
Abstract
Hemoproteins undergo degradation during hypoxic/ischemic conditions, but the prooxidant free heme that is released cannot be recycled and must be degraded. The extracellular heme associates with its high-affinity binding protein, hemopexin (HPX). Hemopexin is shown here to be expressed by cortical neurons and it is present in mouse cerebellum, cortex, hippocampus, and striatum. Using the transient ischemia model (90-min middle cerebral artery occlusion followed by 96-h survival), we provide evidence that HPX is protective in the brain, as neurologic deficits and infarct volumes were significantly greater in HPX−/− than in wild-type mice. Addressing the potential protective HPX cellular pathway, we observed that exogenous free heme decreased cell survival in primary mouse cortical neuron cultures, whereas the heme bound to HPX was not toxic. Heme-HPX complexes induce HO1 and, consequently, protect primary neurons against the toxicity of both heme and prooxidant tert-butyl hydroperoxide; such protection was decreased in HO1−/− neuronal cultures. Taken together, these data show that HPX protects against heme-induced toxicity and oxidative stress and that HO1 is required. We propose that the heme-HPX system protects against stroke-related damage by maintaining a tight balance between free and bound heme. Thus, regulating extracellular free heme levels, such as with HPX, could be neuroprotective.
Introduction
Ischemic stroke is one of the leading causes of morbidity and mortality in Asia and Western countries (Qureshi et al, 2001). Various acute factors have been postulated to promote the toxicity associated with cerebral ischemia, including excitotoxicity, brain edema, and microbleeds. The toxicity of blood components has been associated with the presence of hemoglobin in the extravascular space. Release of ‘free’ heme (iron-protoporphyrin IX) from methemoglobin onto adjacent brain tissue substantially contributes to morbidity (Qureshi et al, 2001). Free heme is also released from various extracellular and intracellular hemoproteins liberated during hemolysis, brain trauma, and ischemia/reperfusion injury. Such heme has prooxidant properties and has been shown to be deleterious in various in vivo and in vitro systems (Wagner et al, 2003; Wang et al, 2006). Heme toxicity has been shown notably in endothelial cells (Alayash et al, 2001) and brain cells (Wang et al, 2006). Newly free heme does not appear to be recycled. Rather, a unique 60-kDa plasma glycoprotein, hemopexin (HPX, also abbreviated as Hx), binds heme with high affinity (Kd < 1 pmol/L), forming a stoichiometric 1:1 complex (heme-HPX) (Alam and Smith, 1992). Formation of this low-spin, bis-histidyl complex prevents heme from generating free radical reactions (Gutteridge and Smith, 1988).
The sequestration of heme by HPX is a primary defense for cells against heme toxicity (Ascenzi et al, 2005). In addition to acting as a heme scavenger, HPX transports heme into cells by endocytosis for catabolism by heme oxygenase isozymes (HO1 and HO2) (Davies et al, 1979). Of the two bioactive isoforms known, HO2 is constitutively expressed in most mammalian tissues, whereas HO1 is often undetectable under basal conditions. However, HO1 is highly inducible by various factors, such as by heavy metals, heme-HPX, and heme itself (Alam and Smith, 1992; Raju et al, 1997). Evidence suggests that HO1 and HO2 protect cells from oxidative stress (Panahian et al, 1999; Wang et al, 2006), in part, through heme catabolites.
The expression of HPX is still being investigated. It has been shown to be synthesized by cells of the hepatic and immune systems, by ganglionic and photoreceptor cells of the retina, by cells of the peripheral nervous system, and by kidney mesangial cells (Camborieux et al, 1998; Hunt et al, 1996; Kapojos et al, 2003). However, its expression in the central nervous system remains to be fully defined. Earlier, the presence of HPX immunologically in human postmortem brain was shown (Morris et al, 1993), and on the basis of its distribution, a role for HPX in brain heme-iron homeostasis was proposed.
In spite of the plethora of evidence for the protective role of heme catabolites generated by HO enzymes in stroke (Panahian et al, 1999; Wang et al, 2006), a question remains regarding the sources of the heme substrate for HO. To explain the beneficial effects of HO enzymes solely in terms of heme catabolites, intracellular heme would need to be present at levels stoichiometrically equivalent to the levels of metabolites shown to be cytoprotective (Doré et al, 1999b). We therefore hypothesize that in pathologic conditions such as stroke, HPX is a principal means to regulate the availability of heme substrate for degradation by HO. Here, to better determine which brain cells synthesize HPX, we first investigated whether mouse brain and primary cultured cortical neurons express HPX. We next addressed the importance of HPX in protection from transient focal cerebral ischemia by determining the infarct size as well as the neurologic deficit outcomes in wild-type (WT) and HPX−/−mice. We also determined whether heme-HPX affects neuronal cell viability during oxidative stress caused by heme or the free radical donor, tert-butyl hydroperoxide (t-BuOOH), and whether the heme-HPX complex mediates cytoprotection of neurons through HO1 induction. To address these questions, we also used a stable cell line overexpressing HO1 as well as cultured neurons from WT and HO1−/− mice.
Materials and methods
Animals
All animal protocols were approved by the Institutional Animal Care and Use Committee of Johns Hopkins University. All mice were maintained and housed in the university's vivarium under controlled conditions (23°C ± 2°C; 12-h light/dark periods), with access to food and water ad libitum. HPX−/− mice were first generated in Dr Tolosano's lab in 1999 (Tolosano et al, 1999). The HPX−/− mice are on a C57BL/6 background, and the size of their litters is normal. No cognition or motor dysfunction is observed. When we examined the gross superficial cerebrovascular anatomy, no detectable changes were observed.
Transient Ischemia Protocol, Neurologic Deficit Scores, and Infarct Size Determination
Male WT (n = 8) and HPX−/− (n = 12) mice were subjected to middle cerebral artery (MCA) occlusion (MCAO) for 90 mins followed by 96 h of reperfusion. Mice were placed under halothane anesthesia (3.0% for induction, 1.0% for maintenance) and ventilated with oxygen-enriched air via a nose cone. Body temperatures were maintained at 37.0°C ± 0.5°C by a heating pad. The mice were subjected to the intraluminal filament technique to produce the MCAO model of transient focal cerebral ischemia, as we have described earlier (Saleem et al, 2008). Relative cerebral blood flow was measured by laser-Doppler flowmetry (Moor Instruments, Devon, England), with a flexible probe affixed to the skull over the parietal cortex supplied by the MCA (2 mm posterior and 6 mm lateral to the bregma). The induction of MCAO was achieved when the relative cerebral blood flow decreased more than 80% from baseline; mice in which the cerebral blood flow did not decrease below that level were excluded from additional experiments. During occlusion, mice were kept in a humidity-controlled, 32°C chamber to help maintain a body core temperature of 37°C. After 90mins of occlusion, mice were briefly reanesthetized, the midline was reopened, and the filament was removed to establish reperfusion. After the incision was sutured, the mice were again placed in the humidity- and temperature-controlled chamber for 6 h and finally returned to their respective cages for survival up to 96 h.
Neurologic function was measured in each mouse at 96 h after reperfusion according to the following graded scoring system: 0 = no deficit; 1 = forelimb weakness and torso turning to the ipsilateral side when held by the tail; 2 = circling to the affected side; 3 = unable to bear weight on the affected side; and 4 = no spontaneous locomotor activity or barrel rolling. After the neurobehavioral assessment, mice were anesthetized and the brains were harvested, sliced into five 2-mm coronal sections, and incubated with 1% 2,3,5-triphenyltetrazolium chloride (Sigma, St Louis, MO, USA) in saline for 20 to 30 mins at 37°C. We used SigmaScan Pro 5 (Systat, Inc., Point Richmond, CA, USA) to measure infarct size by manually outlining the margins of infarcted areas (identified by the absence of stain). Cortical and striatal uncorrected infarct areas and total hemispheric infarct area were calculated separately for each coronal slice. Total cortical and striatal uncorrected infarct volumes were calculated by multiplying the infarct area by the slice thickness and summing the volume of the five slices. A corrected infarct volume was calculated to compensate for the effect of brain edema. Infarct size and volume were calculated and expressed as the percentage of infarct area to total hemispheric area for each slice.
Cell Cultures and Cell Survival
Embryonic cortical neuronal cells were isolated from 17-day embryos of timed pregnant mice, cultured in serum-free Neurobasal medium, and plated onto poly-
Postnatal mouse cortical neurons were obtained and cultured according to a protocol similar to that used for the embryonic cortical neurons with modifications, as detailed earlier (Saleem et al, 2007). Briefly, cerebral cortices were obtained from 1- to 2-day-old mice. High-density cultures (5 × 105 cells, 2,500 cells/mm2) were plated in Neurobasal medium supplemented with 1 mmol/L Glutamax (Invitrogen) and B27 onto 24-well plates coated with poly-
Human embryonic kidney (HEK293) cell lines were cultured in Dulbecco's modified Eagle's medium (Invitrogen) with 10% fetal bovine serum (Hyclone, Logan, UT, USA), penicillin (500 units/mL), streptomycin (500 μg/mL), and glutamine (1 mmol/L) and maintained in growth medium at 37°C in a 95% air/5% CO2-humidified atmosphere. The human isoforms of NADPH-cytochrome P450 reductase (CPR) and HO1 were subcloned into a cytome-galovirus-based expression vector PRK5. The sequences of all constructs were confirmed by nucleotide sequencing. Lipofectamine (Invitrogen) was used to stably transfect the CPR construct into HEK cells (HEK-CPR), which were maintained with the selective agent G418 (BD Sciences, San Diego, CA, USA). The HO1 isoform was subsequently stably expressed into HEK-CPR cells, which were maintained with the selection agent Zeocin (Invitrogen), and resulted in the creation of an HEK-CPR-HO1 cell line.
Primary embryonic neuronal cultures and HEK, HEK-CPR, and HEK-CPR-HO1 cell lines were treated with heme in dimethyl sulfoxide (DMSO), heme-HPX in phosphate-buffered saline (PBS), HPX in PBS, or solvent for 24 h before being assessed for viability. In separate experiments, cellular stress was induced in neurons by 24-h exposure to t-BuOOH (15 μmol/L) or to heme (20 μmol/L). The cells were simultaneously treated with heme-HPX (1, 5, 10, or 20 μmol/L), HPX (20 μmol/L), or the HO inhibitor, tin protoporphyrin IX (SnPPIX, 10 μmol/L). Cell viability was then assessed by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay. After 2-h incubation at 37°C with 0.5mg/mL MTT, living cells containing MTT formazan crystals were solubilized in a solution of anhydrous isopropanol, 0.1 N HC1, and 0.1% Triton X-100. The optical density was determined at 570 nm. Experiments were repeated with at least three separate batches of cultures. Human embryonic kidney cells and neurons were incubated with heme in the absence of serum (plain Dulbecco's modified Eagle's medium and Neurobasal medium, respectively) to prevent heme from binding to serum proteins during the incubation time and to ensure that cells were exposed to precise concentrations of heme.
Reverse Transcription-Polymerase Chain Reaction
Primary cortical neurons (2 × 106) were cultured on six-well plates for 10 to 14 days and treated with heme-HPX or heme for 6h. Wild-type mice were subjected to MCAO and allowed to survive for 96 h. The brains were sliced according to the rodent brain matrix for adult mice (RBM-2000C, ASI Instruments, Warren, MI, USA), and the third to ninth coronal sections were extracted, corresponding to ~6 mm in thickness; the contralateral and ipsilateral portions were isolated. These selected brain slices included significant portions of both the infarct core and the surrounding penumbra. Total RNA was extracted from cells or brain tissue with Trizol (Sigma) and further purified with the RNeasy kit (Qiagen, Valencia, CA, USA). Reverse transcription was carried out with the Superscript First strand synthesis kit (Invitrogen) with oligo (dT) primer. Polymerase chain reaction consisted of 25 to 30 cycles of denaturation at 94°C for 30 secs, annealing at 57°C (68°C for HPX and GAPDH) for 30 secs, and extension at 72°C for 1 min. The PCR primers for HPX, HO1, actin, and GADPH genes were as follows: HPX, 5'-GTCTGGGTGTAC CCTCCTGAAAAG-3' (forward) and 5'-CGTGAATGCATAG GATGGGTGCT-3' (reverse); HO1, 5'-GAGAATGCTGAGTT CATG-3' (forward) and 5'-ATGTTGAGCAGGAAGGC-3' (reverse); actin, 5'-GACTACCTCATGAAGATCCT-3' (forward) and 5'-CCACATCTGCTGGAAGGTGG-3' (reverse); GADPH, 5'-CGGAGTCAACGGATTTGGTCGTAT-3' (forward) and 5'-AGCCTTCTCCATGGTGGTGAAGAC-3' (reverse). The optimal cycle number for each gene under nonsaturating conditions was determined empirically. Polymerase chain reaction products were separated by electrophoresis on a 1 to 1.5% agarose gel containing ethidium bromide.
Immunohistochemistry
Naive WT mice were perfused first with saline and then with 4% paraformaldehyde. The brains were harvested and postfixed in 4% paraformaldehyde for 24 h, transferred to 30% sucrose solution at 4°C, and then stored at −20°C. The brains were sliced by cryostat into 25-μm-thick sections, which were blocked for 1 h in 4% normal goat serum and then incubated overnight at 4°C in the following primary antibodies: anti-Iba1 (Wako Chemicals USA Inc., Richmond, VA, USA), anti-GFAP (DakoCytomation, Carpinteria, CA, USA), and anti-NeuN (Chemicon International, Temecula, CA, USA) at dilutions of 1:200, 1:500 and 1:200, respectively. Finally, the sections were washed and incubated with the biotinylated specific secondary antibody (Vector Laboratories, Burlingame, CA, USA) for 1 h at room temperature. Immunoreactions were developed with the avidin-peroxidase-labeled biotin complex (Vectastain ABC Kit, Elite Rabbit IgG) for 1 h at room temperature and then visualized by treatment of sections with the DAB peroxidase substrate kit (Vector Laboratories). For the second antigen, HPX, a similar protocol was used with a polyclonal goat anti-rat HPX (raised and characterized by A.S.) as the primary antibody. This antibody cross-reacts with mouse HPX and the secondary antibody was selected accordingly (Vector Laboratories). The HPX was finally visualized by developing the sections with the Vector Blue Alkaline Phosphatase Substrate Kit III (Vector Laboratories).
Immunocytofluorescence Staining and Fluorescence Microscopy
Cultured embryonic neurons were grown on poly-
Preparation of Protein Samples and Western Blot Analysis
Three-month-old male C57BL/6 WT littermates of HO1−/− mice were killed and then perfused with saline to clear any residual blood and prevent plasma contamination. Brain cortex, cerebellum, hippocampus, and striatum tissue samples were collected individually and well mixed with lysis buffer (New England Biolabs, Ipswich, MA, USA) supplemented with 0.1 mL of 1 mol/L NaF, 1 mmol/L phenylmethylsulfonyl fluoride, and complete protease inhibitor (Roche, Nutley, NJ, USA). Embryonic cortical neurons were incubated in Neurobasal medium containing B27 minus antioxidant (Invitrogen) for 2h before each experiment. Experiments were terminated by applying 1 x lysis buffer (70 μL/well, 2 × 106 cells/sample). Protein concentration was determined by the BCA kit (Pierce, Rockford, IL, USA). Equivalent amounts of proteins per sample were electrophoretically resolved on 10% polyacrylamide gels. The protein was transferred to a nitrocellulose membrane, which was blocked for 1 h at 22°C with 5% dried milk in PBS containing 0.1% Tween 20. The membranes were incubated overnight at 4°C with primary antibodies to HO1 (Assay Designs, Ann Arbor, MI, USA), actin (the antibody can recognize all three actin isoforms: ±, β, γ; Sigma Co., Milwaukee, WI, USA), or HPX (goat anti-rat HPX raised and characterized by A.S.). Immunocomplexes were visualized by enhanced chemiluminescence detection (Amersham, Pittsburgh, PA, USA). The Western blots shown are representative of at least three separate experiments and were quantified by detecting emitted chemiluminescence with the Image J system (NIH).
Preparation of Heme-Hemopexin Complexes
Intact HPX was isolated from rabbit serum or from rat and human serum, as described earlier (Smith and Morgan, 1984; Vretblad and Hjorth, 1977). Stoichiometric 1:1 heme-HPX complexes (>85% to 90% saturation) were characterized and quantified using extinction coefficients (A mol/L−1cm−1) of 1.1 × 105 at 280 nm for HPX. The nanomoles of heme and HPX were calculated in 1:1 ratio and incubated at 4°C for 30 mins. Then, the complexes were dialyzed against PBS at 4°C before use. Saturation (%) and the final concentration of heme-HPX complexes were determined by absorbance spectrophotometry and published extinction coefficients (Smith and Morgan, 1984). Polyclonal antibodies to purified rat and human HPX were raised in goats.
Statistical Analysis
All data are expressed as means ± s.e.m. One-way ANOVA followed by Tukey's post hoc analysis was used to calculate the difference between groups. Comparisons between two groups were made by Student's t-test. The distribution of neurologic deficit scores was compared across WT and HPX−/− groups using the Fisher exact test. Values of P < 0.05 were considered to be significant.
Results
Hemopexin is Expressed in the Brain
Immunoblotting of mouse brain tissue extracts revealed the HPX protein expression in all regions, including the cerebellum, cortex, hippocampus, and striatum. The levels of HPX were highest in the cortex (Figure 1A, upper panel) relative to the control actin. Brain HPX has a similar molecular weight (Mr) to that in mouse plasma (Figure 1A, lower panel). We also investigated the HPX protein levels in an in vitro culture system. Hemopexin was also expressed by mouse postnatal and embryonic cortical neurons cultured in serum-free medium, sufficiently to be detected immunologically in whole-cell extracts using the polyclonal anti-rat HPX antisera (Figure 1A, lower panel).
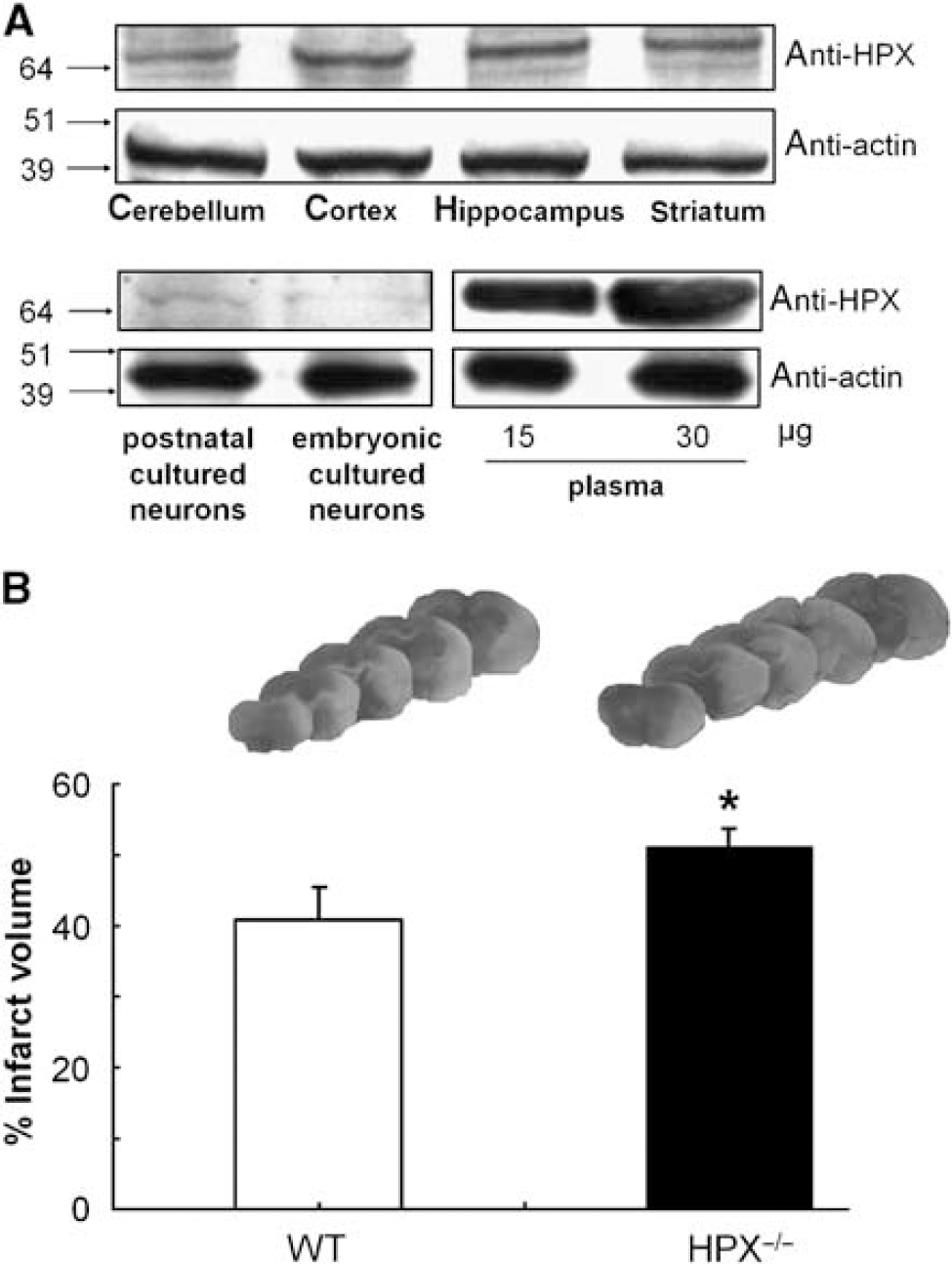
Hemopexin (HPX) is expressed in the mouse brain, and genetic deletion of HPX increases neurologic deficit score and infarct size after transient cerebral ischemia. (
Effect of Hemopexin Deletion on Neurologic Deficit Score and Infarct Volume
We next investigated whether HPX is protective against transient focal cerebral ischemia by comparing the neurologic score and infarct volume in WT and HPX−/− mice. The percent corrected infarct volume was significantly greater (P <0.05) in HPX−/− than in WT mice, as shown in Figure 1B. Accordingly there was a significant increase (P <0.05) in neurologic deficit score in HPX−/− mice compared with that in corresponding controls (Table 1).
Effect of MCAO on neurologic deficit score in WT and HPX−/− mice
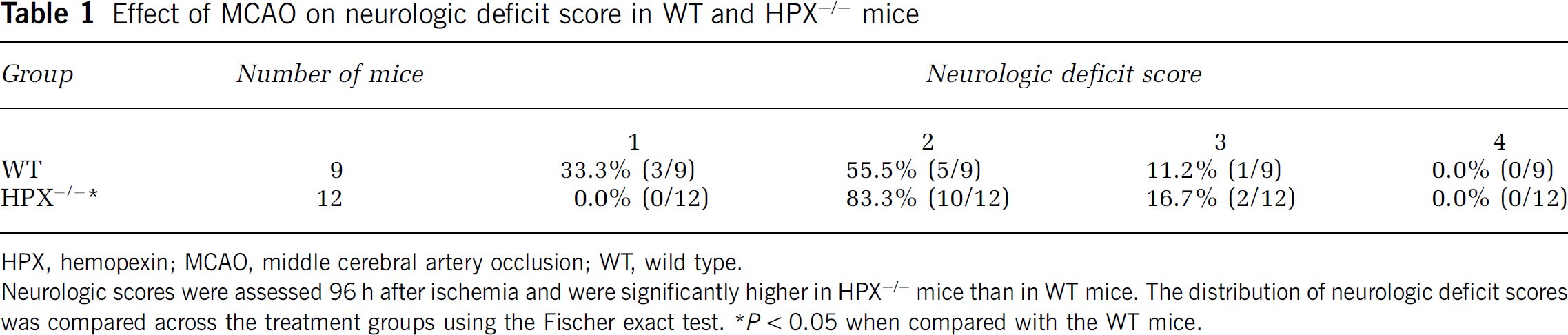
HPX, hemopexin; MCAO, middle cerebral artery occlusion; WT, wild type.
Neurologic scores were assessed 96 h after ischemia and were significantly higher in HPX−/− mice than in WT mice. The distribution of neurologic deficit scores was compared across the treatment groups using the Fischer exact test. *P < 0.05 when compared with the WT mice.
Hemopexin is Expressed by Brain Cells, Including Cortical Neurons
To determine whether the HPX is synthesized locally in the brain cells, we first used indirect immunofluorescence microscopy to show the HPX protein expression in mouse embryonic cortical neurons (Figure 2A). Hemopexin was detected in the neuronal cell body and to a much lesser extent in the dendritic trees and axons. As expected, the neuronal marker, MAP2, was detected in the axon-dendritic compartment but was absent from the soma. The merged image revealed colocalization of HPX with MAP2. Next, using brain slices, we performed double immunostaining to examine in which cell types hemopexin is expressed in the cortex (Figure 2B). Neurons expressed immunoreactive hemopexin (blue), which colocalized with the neuronal marker, NeuN (brown; Figure 2B, left panel), as well as with some astrocytes (brown; Figure 2B, right panel), but not with microglia (brown, Figure 2B, center panel). Overall, these combined data support the conclusion that cortical neurons express HPX.
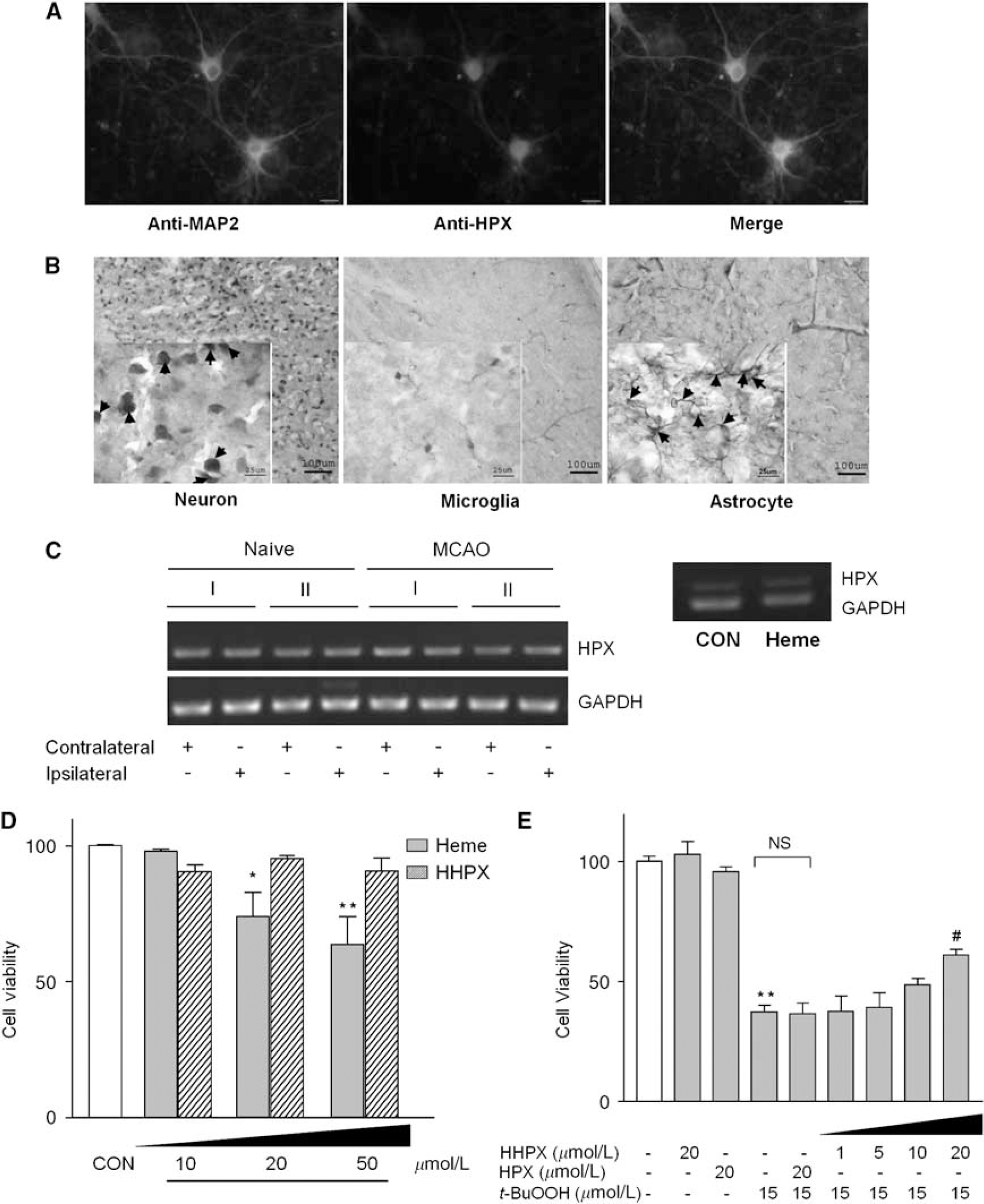
Hemopexin is expressed in brain and cultured cortical neurons and affects neuronal cell viability when complexed with heme. (
Expression of Hemopexin mRNA in Brain Homogenates and Cultured Cortical Neurons after Stress
To determine whether HPX changes in the brain after stroke or in cortical neuronal cells after heme-induced stress, we conducted in vitro and in vivo experiments to examine HPX mRNA levels. Brain samples from mice subjected to 90-min MCAO and 96-h reperfusion revealed no detectable differences in the HPX mRNA levels between the ispilateral and contralateral sides (Figure 2C, left panel) or between ischemic and naive mice. Although the data suggest that HPX mRNA levels are not increased 4 days after stroke, the results do not exclude the possibility that some changes may occur, as HPX is known to be induced by the inflammatory cytokines or chemokines (Baumann et al, 1983). Hemopexin mRNA was also clearly expressed in cultured cortical neurons (Figure 2C, right panel), confirming our findings at the protein level. However, similar to the results from our in vivo experiments, the mRNA level did not change after 6 h incubation with 20 μmol/L heme.
Heme but not Heme-Hemopexin is Toxic to Cortical Neurons
As levels of free heme and HPX are likely to be increased in the cerebrospinal fluid after brain injury such as stroke, here we used an in vitro paradigm. The number of viable cells was determined after incubating primary cortical neurons for 24 h with a range of concentrations of free heme and heme-HPX. Free, non-HPX-bound heme (10 to 50 μmol/L) significantly reduced neuronal cell viability, but the heme-HPX complex (10 to 50 μmol/L) was not toxic to primary neurons (Figure 2D). The 10 to 20 μmol/L heme-HPX complex used here represents a model of a ‘heme load,’ as published earlier (Eskew et al, 1999; Morgan et al, 1993), which can occur, for example, after intracerebral hemorrhage or head trauma. Under these conditions, heme was toxic to cortical neurons at 20 μmol/L or at a higher level. However, heme-HPX was not cytotoxic even at concentrations more than double normal plasma levels (50 μmol/L). Furthermore, HPX was neither toxic nor stimulatory to neurons (data not shown), even at 10 times the normal plasma level. Significantly, these data point to key differences in the ways that neurons respond to heme-HPX and to non-protein-bound heme. They also confirm that neither heme-HPX nor HPX is toxic to primary cultures of mouse cortical neurons.
Heme-Hemopexin Protects Cortical Neurons from Cell Death by Oxidative Stress
Hemopexin levels in the cerebrospinal fluid are increased by systemic inflammation (Saso et al, 1999). Neurons can be exposed to heme-HPX in an environment where there is oxidative stress from the accompanying inflammation. Therefore, to model brain injury with hemolytic sequelae, we next wanted to determine whether heme-HPX could provide cytoprotection to neurons in an in vitro model of oxidative injury. The free-radical donor t-BuOOH (15 μmol/L) was toxic to neurons, causing significant neuronal cell death within 24 h (Figure 2E) (Shah et al, 2007). This finding was also confirmed by the detection of lactate dehydrogenase release into the culture medium (data not shown). However, when primary neurons were incubated simultaneously with t-BuOOH and heme-HPX (1 to 20 μmol/L), 20 μmol/L heme-HPX significantly reduced the t-BuOOH-induced cytotoxicity (Figure 2E). This protection required heme delivery by HPX, because HPX alone was not protective.
Induction of HO1 by Heme-Hemopexin in Mouse Cortical Neurons
Heme oxygenases are the only enzymes that degrade heme derived from hemoproteins when cells are stressed (e.g., during hypoxia from the loss of blood supply). Additional heme for HO is derived from extracellular hemoglobin during normal and abnormal red blood cell metabolism and can also be from degradation of heme-containing proteins during stroke. Evidence indicates that both HO1 and HO2 play important roles in protecting cells of the central nervous system from heme toxicity or oxidative stress in a variety of animal models of stroke, including transient and permanent focal cerebral ischemia (Doré et al, 1999b; Panahian et al, 1999). As HO1 is cytoprotective and heme-HPX induces transcription of the HO1 gene in nonneuronal cells and in retinal cells (Hunt et al, 1996), we next investigated whether HO1 expression is induced when primary cultures of embryonic neurons are incubated with heme-HPX. HO1 mRNA and protein levels were induced dose dependently in cortical neurons 6h after the addition of heme-HPX (1 to 20 μmol/L) to the medium (Figure 3). To our knowledge, this is the first demonstration that HO1 is induced by heme-HPX in differentiated cells and shows that cortical neurons respond to heme-HPX.
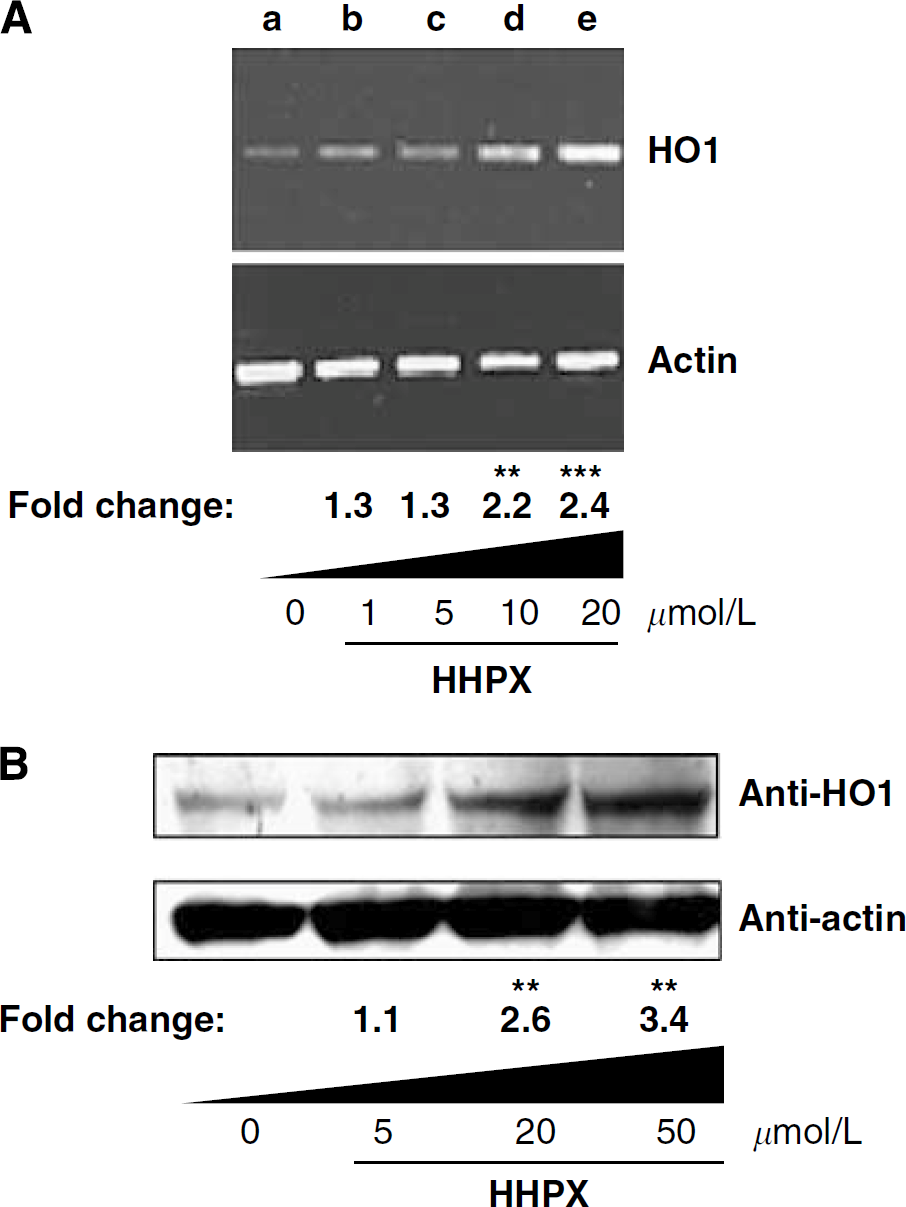
Heme-HPX induces HO1 expression in primary cortical neurons. Embryonic cortical neurons were incubated for 6 h with HHPX. HO1 induction was measured by RT-PCR (
Overexpression of HO1 Reduces Heme-Induced Cytotoxicity in Human Embryonic Kidney Cells
Exogenous free heme induced HO1 in primary neurons (Figure 4A) but was nevertheless toxic not only to primary neurons (Figure 4B) but also to HEK293 cells (Figure 4B). However, when HO1 level was increased, ~70% of HEK-CPR-HO1 cells survived a formerly toxic concentration of exogenous heme (50 μmol/L; Figure 4B). Significantly, more HEK-CPR-HO1 cells survived than did untransfected HEK293 cells or those expressing only CPR (HEK-CPR). Furthermore, when cortical neurons were incubated with a toxic concentration of heme but in the presence of the HO inhibitor, SnPPIX, at 10 μmol/L, heme toxicity (20 μmol/L) was exacerbated (Figure 4D), indicating that the HO1 enzymatic activity was required for protection. High concentrations of heme-HPX (5, 20, and 50 μmol/L; Figure 4C) or up to 10 times normal plasma levels of HPX (data not shown) were not toxic to any of these HEK cell lines, which is consistent with a protective role for heme-HPX. These data support the concept that once HO1 has been induced, and with sufficient CPR activity, cells are potentially protected from heme toxicity.
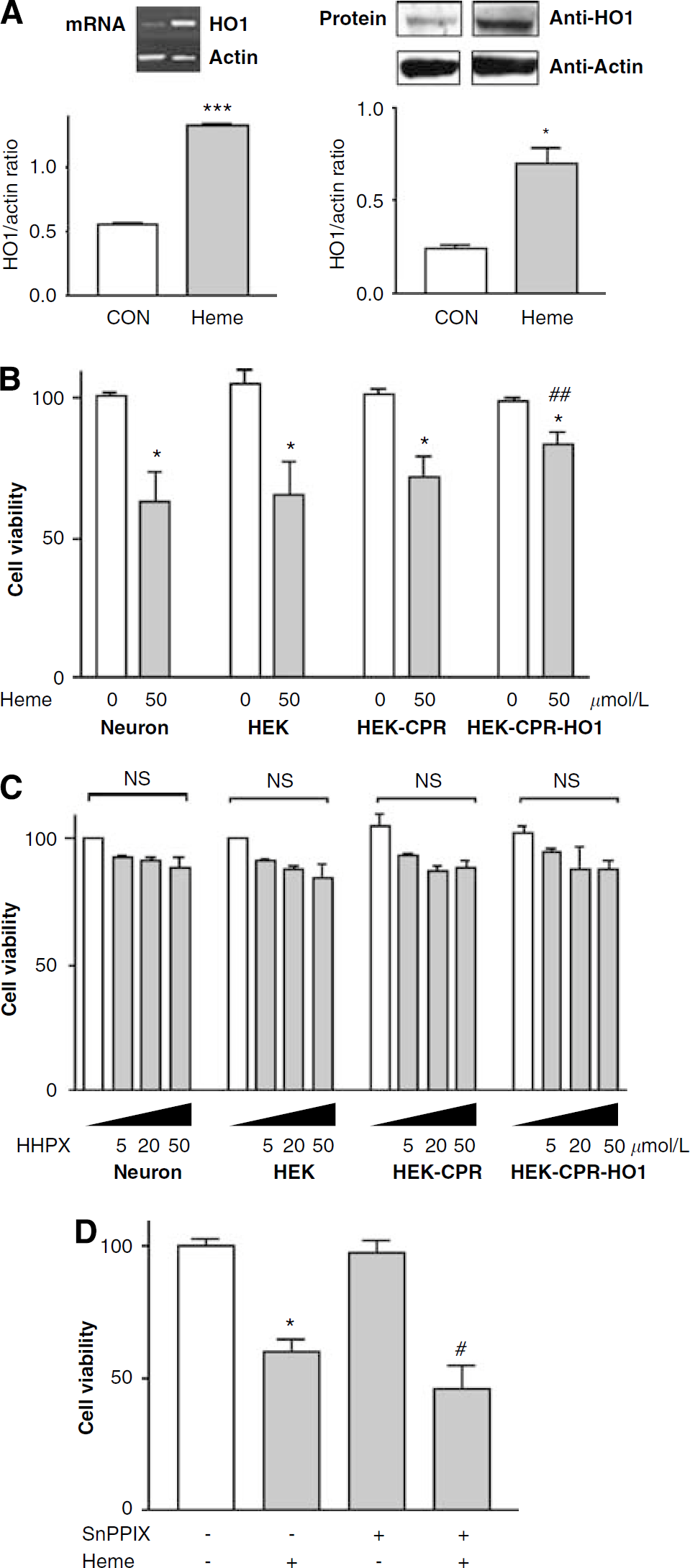
Elevated HO1 levels protect against heme-induced cytotoxicity. (
Genetic Evidence Shows that the Protection of Neurons by Heme-Hemopexin against both Heme Toxicity and Oxidative Stress Requires HO1
To show that HO1 is needed for heme-HPX protection of neuronal cells during oxidative stress caused by heme or t-BuOOH, we compared the protective effects of heme-HPX on postnatal neurons isolated from WT and HO1−/− mice. Each of heme (20 μmol/L) and t-BuOOH (15 μmol/L) significantly increased neuronal cell death in neurons from both WT and HO1−/− mice. Heme-HPX (10 μmol/L) had no effect on baseline viability of WT or HO1−/− neurons. Importantly, heme-HPX significantly reduced the neuronal cell death induced by both heme (Figure 5A) and t-BuOOH (Figure 5B) in WT neurons, but did not protect HO1−/− neurons. To mimic the situation in vivo after a hematological/oxidative event in the brain, neurons were incubated with a toxic concentration of heme in the presence or absence of heme-HPX. More neurons survived this heme exposure when heme-HPX was present simultaneously. Furthermore, HO enzymatic activity was required, as the protection was prevented by SnPPIX (Figure 5C). Overall, these data show that heme-HPX contributes to neuronal cell survival against both heme toxicity and oxidative stress from reactive oxygen species through a pathway that includes HO1 induction and heme catabolites.
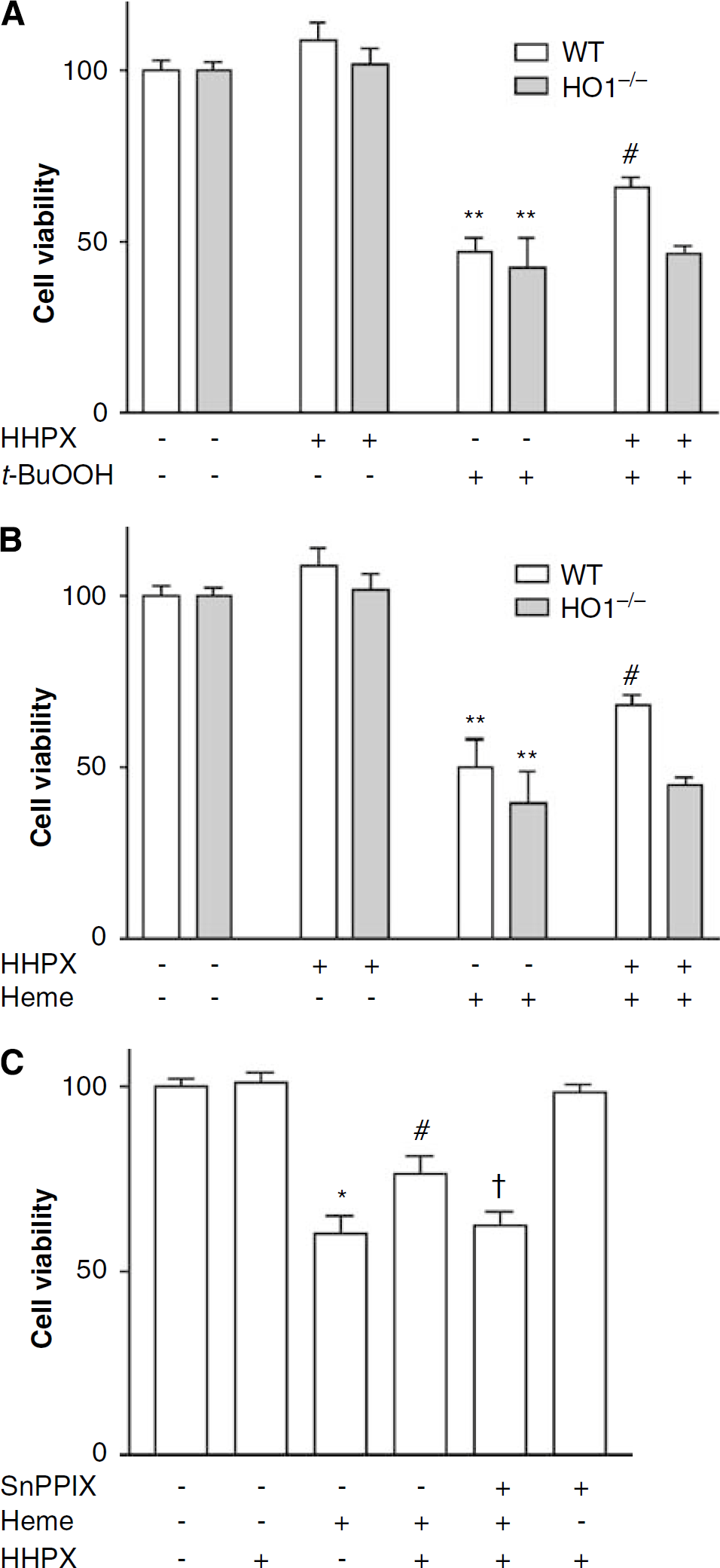
Cytoprotection by heme-HPX requires HO1. (
Discussion
Here, we have documented for the first time a unique and significant protective ability of HPX to modulate brain infarct size and its associated neurologic deficits by comparing the outcomes in WT and HPX−/− mice subjected to a model of transient ischemia. We further report that HPX protein is present in various mouse brain regions and that HPX mRNA and protein are detectable in cultured cortical neurons from mouse brain. To better address the cellular protective pathway, we showed that HPX was sufficient to protect against extracellular heme-induced cytotoxicity. Indeed, the heme-HPX complex protected cultured neurons against the toxicity of both heme and oxidant stress (i.e., t-BuOOH). Finally, this cellular neuroprotective pathway appears to require HO1 activity, as the protection was prevented by an HO inhibitor and did not occur in neuronal cells from HO1−/− mice. Taken together, these data provide novel information and evidence that HPX has a protective role in the central nervous system.
Hemopexin has been proposed to play a role in the transfer of heme to various brain cells (Morris et al, 1993). It is present in the cerebrospinal fluid (Castano et al, 2006; Davidsson et al, 2002; Ramstrom et al, 2003; Saso et al, 1999) and upregulated during inflammation in a rodent model (Saso et al, 1999). Hemopexin is also increased in the cerebrospinal fluid of Alzheimer's patients (Castano et al, 2006), a finding that supports the possibility that it is affected by neurodegeneration. Although HPX is normally abundant in plasma, circulating levels are induced by schizophrenia (Maes et al, 1997) and type II diabetes (Van Campenhout et al, 2006). It is also worth noting that, as HPX has a much higher affinity for heme (Kd ~pmol/L) than does albumin, it may function physiologically to regulate the balance between free heme and bound heme, and/or regulate heme degradation.
Our findings with cultured cortical neurons support the premise that HPX protects neuronal cells by neutralizing the prooxidant properties of free heme. In addition, the data presented here indicate that HPX is produced locally by certain brain cells, such as neurons. During trauma or hemorrhage, release of blood and formation of heme-HPX induce HO1 in neurons, as observed in our in vitro model. The existence of this coordinated action of heme-HPX-HO1 is evidenced by the fact that heme-HPX decreased the cytotoxicity of both heme- and t-BuOOH-mediated oxidative stress in WT, but not in HO1−/−, cortical neurons. The biologic and regulatory effects of heme-HPX also protected primary neurons in the presence of toxic levels of heme. Protection by heme-HPX against this heme toxicity appears to require HO1 activity, as the HO inhibitor, SnPPIX, exacerbated neuronal cell death. This requirement for HO1 observed in neurons with heme-HPX was further confirmed by studies in HEK cells that stably overexpressed HO1. Taken together, these data support the concept that heme-HPX potentially provides primary defense against heme-mediated oxidative stress in the nervous system through the cytoprotection of neurons and that the induction of HO1 by heme-HPX is part of this protective mechanism.
Our present results with the transient MCAO model and primary neurons extend our earlier observations (Doré et al, 1999a) and those of other investigators (Chang et al, 2003; Panahian et al, 1999) that HO isozymes protect the central nervous system from ischemic and traumatic insults. In this case, our data provide direct evidence for the protective effect of heme-HPX through HO1. HO1 induction in brain cells has been described earlier as having a ‘Janus,’ or double-edged, effect (Matsuoka et al, 1999) that we believe is reflected in the differences in cell type as well as in the in vitro conditions used. In astrocytes, increasing HO1 expression by adenoviral gene transfer before heme exposure attenuates oxidative stress and cell death (Chen and Regan, 2005). In fibroblasts, a moderate overexpression of HO1 is protective, whereas uncontrolled (beyond threshold) HO1 levels can be detrimental (Abate et al, 2005). Preconditioning with low heme levels to induce hepatic HO1 prevented heme overload-induced liver damage (Vinchi et al, 2008). Factors that will be important in vivo include the relative extracellular concentrations of HPX, haptoglobin, and other proteins involved in the regulation of heme/iron levels (Alam and Smith, 1989; Davies et al, 1979; Fagoonee et al, 2006); the concentration of heme-HPX and hemoglobin-haptoglobin complexes; the cellular expression of HPX and HPX-binding proteins; and the regulation of additional protective proteins by heme-HPX compared with non-protein-bound heme (Eskew et al, 1999).
Considerable evidence also indicates that HPX recycles after endocytosis and heme delivery (Smith and Morgan, 1978). Heme-HPX complexes are taken into cells by a high-affinity, low-capacity process, likely through LRP1/CD91, which binds heme-HPX tightly (Hvidberg et al, 2005), or by a second HPX receptor that is involved in HO1 induction for a low-affinity, high-capacity heme-uptake ‘selective’ process (Smith and Morgan, 1978). Additional work is warranted to fully understand the various steps leading to the various functions of HPX and the heme-HPX complexes.
We conclude that genetic deletion of HPX significantly increases the severity of the brain damage from ischemic stroke, that heme-HPX protects cells and particularly neurons against both heme- and oxidative stress-induced toxicity, and that this neuroprotection requires HO1. We propose that heme-HPX-mediated HO1 induction, which also requires endocytosis (Flaherty et al, 2008), plays a protective role in neuronal survival, at least for short-term cell defense. Oxidative stress is known to contribute to the pathology of neurodegenerative disorders, such as Alzheimer's disease, stroke, and aging. Therefore, a better understanding of the intricate molecular mechanisms regulated by HPX in an ischemic environment will provide insight into the discovery of drugs for the treatment of acute and potentially chronic neurologic disorders. The research presented here begins to provide a characterization of the neuroprotective activity of HPX and will potentially lead to the development of innovative tools for which clinical applications can be tested.
Footnotes
Acknowledgements
We thank Kim Push for the isolation of HPX, David L Greenberg and Xiaoling Li for cell cultures, Salahaldin Rifat and John Langer for brain tissue preparation, Jennifer Mytar for genotyping and animal colony maintenance, Fumin Chang for microscopy help, Claire Levine for editorial assistance, and all the members of the Doré lab for their insightful comments.
The authors state no conflict of interest.