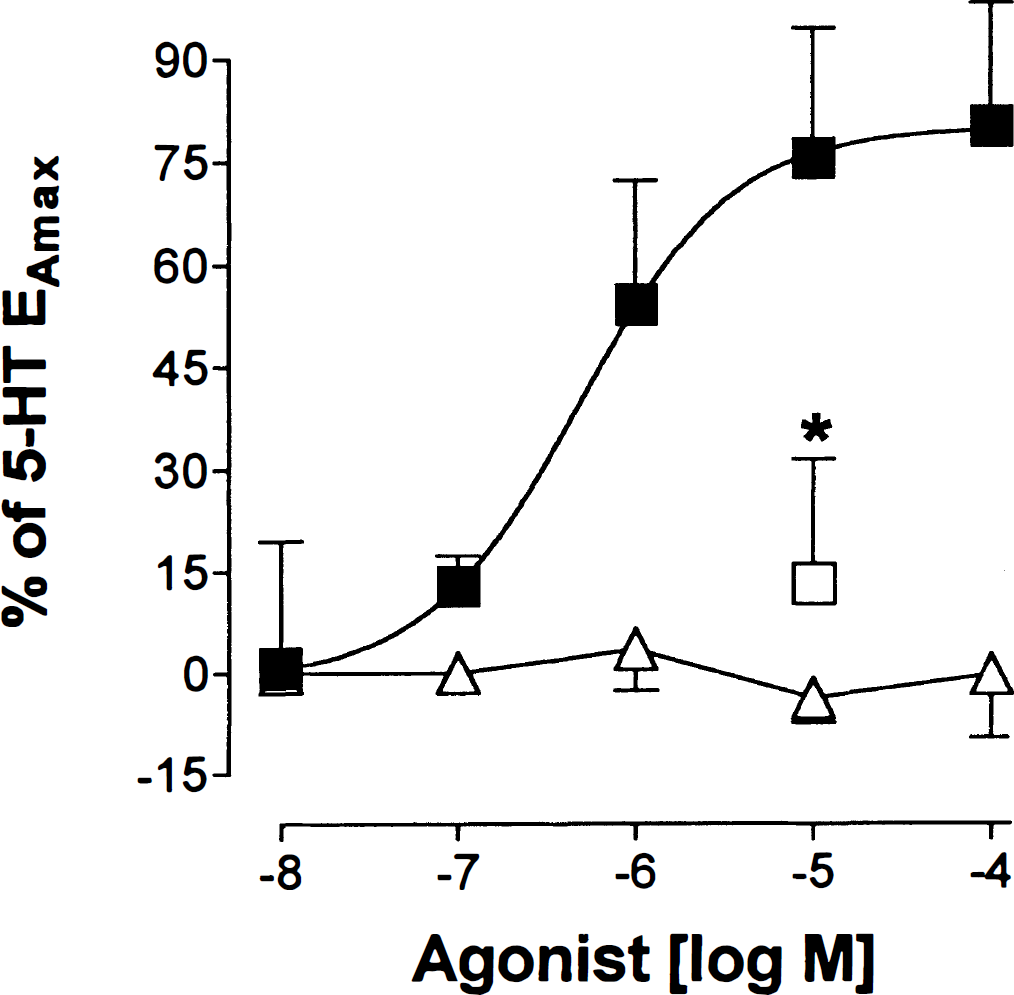
Concentration-response curves of the effects of acetylcholine (squares) and nicotine (triangles) in bovine intracortical arterioles preconstricted with 10−6 mol/L serotonin (5-HT). The inhibitory effect of 10−5 mol/L Nω-nitro-L-arginine (L-NNA; square) on the dilation induced by 10−5 mol/L acetylcholine is also illustrated (n = 5, *P < 0.05).
RESULTS
At 37°C, pH 7.4, and 60 mm Hg intraluminal pressure, the vessel luminal diameter at spontaneous tone was 46.9 ± 19.5 μm in bovine (n = 56) and 47.4 ± 19.1 μm in human (n = 14) microvessels. This spontaneously developed tone represented constrictions of 37.8 ± 12.4% (n = 17) in bovine and 41.0 ± 7.6% (n = 5) in human arterioles from the passive diameter as determined by application of papaverine (10−4 mol/L). Application of the nonselective NOS inhibitor L-NNA (10−5 mol/L) resulted in a gradual and significant decrease in diameter that amounted to 20.6 ± 13.3 and 19.0 ± 14.0% from spontaneous tone in bovine (n = 10, P < 0.05, analysis of variance) and in human (n = 4, P < 0.05, analysis of variance) vessels, respectively. This response developed gradually over a 25-minute period and in bovine vessels, as demonstrated by a vehicle control, was not due to any other time-dependent changes in the vessel (Fig. 1).
At spontaneous tone, ACh (10−11 to 10−4 mol/L) dilated bovine and human intracortical arterioles (Fig. 2), whereas nicotine (10−8 to 10−4 mol/L) failed to significantly affect bovine arteriole diameters (Fig. 2A). The ACh pD2 values for bovine and human arterioles were 6.4 ± 0.3 (n = 8) and 7.2 ± 0.3 (n = 9), respectively, and the EAmax corresponded to 65.0 ± 26.8 and 43.2 ± 30.1%, respectively, of the maximal dilation obtained with papaverine (10−4 mol/L). L-NNA dose dependently inhibited the ACh-mediated dilation in bovine vessels, with a significant shift of the curve to the right at 10−5 mol/L (pA2 value of 5.4 ± 0.4; P < 0.001) and a virtually complete abolition at 10−4 mol/L (P < 0.001; Fig. 2A). In human vessels, 10−5 mol/L L-NNA was sufficient to significantly abolish the ACh dilation (P < 0.05; Fig. 2B).
In preconstricted bovine arterioles, ACh (10−8 to 10−4 mol/L) also elicited a dose-dependent dilation (pD2 = 6.3 ± 0.1, EAmax = 80.0 ± 17.9% of 5-HT-induced tone; Fig. 3), and again, nicotine (10−8 to 10−4 mol/L) was devoid of any vasomotor effects. In such vessels, the dilation induced by ACh (10−5 mol/L) was potently inhibited (86.7 ± 18.2%; P < 0.05) by 10−5 mol/L L-NNA (Fig. 3).
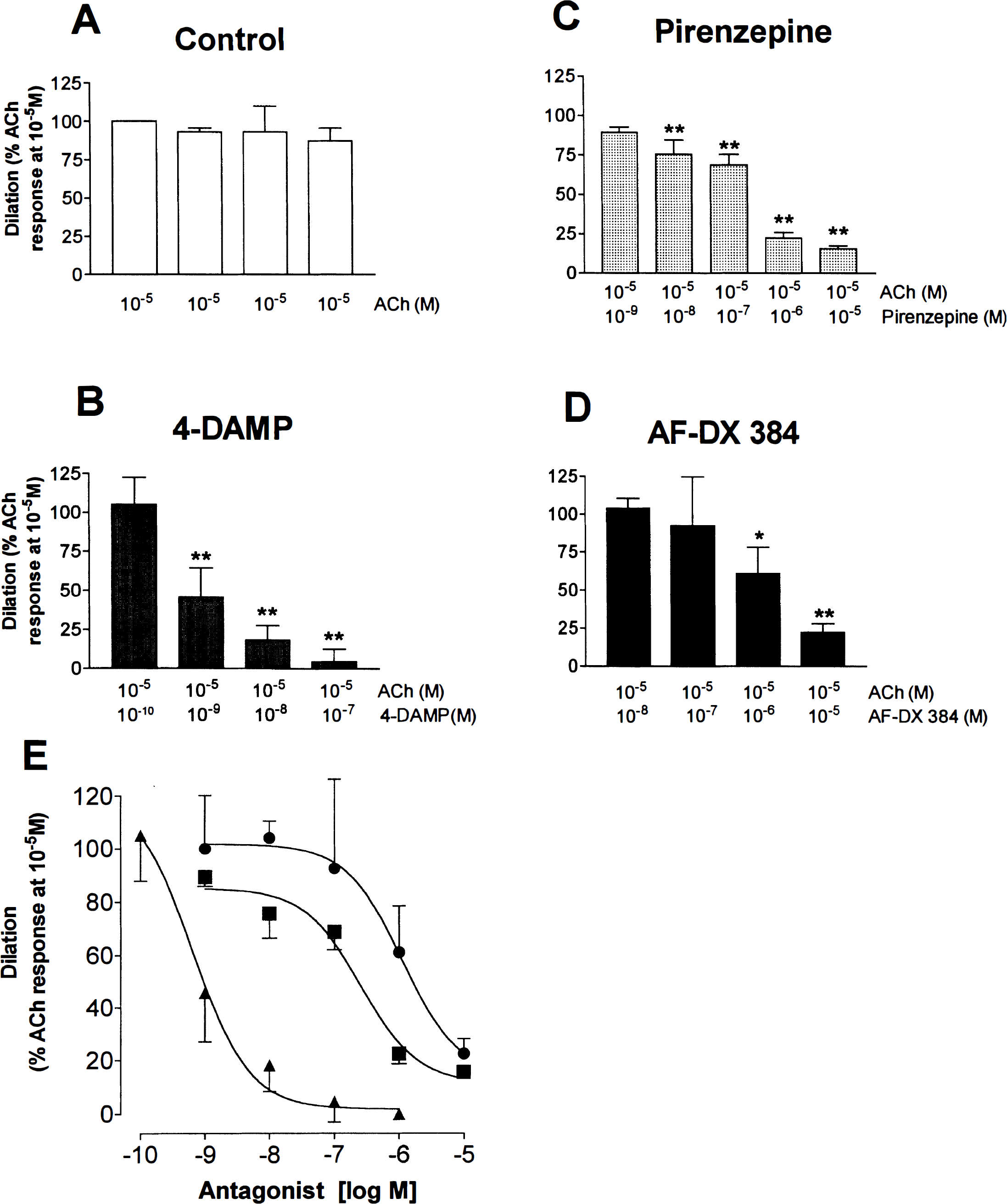
Repeated (up to four times) application of ACh (10−5 mol/L) to preconstricted bovine arterioles resulted in a reproducible dilation with no significant differences in efficacy between trials (see Fig. 4A). 4-DAMP potently inhibited the ACh-mediated dilation at concentrations as low as 10−9 mol/L, with almost complete blockade (95.5 ± 7.7%; P < 0.01) of the response at 10−7 mol/L (Fig. 4B). In contrast, the mAChR antagonist pirenzepine significantly inhibited the ACh-induced vasodilation only at relatively high concentrations with a maximal inhibition (84.4 ± 1.9%; P < 0.01) obtained at 10−5 mol/L pirenzepine (Fig. 4C). The M2/M4 mAChR antagonist AF-DX 384 was also a weak inhibitor of the ACh-induced dilation (Fig. 4D). A significant (P < 0.05) decrease was observed only at AF-DX 384 concentrations of ≥ 10−6 mol/L, with maximal inhibition (77.5 ± 5.6%; P < 0.01) occurring at 10−5 mol/L. Calculated pIC50 values from the inhibition curve for each antagonist (Fig. 4E) yielded the following rank order of potency: 4-DAMP (pIC50 = 9.2 ± 0.3) ≫ pirenzepine (pIC50 = 6.7 ± 0.4) > AF-DX 384 (pIC50 = 5.9 ± 0.2). Although limited to three values, linear regression analyses indicated a positive and significant correlation (P < 0.05) between antagonist potencies and published affinities only with the M5 mAChR (r = 0.997, slope = 0.80 ± 0.06).
DISCUSSION
The present study demonstrates that ACh can induce an NO-mediated dilation in bovine and human intracortical arterioles in vitro. The data further indicate that this response is mediated uniquely, at least in bovine microvessels, by muscarinic receptors because nicotine failed to affect microvascular tone. Moreover, the rank order of potency and affinity profile of discriminative mAChR antagonists suggest that the ACh-induced microvascular dilation is mediated by a receptor pharmacologically similar to the M5 mAChR subtype. Altogether, these findings suggest that ACh released perivascularly can regulate the tone of neighboring microvessels and thus modify local CBF.
A consistent and reproducible dose-dependent dilation was induced by ACh in both bovine and human intracortical arterioles. In agreement with other studies in rat intracerebral arterioles (Dacey and Bassett, 1987; Sagher et al., 1993) and in bovine and human major cerebral arteries (Fischer-Nakielski and Schrör, 1990; Tsukahara et al., 1989; Conde et al., 1991), we did not observe any vasoconstriction with ACh, a response previously reported in intact cat (Dauphin et al., 1991) and endothelium-denuded mouse (Shimizu et al., 1993) pial vessels. Dacey and Basset (1987) were the first to demonstrate an ACh-mediated dilation of pressurized intracerebral arterioles, albeit in rat preconstricted vessels. Acetylcholine (10−5 mol/L) also dilated norepinephrine-preconstricted intracerebral microvessels studied in a brain slice preparation (Sagher et al., 1993). In the present study, both bovine and human intracortical arterioles dilated to ACh at spontaneous tone and in the bovine vessels after preconstriction with 5-HT. The failure of ACh to vasodilate rat pressurized intracerebral arterioles without a pharmacologically induced tone (Dacey and Bassett, 1987) might be partly explained by the weaker spontaneous tone in these rat vessels, as compared with those in bovine and human vessels. Indeed, the percentage of constriction from passive diameter to spontaneous tone was slightly but significantly (P < 0.05, unpaired Student's t-test) larger in bovine (38%) and human (41%) vessels than that reported (30%) in the rat (Dacey and Duling, 1982). However, the possibility that the cholinergic response may vary based on species and vessel types or be unmasked under certain conditions (Shimizu et al., 1993) should not be excluded. In this respect, we found that ACh did not dilate rat intracerebral lentiform arteries regardless of whether or not they were preconstricted (A. Elhusseiny and E. Hamel, unpublished data).
The finding that the pressure-induced spontaneous tone developed by the arterioles in vitro is sufficient to allow neuromediator-induced dilation to take place is compatible with the suggestion that, under resting tone in vivo, perivascular release of ACh may result in local vasodilation of brain microvessels. Interestingly, the potency of ACh in inducing dilation was very similar at both spontaneous and pharmacological tone, an indication of the fidelity of the ACh response under different conditions of microvascular tone. Such a property could be highly relevant in vivo due to the presence of multiple neuromediators with vasoactive properties in the perivascular space. Additionally, the affinity values obtained for ACh in inducing dilation in intracortical arterioles (pD2 values of 6.4 and 7.2 in bovine and human vessels, respectively) compared very well with those (6.1 to 7.5) reported in other vessels, such as the pial and major cerebral blood vessels of several species, including humans (Dauphin et al., 1991; Tsukahara et al., 1989; Edvinsson et al., 1977; Kanamaru et al., 1989).
The ACh-induced dilation in intracortical arterioles at spontaneous tone (bovine and human) or after preconstriction (bovine) was potently inhibited by L-NNA, a strong indication of the involvement of NO in the mediation of this response. We cannot ascertain the exact source (endothelial or neuronal) of NO in our arterioles due to the lack of selective inhibitors for the various forms of NOS and the difficulty in removing the endothelium of these small arterioles without damaging the smooth muscle layer. However, it has previously been shown that the ACh dilatory response is abolished in endothelium-denuded human (Conde et al., 1991; Kanamaru et al., 1989) and bovine cerebral arteries (Fischer-Nakielski and Schrör, 1990). Additionally, physiological studies have indicated that endothelial NO is involved in the hyperemia observed after basal forebrain stimulation in rats (Zhang and Iadecola, 1995). We therefore suggest that in these isolated arterioles, endothelial NO synthesis and release are the most likely contributors of the ACh-mediated dilation. Our finding that inhibition of NOS with L-NNA elicited a spontaneous contractile response in both human and bovine intracortical arterioles further agrees with previous studies that suggested that cerebrovascular tone is in part mediated by constitutively active endothelial NOS (Rosenblum et al., 1990; Faraci, 1991; Kimura et al., 1994; Fergus et al., 1996).
The NO-dependent dilation described above could be mediated by either one of two types of ACh receptors identified in brain microvasculature: nicotinic (Kalaria et al., 1994) and/or muscarinic (Estrada et al., 1983). The failure of nicotine to elicit any vasomotor response(s) in bovine arterioles, in addition to the potent inhibition and/or abolition of the ACh response by selective mAChR antagonists (especially 4-DAMP), suggests that vascular nicotinic receptors are not involved in the ACh-mediated dilation. This finding would agree with the previous suggestion that the efficacy of nicotinic antagonists in attenuating the cortical vasodilation elicited by stimulation of the basal forebrain (Biesold et al., 1989) is most likely mediated by neuronal mechanisms such as the inhibition of nicotinic receptors located on basal forebrain neurons (Linville et al., 1993) or on perivascular nerve terminals releasing NO or calcitonin gene-related peptide, as documented in large cerebral arteries (Ayajiki et al., 1994).
In comparison, selective mAChR antagonists blocked the ACh-mediated dilation with varying degrees of efficacy depending on their selectivity for the different mAChR subtypes (Dörje et al., 1991; Alexander and Peters, 1998; Caulfield and Birdsall, 1998). The mAChR antagonist 4-DAMP was the most potent inhibitor of the ACh dilation, followed by the respective M1 and M2/M4 mAChR antagonists pirenzepine and AF-DX 384, which were much weaker antagonists of this response. Such a rank order of antagonist potency (4-DAMP ≫ pirenzepine > AF-DX 384), which best corresponded to that of the M5 subtype, and the significant correlation between their microvascular potencies and those at the M5 mAChR subtype together with the recent findings of only M2 and M5 mAChR mRNA expression in cerebromicrovascular endothelial cells, albeit in humans (Elhusseiny et al., 1999), all converge to suggest that the M5 subtype is the most likely candidate to mediate the NO-dependent dilation in intracortical arterioles. Such a hypothesis is further supported by the potent coupling of the M5 mAChR subtype to NO synthesis and release (Wang et al., 1994) and its shared pharmacological similarities to the M3 mAChR, a subtype previously suggested to mediate the endothelium-dependent dilation of large cerebral (Dauphin and Hamel, 1990) and small pial (Shimizu et al., 1993) arteries. These characteristics, together with the ubiquitous presence of M5 mAChR in peripheral (Phillips et al., 1997), cerebral, and cerebromicrovascular tissues, have already led to the suggestion for its role in the ACh-mediated dilation (Hamel et al., 1994; Linville and Hamel, 1995; Elhusseiny et al., 1999; Phillips et al., 1997). The availability of a selective agonist or antagonist at this site will be necessary to unequivocally confirm this suggestion.
In conclusion, this study provides new and important insights into the physiological and pharmacological properties of vessels that are known to contribute significantly to local cerebrovascular resistance. The data show that brain intracortical arterioles dilate via an NO-dependent mechanism in response to perivascular ACh application, putatively by direct activation of an endothelial mAChR corresponding best to the M5 subtype. As these vessels are located within the cortical subdivisions in which changes in CBF have been observed after stimulation of the basal forebrain in the rat (for review, see Sato and Sato, 1995), we hypothesize that the cholinergic-induced changes in flow can occur in part directly at the level of the microvessels. These findings may have implications for the regulation of CBF in pathological conditions such as Alzheimer's disease, which alter brain cholinergic basal forebrain neurons and their cortical neurovascular projections (Tong and Hamel, 1999).
Footnotes
Abbreviations used
Acknowledgements
The authors are indebted to Dr. M. Ward (McGill University) and Drs. K. S. Lee and B. R. Duling (University of Virginia) for their invaluable assistance in the development of techniques. The authors also thank Dr. A. Olivier for provision of human tissues, Mr. D. Bélanger (Ecolait Ltée) for the generous gift of bovine brains, and Ms. Linda Michel for preparing the manuscript.