
Abstract
Recent observational studies have reported that patients with low circulating levels of vitamin D experience larger infarct volumes and worse functional outcomes after ischemic stroke compared to those with sufficient levels. However, it is unknown whether a causal relationship exists between low vitamin D levels and poor stroke outcome. This study aimed to assess the effect of vitamin D deficiency on acute outcomes post-stroke. Male C57Bl6 mice (six week old) were assigned to either a control or vitamin D deficient diet for four weeks prior to stroke. Stroke was induced by 1 h middle cerebral artery occlusion (MCAO) with reperfusion. At 24 h, we assessed functional outcomes, infarct volume, quantified immune cells in the brain by immunofluorescence and examined susceptibility to lung infection. ELISAs showed that the plasma level of hydroxyvitamin D3 was 85% lower in mice fed the vitamin D-deficient diet compared with the control group. Despite this, vitamin D deficiency had no impact on functional outcomes or infarct volume after stroke. Further, there were no differences in the numbers of infiltrating immune cells or bacterial load within the lungs. These data suggest that diet-induced vitamin D deficiency has no effect on acute post-stroke outcomes.
Introduction
Stroke is the world’s second leading cause of death after only ischemic heart disease accounting 6.7 million deaths annually. 1 It is also the leading cause of adult morbidity, with one-third of all stroke survivors being left with major neurological and functional impairment. 2 Further, with the annual number of strokes increasing due to the aging population, an increasingly greater financial and social burden will be placed on the survivors and the community. 3 Therefore, major advances in understanding factors, which contribute to stroke pathogenesis, as well as novel effective ways to treat and/or prevent stroke, are desperately needed.
Vitamin D deficiency, which is prevalent in almost half the healthy population of developed countries, 4 is particularly common in patients with cardiovascular disease4–7 and in those that have suffered a stroke.8–10 Clinical and epidemiological studies have reported that low vitamin D levels are not only associated with an increase in the risk of a stroke occurring, but also to the likelihood of a poor outcome from stroke. For example, there is a stepwise increase in stroke incidence for decreasing vitamin D level quartiles. 11 Additionally, a meta-analysis pooling the results of 7 prospective observational studies found that low vitamin D levels are associated with a 52% increase in risk of incident stroke. 12 Indeed, low levels of vitamin D have now been proposed as an independent prognostic biomarker for death and functional outcome in stroke patients.8–10,13–15 Specifically, a low serum vitamin D level is independently associated with greater stroke severity at admission,13,15 a larger infarct volume,10,14 a poorer functional outcome at discharge,13,15 a higher risk of death at one year 15 and also a greater risk of recurrent stroke. 16 However, lower levels of vitamin D are also associated with a number of stroke pre-morbidities including endothelial dysfunction,17–19 hypertension,20,21 hyperglycemia,22,23 cerebral small vessel disease, 24 advanced age, 25 cognitive decline26,27 and low-grade systemic inflammation19,28–30 which are similarly associated with a poorer outcome after stroke.2,31–42 Thus, it is currently unknown as to whether a casual relationship exists between low circulating levels of vitamin D and a poor outcome after stroke.
The active form of vitamin D, 1,25-dihydroxyvitamin D3, exerts its main biological effects by binding to and activating the vitamin D receptor (VDR), which is expressed on wide range of cell types including endothelial cells, vascular smooth muscle, leukocytes, astrocytes and neurons. 5 While, best-characterized effect of vitamin D is the promotion of calcium absorption from the intestine, more recent evidence suggests that the effects of vitamin D extend beyond this. Specifically, vitamin D has been shown to control expression of a large number of genes including those responsible for cell proliferation, differentiation, apoptosis, inflammation and oxidative stress.5,43 Indeed application of exogenous vitamin D to cultured neurons can provide direct protection from excitotoxic injury and oxidative stress.44–46 Moreover, it is known that vitamin D can modulate the immune response to injury by directly polarizing CD4+ T cells towards an anti-inflammatory phenotype and preventing the release of pro-inflammatory cytokines from monocytes and dendritic cells.47–52 It has also been reported that vitamin D has anti-thrombotic properties53,54 and may promote vasodilatation by improving endothelial cell function55–57 and negatively regulating the renin-angiotensin system. 58 Given these known actions of vitamin D and recent clinical evidence demonstrating an association between low levels of vitamin D and poor stroke outcome, it is conceivable that vitamin D may offer protection in the setting of stroke. Thus, it was hypothesized that vitamin D deficiency, which is easily identifiable and treatable, would exacerbate stroke outcome, and thus could prompt clinical investigation of vitamin D supplementation for the treatment of acute ischemic stroke.
Methods
Animals
This study fully adheres to ARRIVE guidelines. 59 All experiments were approved by Monash University Animal Ethics Committee (project number MARP 2016/038) and conducted according to the National Health and Medical Research Council of Australia guidelines for the care and use of animals in research. A total of 51 male C57Bl6 mice (10–12-week-old; 25–35 g) were used for this study. Mice were housed in specific pathogen-free cages and had free access to water and food pellets. The rooms that mice were housed in were maintained at 21℃ on a 12-h light/dark cycle (lights on 7:00). Mice were excluded from the study if: during the surgical procedure to induce middle cerebral artery occlusion: (1) >0.2 ml of blood was lost (n = 1 control diet mouse); (2) the occluding clamp on the common carotid artery was in place for ≥7 min (n = 1 vitamin D-deficient mouse); (3) <70% rCBF reduction (n = 4; 1 control mouse and 3 vitamin D-deficient mice); (4) if no reperfusion occurred following removal of the filament (n = 1 vitamin D-deficient mouse); (5) if subarachnoid hemorrhage occurred (n = 2; 1 mouse from each group); (6) if death occurred prior to the designated time for euthanasia (n = 3 control mice).
Diet-induced vitamin D deficiency
At approximately six weeks of age, mice were randomly placed on either a vitamin D deficient (0 IU/kg; Specialty Feeds, Australia; SF05-033; n = 25) or control (2195 IU/kg; Specialty Feeds, Australia; SF05-034; n = 26) diet for at least four weeks prior to stroke. The vitamin D deficient diet was supplemented with 2% calcium and 1.2% phosphorus to maintain similar serum calcium and phosphorus levels between the two groups. The caloric content was also manipulated (wheat starch content 34.0 g/100 g deficient versus 37.8 g/100 g control) to ensure similar energy intake.
Middle cerebral artery occlusion
Ischemic stroke was induced by transient intraluminal occlusion of the middle cerebral artery (MCAO) as previously described.60,61 Ischemia was produced in anesthetized mice (ketamine: 80 mg/kg plus xylazine: 10 mg/kg i.p.) by occlusion of the middle cerebral artery (MCA) using a 6.0 silicone-coated monofilament (Doccol Corporation, Redlands, CA, USA). Rectal temperature was monitored and maintained at 37.0 ± 0.5℃. MCA occlusion (MCAO) was sustained for 60 min, and the filament then retracted to allow reperfusion. Both successful occlusion (>70% reduction in cerebral blood flow; CBF) and reperfusion (>80% return of CBF to pre-ischemic levels) were confirmed by transcranial laser-Doppler flowmetry (PeriMed, Sweden; 2 mm posterior and 5 mm lateral to bregma). Neck and head wounds were then closed, and the animals were allowed to recover from anaesthesia. Animals were returned to clean cages once they had regained consciousness. All surgeries were performed between 7:00 and 12:00. Close monitoring was performed and if an animal experienced severe clinical signs such as laboured breathing, immobility when lifted by the tail, unconsciousness or bleeding from the wound, they were euthanized immediately. No mice in this study required early euthanasia. Immediately after surgery, all animals received 1 ml of sterile saline (s.c.).
Functional assessment
Mice were assessed for functional deficits at approximately 30 min prior to euthanasia. This comprised a six-point scoring system for neurological deficits: 0 = normal motor function, 1 = flexion of torso and contralateral forelimb when lifted by the tail, 2 = circling to the contralateral side when held by the tail on a flat surface but normal posture at rest, 3 = leaning to the contralateral side at rest, 4 = no spontaneous motor activity, 5 = death. A hanging grip test was performed as a measure of grasping ability and forelimb grip strength in which mice were suspended by their forelimbs on a wire between two posts 60 cm above a soft pillow for up to 60 s. The time until the animal fell was recorded (a score of 0 s was assigned to animals that fell immediately and a score of 60 s was assigned to animals that did not fall) and the average time of three trials with 5 min rests in between was calculated. Locomotor activity was assessed using a parallel rod floor apparatus using ANY-maze software (Stoelting Co., Wood Dale, IL, USA) coupled to an automated video-tracking system as previously described. 62
Quantification of infarct volume
Infarct volume was the primary end-point of this study. At 24 h, post-stroke mice were killed by inhalation of isoflurane, followed by decapitation. Brains were immediately removed, snap-frozen in liquid nitrogen, and stored at −80℃. Coronal brain sections (30 µm) were collected every 420 µm throughout the brain and stained with 0.1% thoinin (Sigma, St Louis, MO, USA) to delineate the infarct. Images of the sections were captured with a charge-coupled device camera and analysed using Image J software (NIH, Bethesda, MD, USA). Cerebral infarct and oedema volumes were determined as previously described. 60 Total, cortical and subcortical infarct volumes and the anatomic distribution of the infarcts were quantified individually.
25-hydroxyvitamin D3 ELISA and calcium detection assay
Plasma (EDTA/heparin) samples were collected from vitamin D deficient and control animals at the time of euthanasia (i.e. 24 h post-stroke). Levels of 25-hydroxyvitamin D (25(OH)D3) and calcium were quantitatively measured using an ELISA kit (Immunodiagnostic Systems Ltd, UK) and colorimetric calcium detection kit (Abcam, Cambridge, MA, USA) as per manufacturers instructions.
Immunofluorescence
Six serial coronal sections (10 µm thick) per animal were collected at six regions: bregma +0.06 mm; −0.78 mm; −1.2 mm; −1.62 mm; −2.04 mm; −2.46 mm. Frozen brain sections (10 µm) were fixed in 4% paraformaldehyde for 15 min and washed in 0.01 M phosphate-buffered saline (PBS; 3 × 10 min). Sections were then blocked using 10% goat-serum (Sigma) for 60 min to block non-specific binding of the secondary-antibody. Next, brain sections were incubated overnight at 4℃ with one of the following primary antibodies: rabbit anti-CD3 (1:200), rabbit anti-myeloperoxidase (1:100; both from Abcam, Cambridge, MA, USA), rat anti-F4/80 (1:100; Biorad; Hercules, CA, USA). The following day, sections were washed (PBS; 3 × 10 min) and then incubated for 2 h in either a goat anti-rabbit Alexa Fluor 594 (1:500) or goat anti-rat Alexa Fluor 594 (1:500; both from Thermofisher Scientific, Waltham, MA, USA). Finally, sections were washed, mounted with Vectashield mounting medium containing 4,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, Burlingame, CA, USA) and a coverslip was applied. All tissue-mounted slides were viewed, analyzed and photographed with an Olympus fluorescence microscope (Olympus, Japan) and counted manually.
Bacterial analysis in lungs
Mice were euthanized by inhaled isoflurane overdose and the skin washed with 70% ethanol under sterile conditions. Lungs were removed and homogenized in sterile PBS. To determine colony forming units (CFU), 10 µL of tissue homogenate was serially diluted, plated onto brain heart infusion (BHI) agar plates (Department of Microbiology, Monash University, Australia), supplemented with 5% sheep blood, incubated at 37℃ for 18 h, and bacterial colonies were counted. Numbers of bacterial colonies were normalized to lung weight.
Statistical analysis
Data are typically presented as a combined scatter plot with mean ± standard error, with the exception of neurological deficit scores, which are presented as a scatter plot with median. All assessments were performed by an investigator blinded to experimental treatment and protocol. Mice were assigned to an experimental group by a coin toss or equivalent randomisation protocol. Studies were powered to detect a 20% difference in the primary endpoint measure (infarct volume: alpha = 5%; beta = 50%; standard deviation = 14 mm3; group size = 17) using DSS Research online tool. Statistical analyses were performed using GraphPad Prism version 6.0 (GraphPad Software Inc., La Jolla, CA, USA). Between-group comparisons were made using a Student’s unpaired t-test. Neurological deficit scores were compared using a Mann–Whitney test. If there were two independent variables, data were compared using a two-way ANOVA. No data were excluded. Statistical significance was accepted as P < 0.05.
Results
Model characteristics
Vitamin D deficiency was generated via dietary manipulation as previously described.
63
ELISAs revealed that the plasma level of hydroxyvitamin D3, the major circulating precursor, was ∼85% lower in mice fed the vitamin D deficient diet for four weeks compared to those fed the control diet (Figure 1(a); 5.2 ± 0.5 ng/ml versus 29.6 ± 0.8 ng/ml respectively). Moreover, we found that the levels of plasma calcium were similar between the two groups (Figure 1(b)). There were no differences in body weight prior to stroke between the vitamin D deficient and control groups (Figure 1(c)), suggesting that food consumption and energy intake were similar. During ischemia, rCBF was reduced to ∼85% of the pre-ischemic level in each group and was increased similarly upon reperfusion (Figure 1(d))
Model characteristics. Plasma levels of (a) hydroxyvitamin D and (b) calcium from vitamin D deficient and control animals. (c) Pre-stroke body weight and (d) cerebral blood flow recorded during and immediately after middle cerebral artery occlusion from vitamin D deficient and control animals. Control: n = 14–17 and vitamin D deficient: n = 14–18, data are presented as mean ± SEM. ***P < 0.001, Student’s unpaired t-test, where appropriate.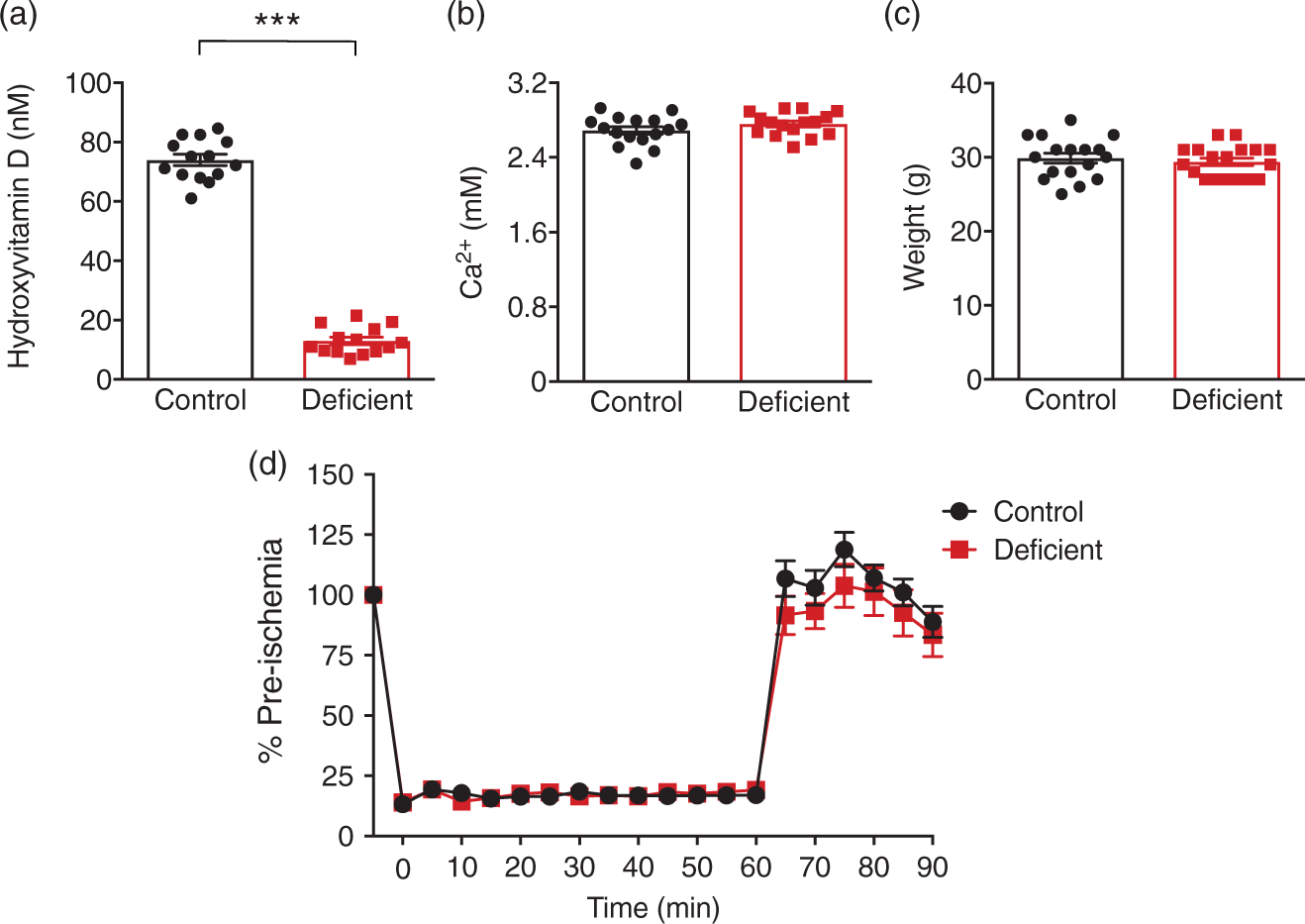
Vitamin D deficiency has no impact on cerebral infarct volume
Vitamin D deficient mice had a similar total (Figure 2(b)), subcortical (Figure 2(c)) and cortical infarct (Figure 2(d)) volume compared to controls at 24 h post-stroke. Similarly, the distribution of total infarct throughout the ischemic hemisphere did not differ between vitamin D deficient and control animals (Figure 2(e)). Moreover, we observed no difference in cerebral swelling volume between the two groups (Figure 2(f)).
Diet-induced vitamin D deficiency does not affect infarct volume at 24 h post-stroke. (a) Representative coronal brain sections from vitamin D deficient and control animals (infarct area is outlined in white). (b) Total infarct volume, (c) subcortical infarct volume, (d) cortical infarct volume, (e) distribution of total infarct area and (f) oedema volume from vitamin D deficient and control animals. Control: n = 17 and vitamin D deficient: n = 18, data are presented as mean ± SEM.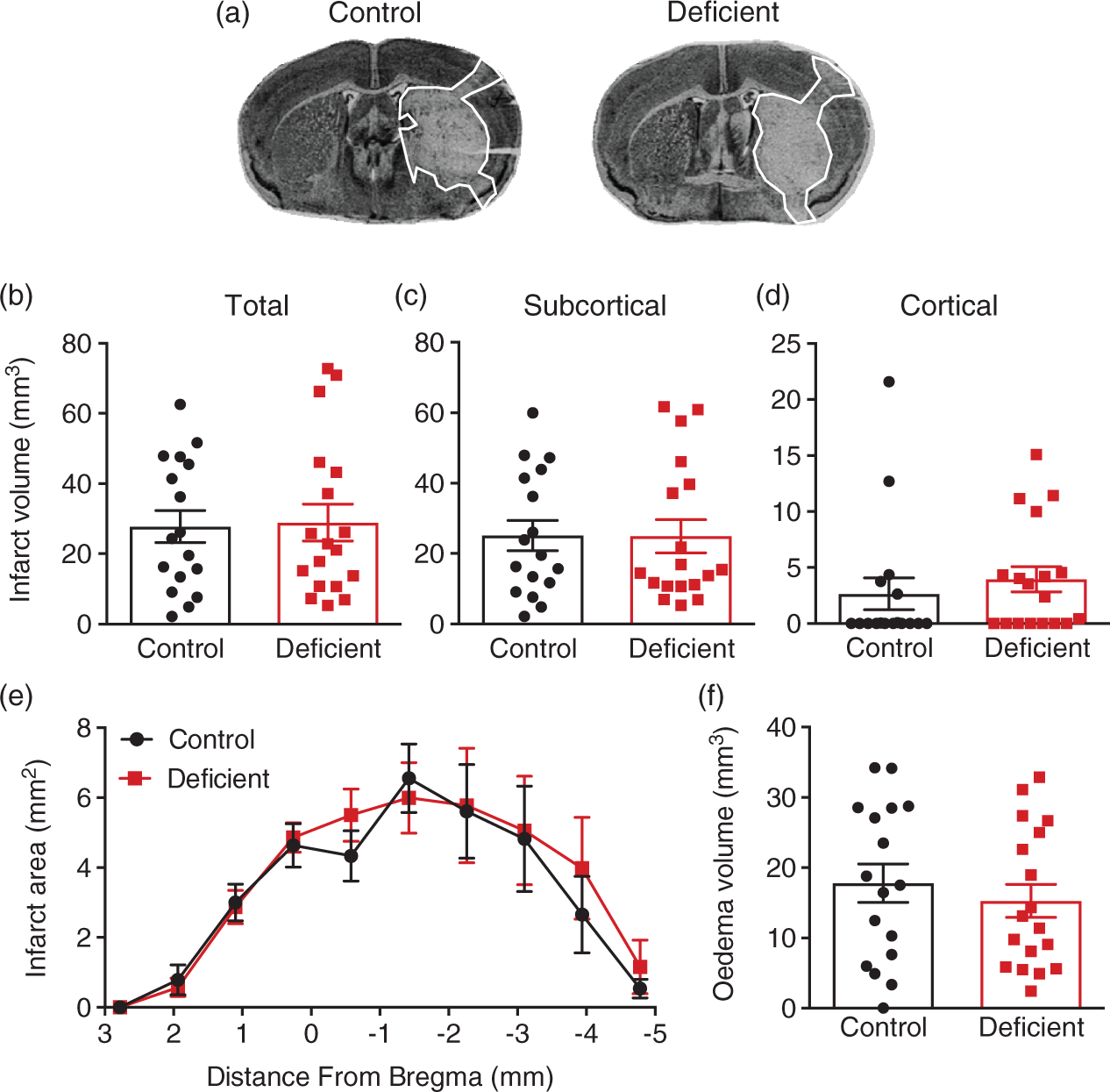
Vitamin D deficiency does not influence post-stroke functional outcome
When assessing functional outcome at 24 h post-stroke, we found there was no difference in median neurological deficit score (Figure 3(a)) or mean hanging grip time (Figure 3(b)) between vitamin D deficient and replete animals. Examining locomotor activity, there appeared to be a trend for vitamin D deficient animals to travel a greater distance (Figure 3(c)), spend more time mobile (Figure 3(d)) and move at a greater average speed (Figure 3(e)), compared to control animals; however, these data were not statistically different.
Vitamin D deficiency has no effect on functional outcome at 24 h post-stroke. (a) Neurological deficit scores and (b) latency to fall on hanging grip test of vitamin D deficient and control animals. Parallel rod floor test examining (c) total distance travelled (d) percentage time mobile and (e) average speed of vitamin D deficient and control animals. Control: n = 17 and vitamin D deficient n = 18, data are presented as mean ± SEM, with the exception of neurological deficit scores which are presented as median.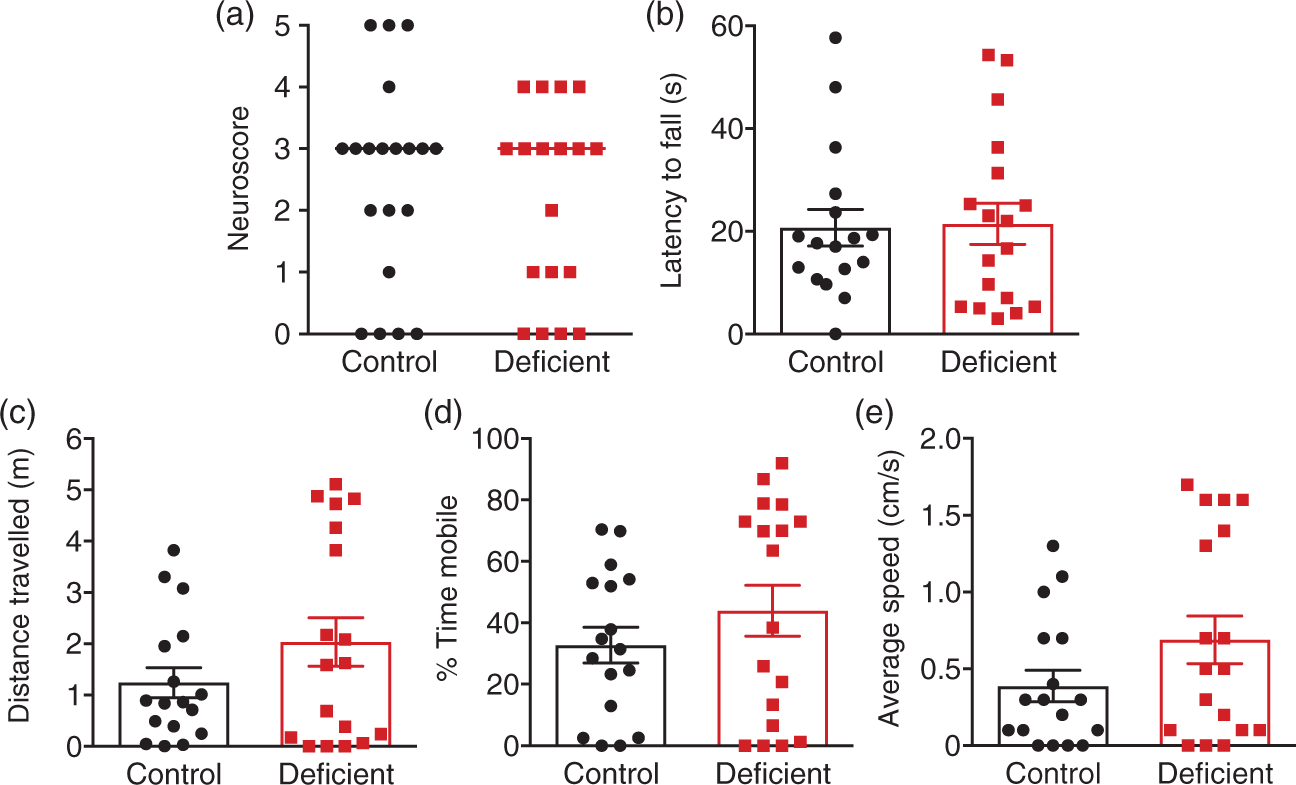
Vitamin D deficiency has no effect on leukocyte numbers in the brain post-stroke
Vitamin D has been shown to elicit anti-inflammatory actions in various different disease settings, and50,52,64,65 thus we examined the effect of vitamin D deficiency on immune cell numbers in the brain at 24 h post-stroke. We found that there was no difference in the numbers of infiltrating neutrophils (Figure 4(a)) and T cells (Figure 4(c)) between vitamin D deficient and control animals. Moreover, we found there was no difference in the numbers of macrophages (Figure 4(b)) between the two groups.
Vitamin D deficiency does not affect leukocyte infiltration into the brain following stroke. Immunofluorescence was used to quantify numbers of (a) myeloperoxidase (MPO) + cells (b) F4/80 + cells and (c) CD3+ cells per right ischemic hemisphere in control and vitamin D deficient animals. Control: n = 10–12 and vitamin D deficient: n = 10–12, data are presented as mean ± SEM. Scale bar represents 20 µm, white arrows indicate positive cells.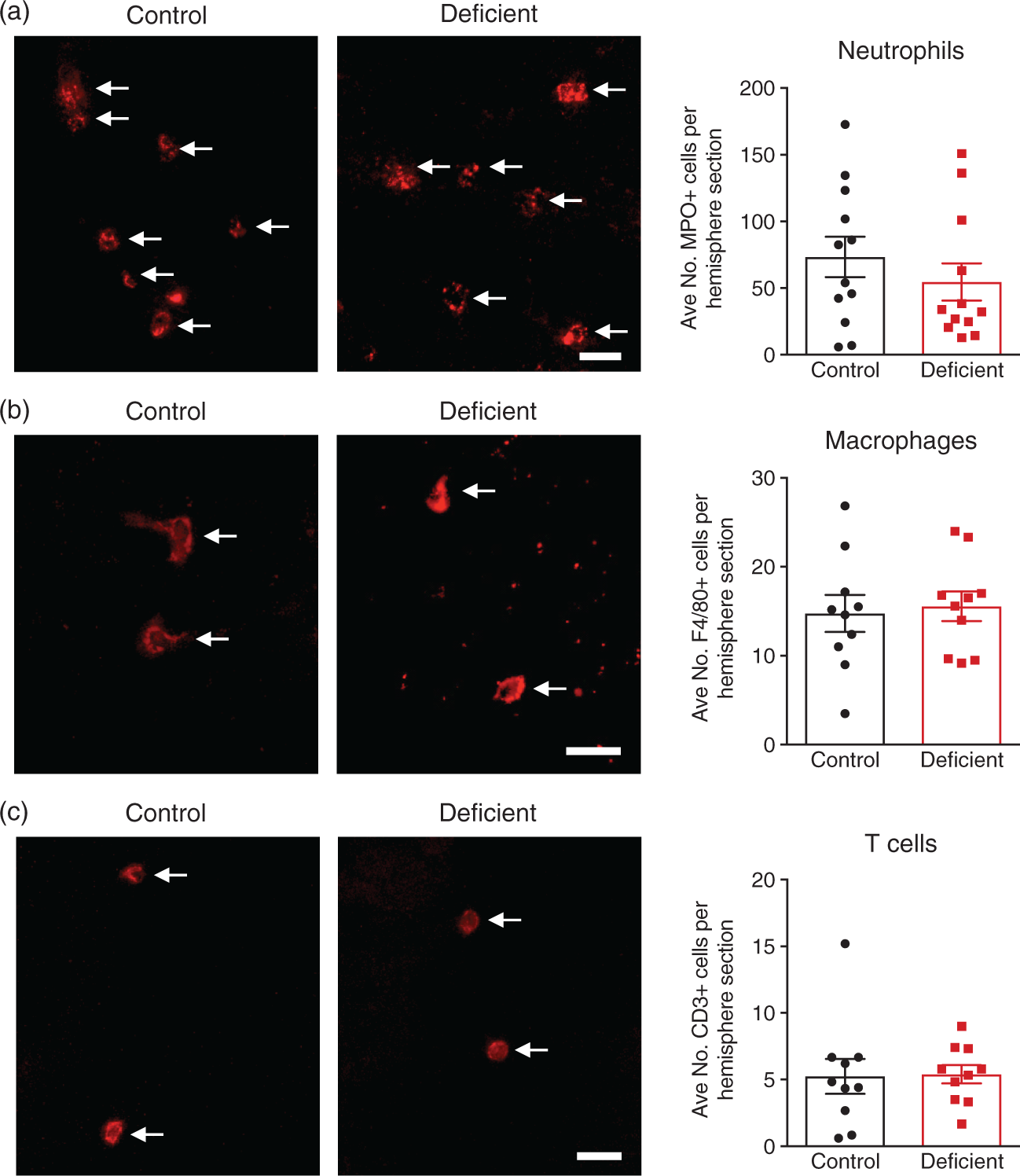
Vitamin D deficiency has no impact on post-stroke lung infection
Infection is the single greatest cause of death after the first 24 h in stroke patients
66
and is also a significant cause of morbidity.
67
Therefore, we examined the effect of vitamin D deficiency on bacterial lung infection at 24 h post-stroke. We found there was no difference in aerobic lung infection (Figure 5(a)) or lung weight (Figure 5(b)) between vitamin D deficient and control animals.
Vitamin D deficiency does not influence susceptibility to post-stroke lung infections. (a) Bacteriological analysis of lungs 24 h post-stroke. Data are expressed as Log10 of mean colony forming units (CFU)/mg tissue weight. (b) Lung weight of vitamin D deficient and control animals. Control: n = 17 and vitamin D deficient: n = 18, data are presented as mean ± SEM.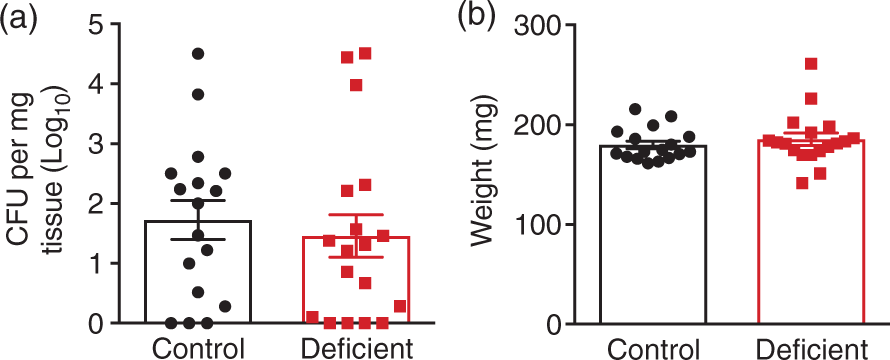
Discussion
Vitamin D deficiency is common amongst stroke patients8–10 and recent clinical studies have reported that patients with lower circulating levels of vitamin D experience a worse outcome after stroke.8–10,13–15 However, it is unknown as to whether a causal relationship exists between lower circulating levels of vitamin D and a poor outcome after ischemic stroke. In this study, we used a dietary approach to induce vitamin D deficiency in mice and evaluated the impact of vitamin D deficiency on outcomes acutely after cerebral ischemia. We found that vitamin D deficiency had no effect on the extent of brain injury or functional outcome following stroke, suggesting that lower levels of vitamin D may not directly contribute to a worse outcome.
Mice that were placed on the vitamin D deficient diet were found to be vitamin D deficient after four weeks as indicated by a plasma hydroxyvitamin D3 level <20 ng/ml. Despite this, we found that vitamin D deficiency had no impact on total, subcortical or cortical infarct volume at 24 h post-stroke. Moreover, deficiency of vitamin D did not influence functional outcomes after stroke as we found that neurological deficit scores and hanging grip times were similar between vitamin D deficient and replete animals. While there appeared to be a trend for improved locomotor activity in vitamin D deficient animals, this was not statistically significant.
It is known that inflammation and the infiltration of circulating immune cells into the brain can not only contribute to secondary brain damage68–71 but also impede functional recovery.72,73 Further, the VDR is expressed on most immune cells 5 and activation of this receptor by the active form of vitamin D can have immunomodulatory actions47,49,64 and prevent leukocyte infiltration into damaged tissue.50,52,74 Therefore, we examined the effect of vitamin D deficiency on leukocyte numbers in brain following ischemic injury. Despite the known immunomodulatory effects of vitamin D, we found that vitamin D deficiency had no effect on the numbers of neutrophils, T cells or macrophages within the brain at 24 h post-stroke. We also examined susceptibility to post-stroke infection, as infections are a major cause of stroke-related death and disability after the first 24 h and are generally a result of a dampened systemic immune system.67,75,76 However, vitamin D deficiency had no impact on this parameter either.
To date only one other experimental study has examined the effect of vitamin D deficiency on stroke outcome. 77 Using a small cohort of rats, it was reported that vitamin D deficiency resulted in a larger infarct volume at five days but not three days post-stroke. Moreover, these larger infarct volumes were associated with a worse functional outcome and a reduction in anti-inflammatory cytokines within the brain. 77 These data contrast to the current study where we found that vitamin D deficiency had no impact on infarct volume, functional deficits or cerebral inflammation after stroke in a relatively large cohort of mice. While, we only examined outcome at one day post-stroke, a number of previous studies from our laboratory have shown that infarct volume is maximal at this time-point60,61 and so it is unlikely that vitamin D deficiency would affect infarct volume at a later time-point. Nevertheless, we do acknowledge the importance of evaluating the effect of vitamin D deficiency at later time-points after stroke, particularly on functional recovery and endogenous brain repair mechanisms. Another notable difference between the two studies that is that in our study we used male mice, whereas the previous study used female rats. Aside from the species difference, studies have observed that sex-differences can exist to vitamin D deficiency and males and females respond differently to vitamin D.65,78–81 Moreover, it has been reported females are more likely to be vitamin D deficient than males, particularly after menopause. 82 As stroke is a sexually dimorphic disease, 83 where females tend to have a worse outcome than males, 2 further studies should determine whether vitamin D deficiency might affect males and females differently after stroke.
A key difference between our study and the clinical situation is that were able to prevent hypocalcaemia in the vitamin D deficient animals as the deficient diet was supplemented with 2% calcium (versus 1% in the control diet). Indeed, we confirmed there were no differences in serum calcium levels between vitamin D deficient and control animals. Thus, the diet we used enabled us to examine the effects of vitamin D deficiency independent of serum calcium levels. However in the clinic, vitamin D deficiency can lead to hypocalcaemia, which consequently causes secondary hyperparathyroidism. 5 While parathyroid hormone acts to normalize serum calcium levels, it may also have detrimental effects on cardiovascular health independent of vitamin D levels. For instance, parathyroid hormone has direct pro-inflammatory actions and can stimulate cytokine release from lymphocytes and smooth muscle cells.84,85 Moreover, parathyroid hormone is associated with hypertension, endothelial dysfunction, increased risk of stroke and death due to cardiovascular disease.86–90 However, it is unknown as to what contribution parathyroid hormone may play in influencing outcome after stroke in vitamin D deficient patients. Future pre-clinical studies should therefore be designed to examine vitamin D deficiency together with hyperparathyroidism to more closely mimic the clinical scenario and examine the potential contribution of parathyroid hormone to post-stroke outcome.
Despite emerging evidence from the clinic suggesting that lower levels of vitamin D are associated with poorer outcomes after stroke, lower levels of vitamin D are also associated with higher rates of stroke pre-morbidities such as endothelial dysfunction, hypertension, cerebral small vessel disease, hyperglycemia, low-grade systemic inflammation, cognitive decline and depression.17–24,26,27,91,92 While current studies have adjusted for some of these pre-morbidities, others are likely more difficult to control for such as cognitive decline, baseline inflammatory status and endothelial dysfunction therefore making it hard to determine causality. Further, it is also been postulated that vitamin D status may be a marker for other factors, which contribute to cardiovascular health such as diet and sunlight exposure. 93 For instance, sunlight or ultra-violet light (UV) exposure on the skin has been shown to not only stimulate the synthesis of vitamin D but also enhance the mobilization of cutaneous nitric oxide (NO) stores.94,95 Increased NO bioavailability can be protective in the setting of cerebrovascular disease and stroke, by promoting vasodilatation and helping preserve cerebral blood flow during ischemia. 96 While we included relatively young mice (6–10 weeks old) in this study, it is possible that vitamin D deficiency might produce more overt detrimental effects in aged and/or hypertensive animals. Furthermore, it is conceivable that a duration of vitamin D deficiency much longer than four weeks might predispose animals to a more severe outcome following stroke.
A limitation of this study is the variability of some of the outcome measures. Nevertheless, we have carefully applied inclusion/exclusion criteria such that we are confident that all mice were subjected to similar levels of cerebral ischaemia. We were also careful to ensure the data are strongly powered with relatively large group sizes per endpoint, and we have used littermate controls equally assigned to the two different diets. We are thus confident that our comparative group data are reliable and meaningful. We can only speculate as to why this greater variability may have occurred. All mice here were on a special diet (either vitamin D-deficient or calorie-matched control) that contained 23% more calories than our normal laboratory chow (15.8 MJ/kg vs. 12.8 MJ/kg) for four weeks, and consequently at 10 weeks of age, the mice weighed 30–35 g, which is up to 15% heavier than is commonly the case for mice at this age in our laboratory. With body growth therefore having been greater/faster than usual in some mice on these diets, it is possible that there may have been more variability in the growth of cerebral artery collaterals, which would result in smaller infarcts in such cases.
In conclusion, these data suggest that vitamin D deficiency may not directly contribute to a worse stroke outcome, at least in the acute phase after stroke. However, future studies are warranted to investigate the effect of vitamin D deficiency on longer term outcomes and also examine other factors associated with vitamin D status such as UV-light exposure and hyperparathyroidism on outcomes after stroke.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: We wish to acknowledge the support from Monash Faculty Postgraduate Scholarship (MAE), NHMRC CJ Martin Post-Doctoral Fellowship (TMDS), NHMRC Senior Research Fellowships (GRD and CGS).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
MAE conducted experiments, data analysis and manuscript drafting. CGS, BRSB, GRD and GRZ developed the study concept and design and contributed to data interpretation and manuscript writing. HK and TMDS contributed to data interpretation.