
Abstract
The blood–brain barrier (BBB) exhibits a highly selective permeability to support the homeostasis of the central nervous system (CNS). The tight junctions in the BBB microvascular endothelial cells seal the paracellular space to prevent diffusion. Thus, disruption of tight junctions results in harmful effects in CNS diseases and injuries. It has recently been demonstrated that glucocorticoids have beneficial effects on maintaining tight junctions in both in vitro cell and in vivo animal models. In the present study, we found that dexamethasone suppresses the expression of JMJD3, a histone H3K27 demethylase, via the recruitment of glucocorticoid receptor α (GRα) and nuclear receptor co-repressor (N-CoR) to the negative glucocorticoid response element (nGRE) in the upstream region of JMJD3 gene in brain microvascular endothelial cells subjected to TNFα treatment. The decreased JMJD3 gene expression resulted in the suppression of MMP-2, MMP-3, and MMP-9 gene activation. Dexamethasone also activated the expression of the claudin 5 and occludin genes. Collectively, dexamethasone attenuated the disruption of the tight junctions in the brain microvascular endothelial cells subjected to TNFα treatment. Therefore, glucocorticoids may help to preserve the integrity of the tight junctions in the BBB via transcriptional and post-translational regulation following CNS diseases and injuries.
Introduction
The blood–brain barrier (BBB) maintains the homeostasis of the central nervous system (CNS) by regulating the permeability between the CNS interstitial fluids and the circulating blood.1,2 The BBB consists of a monolayer of capillary endothelial cells connected by intercellular junction complexes containing tight junctions, adherens junctions, and desmosomes. Tight junctions, which prevent paracellular diffusion, mainly consist of transmembrane proteins, such as claudins and occludin, along with cytoplasmic accessory proteins, such as Zonula Occludens 3 and cingulin.
Claudins and occludin are expressed abundantly in brain endothelial cells. 4 Claudins, a family of 24 transmembrane proteins, consist of four transmembrane domains, two extracellular loops, and a cytoplasmic N and C-terminus. 5 The cytoplasmic scaffold proteins, such as ZOs, multi-PDZ domain protein (MUPP)-1, and PALS-1-associated TJ protein (PATJ), interact with the PDZ motif within the C-terminal of claudins. In addition, homotypic and heterotypic interactions between the extracellular loops of claudins provide a major backbone of a tight junction. Claudin 5 has been shown to be the dominant claudin in the endothelial cells of the BBB.6,7 Moreover, claudin 5-deficient mice demonstrated an impaired BBB in a size-selective manner. 8 Occludin also consists of four transmembrane domains, two extracellular loops, and a cytoplasmic N and C-terminus. 9 Deletion of the N-terminal and extracellular domain results in an impaired tight junction assembly and barrier permeability. 10 Occludin-deficient mice showed an intact tight junction, indicating that other member of TAMP (tight junction-associated MARVEL protein) may partially compensate for occludin and the function of occludin may be more complicated. 11 In fact, knockdown of occludin results in a redistribution of tricellulin from tricellular tight junction to bicellular tight junction. 12 In addition, knockdown of combinations of TAMPs indicates overlapping but distinct functions of TAMPs at the tight junction. 13 The phosphorylation of occludin has also been found to regulate the tight junction permeability in a G protein-dependent or – independent manner. 14
The dysregulation of tight junction proteins is frequently associated with BBB disruption in various neurological diseases, including meningitis, epilepsy, Alzheimer's, Parkinson's, and multiple sclerosis, as well as traumatic CNS injury and ischemia.15–17 BBB disruption leads to blood extravasation and the infiltration of circulatory inflammatory cells. These events further contribute to harmful effects, such as neuronal death in CNS diseases and injuries. Therefore, a number of studies have focused on the development of the effective therapeutic intervention to prevent or reduce BBB disruption. Since nuclear hormone receptors are expressed in endothelial cells, the therapeutic effect of hormones on tight junctions or the BBB has already been explored.18–21
In particular, glucocorticoids have been investigated as a therapeutic intervention for protecting tight junctions and avoiding BBB disruption in a number of cells and animal models. Glucocorticoids, such as dexamethasone, hydrocortisone, and corticosterone, induce improved tight junctions through an increased level of tight junction proteins in vascular endothelial cells.19,22–27 This improvement in the tight junctions by glucocorticoids is also associated with a rearrangement of the cytoskeleton.22,25,27 Moreover, dexamethasone induces transcriptional activation of TIMP-1 (tissue inhibitor of metalloproteinase-1) and TIMP-3, which may block MMP-9-mediated degradation of tight junction proteins.28,29 It has also been reported that annexin A1, which is up-regulated by glucocorticoids in the cerebral endothelium, plays an important role in the BBB integrity via regulation of the tight junction protein expression and actin cytoskeleton. 30
In the present study, we found that dexamethasone suppresses the expression of JMJD3, a histone H3K27 demethylase, via the binding to glucocorticoid receptor α (GRα) and nuclear receptor co-repressor (N-CoR) to the negative glucocorticoid response element (nGRE) in the upstream region of the gene. As a result, MMP-2, MMP-3, and MMP-9 expression were decreased, thereby attenuating the disruption of the tight junctions in the brain microvascular endothelial cells subjected to TNFα treatment.
Materials and methods
Cell culture and drug treatment
Mouse brain microvascular endothelial cell line bEnd.3 (ATCC) maintained in DMEM supplemented with 10% fetal bovine serum and antibiotics. Human brain microvascular endothelial cell line hCMEC/D3 (EMD Millipore) maintained in EndoGro-MV complete media (EMD Millipore) supplemented with 5% fetal bovine serum, 1 ng/ml FGF-2, and antibiotics. Cells were treated with 100 nM TNFα (BIO BASIC) alone and in combination with 200 nM dexamethasone (Sigma Aldrich) for 4 h. To inhibit GR activation, cells were treated with 1 μM mifepristone (RU486, Sigma Aldrich) for 4 h.
RNA isolation and quantitative PCR analysis
Total RNA was isolated and real-time PCR analysis was performed as described previously. 31 PCR amplification was achieved using oligonucleotide primers described in Supplementary Table S1.
Promoter reporter assay
Mouse JMJD3 gene promoter was amplified and cloned into pGL4.15 (luc2P/hygro) vector (Promega). Deleted promoter reporter constructs were generated by PCR. The oligonucleotide primers for cloning and deleted constructs are described in Supplementary Table S2. Mutation of nGRE in JMJD3 gene promoter was performed by PCR using oligonucleotide primers described in Supplemental Table 2. Mouse bEnd.3 cells were transfected transiently with JMJD3 gene promoter-driven firefly luciferase (1 µg) in conjunction with control thymidine kinase promoter-driven renilla luciferase (100 ng). Following transfection for 36 h, cells were subjected to drug treatment for 4 h and harvested for luciferase activity using the Dual-Luciferase Assay System (Promega) with a Lumat BL 9507 luminometer (Berthold technologies). Renilla luciferase activity served as internal control for normalizing firefly luciferase activity of gene promoter.
Nuclear and cytoplasmic fractionation
Cells grown in 10 cm dish were washed with ice-cold PBS and harvested. After centrifugation, cell pellet was gently resuspended in 500 μl hypotonic buffer [20 mM Tris-HCl (pH 7.4), 10 mM NaCl, 2 mM MgCl2] in ice for 15 min. Resuspended cells were treated with NP-40 (final concentration: 0.5%), vortexed, and centrifuged for 10 min at 3000 r/min. Supernatant was used as cytoplasmic fraction. To obtain nuclear fraction, cell pellet was washed repeatedly with hypotonic buffer more than five times and lysed in lysis buffer [25 mM Tris-HCl (pH 8.0), 10% glycerol, 0.5 mM EDTA, 0.1% NP40, 150 mM NaCl, 1 mM DTT] using sonication.
Western blot analysis and immunoprecipitation
Western blotting and immunoprecipitation were performed as described previously. 31 Cells were harvested for protein extraction. Anti-GRα (1:10,000, sc1004, Santa Cruz Biotechnology), anti-phospho-GR (1:1000, 4161, Cell Signaling), anti-JMJD3 (1:5000, ab38113, Abcam), anti-N-CoR (1:5000, cs207360, Millipore), anti-MMP-2 (1:5000, ab79781, Abcam), anti-MMP-3 (1:5000, ab53015, Abcam), anti-MMP-9 (1:5000, 1AB19016, Millipore), Histone H3 (1:5000, LF-MA10138, Abfrontier) and anti-β actin antibodies (1:5000, YF-MA10008, Abfrontier) were used. For immunoprecipitation, cell lysates were immunoprecipitated with 1 µg of anti-GRα antibody (sc1004, Santa Cruz Biotechnology). Normal IgG (sc2027, Santa Cruz Biotechnology) was used as control. Western blots were analyzed quantitatively using ImageJ software (NIH).
Zymography
The activities of MMP-2 and MMP-9 were examined using gelatin zymography as previously described. 32 To determine MMP-3 activities, casein zymography was performed as previously described. 33
Immunocytochemistry
Cells were fixed and immunocytochemistry was performed as described previously. 31 Anti-JMJD3 (1:500, ab38113, Abcam), anti-claudin 5 (1:500, ab53765, Abcam), and anti-occludin antibodies (1:500, ab31721, Abcam) were used. Nuclei were identified using DAPI staining.
RNA interference (siRNA)
Small interfering RNA (siRNA) against mouse GRα (M-045970-01-0005, Dharmacon) and N-CoR (1390518, Bioneer) were used. Control siRNA (sc37007, Santa Cruz Biotechnology) was used for negative control. Following 50 μM siRNA transfection in six-well plate using the X-treamGENE siRNA transfection reagent (Roche) for 48 h, cells were subjected to drug treatment. The efficiency of knockdown of specific gene was confirmed using real time-PCR.
Chromatin immunoprecipitation
Cells were harvested and Chromatin immunoprecipitation (ChIP) was performed as described previously. 31 Soluble chromatin from cells grown in 10 cm dish was immunoprecipitated with 2 µg of antibodies. Anti-GRα (sc1004, Santa Cruz Biotechnology), anti-N-CoR (cs207360, Millipore), anti-H3K27me1 (07-448, Millipore), and anti-H3K27me3 antibodies (07-449, Millipore) were used. Normal IgG (sc2027, Santa Cruz Biotechnology) was used as ChIP control. Quantitative PCR was performed using oligonucleotide primers described in Supplementary Table S3.
Measurement of trans-endothelial electrical resistance
Endothelial permeability was determined by measuring trans-endothelial electrical resistance (TEER) as described previously. 34 TEER across the monolayers grown on filter membranes was measured using the Millicell ERS Voltohmmeter (Millipore), and the values are shown as Ω·cm2 based on culture inserts. The TEER of cell-free inserts was subtracted from that of filters with cells.
Statistical analyses
All quantitative data are presented as mean ± S.E.M. (standard error of the mean) for three independent experiments. The differences between two groups were evaluated by a paired t-test. The significance of multiple comparisons was determined one or two-way ANOVA followed by post hoc t-test. Significance values were *P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.005.
Results
Dexamethasone suppresses up-regulation of JMJD3 expression in brain microvascular endothelial bEnd.3 cells subjected to TNFα treatment
It is already known that glucocorticoids improve the integrity of the BBB in both in vitro cell and in vivo animal models.19,22–27,35–37 We recently suggested that histone H3K27me3 demethylase JMJD3 may be an important epigenetic factor, which regulates the integrity of the BBB following CNS injury.31,38 Thus, we examined the effect of dexamethasone (Dex), a synthetic glucocorticoid, on JMJD3 gene expression in mouse brain microvascular endothelial bEnd.3 cells subjected to TNFα treatment, which causes BBB disruption.39–44 The expression and nuclear translocation of JMJD3 increased in the TNFα-treated bEnd.3 cells (Figure 1(a) to (d)). However, 200 nM Dex treatment significantly suppressed the TNFα-induced JMJD3 expression and nuclear translocation (P < 0.005). Next, we examined the reporter activity using the 2.394 kb upstream region of the JMJD3 gene. As expected, Dex suppressed the TNFα-induced activation of JMJD3 gene activity in the bEnd.3 cells (Figure 1(e)). Dex-mediated suppression of JMJD3 gene activation was also confirmed in TNFα-treated human brain microvascular endothelial hCMEC/D3 cells (Supplementary Figure S1).
Dexamethasone suppresses up-regulation of JMJD3 gene expression in brain microvascular endothelial bEnd.3 cells subjected to TNFα treatment. Expression of JMJD3 is up-regulated in bEnd.3 cells subjected to TNFα treatment. However, dexamethasone (Dex) suppresses TNFα-induced up-regulation of JMJD3 expression. (a) Transcripts of JMJD3 and GAPDH were determined by quantitative PCR (n = 3). (B) Lysates were immunoblotted with anti-JMJD3 antibody (n = 3). Anti-β-actin antibody was used as loading control. Western blots were analyzed quantitatively. (c) Representative photomicrographs of JMJD3 expression in bEnd.3 cells. Cells were immunostained using anti-JMJD3 antibody (n = 3). Nuclei were identified using DAPI staining. Scale bar, 25 µm. (d) Increased nuclear translocation of JMJD3 in TNFα-treated bEnd.3 cells. However, Dex suppresses nuclear translocation of JMJD3. Immunoblotting with anti-JMJD3 antibody using nuclear and cytoplasmic fractions (n = 3). Anti-β-actin and anti-histone H3 antibodies were used as loading controls. Western blots were analyzed quantitatively. (e) Cells were transiently transfected with 2.394 kb upstream region of JMJD3 gene-driven firefly luciferase reporter vector in conjunction with control renilla luciferase expression vector (n = 3). Reporter activity is represented as fold activation relative to renilla luciferase activity. All data represent mean ± S.E.M. Significance value was ***P ≤ 0.005.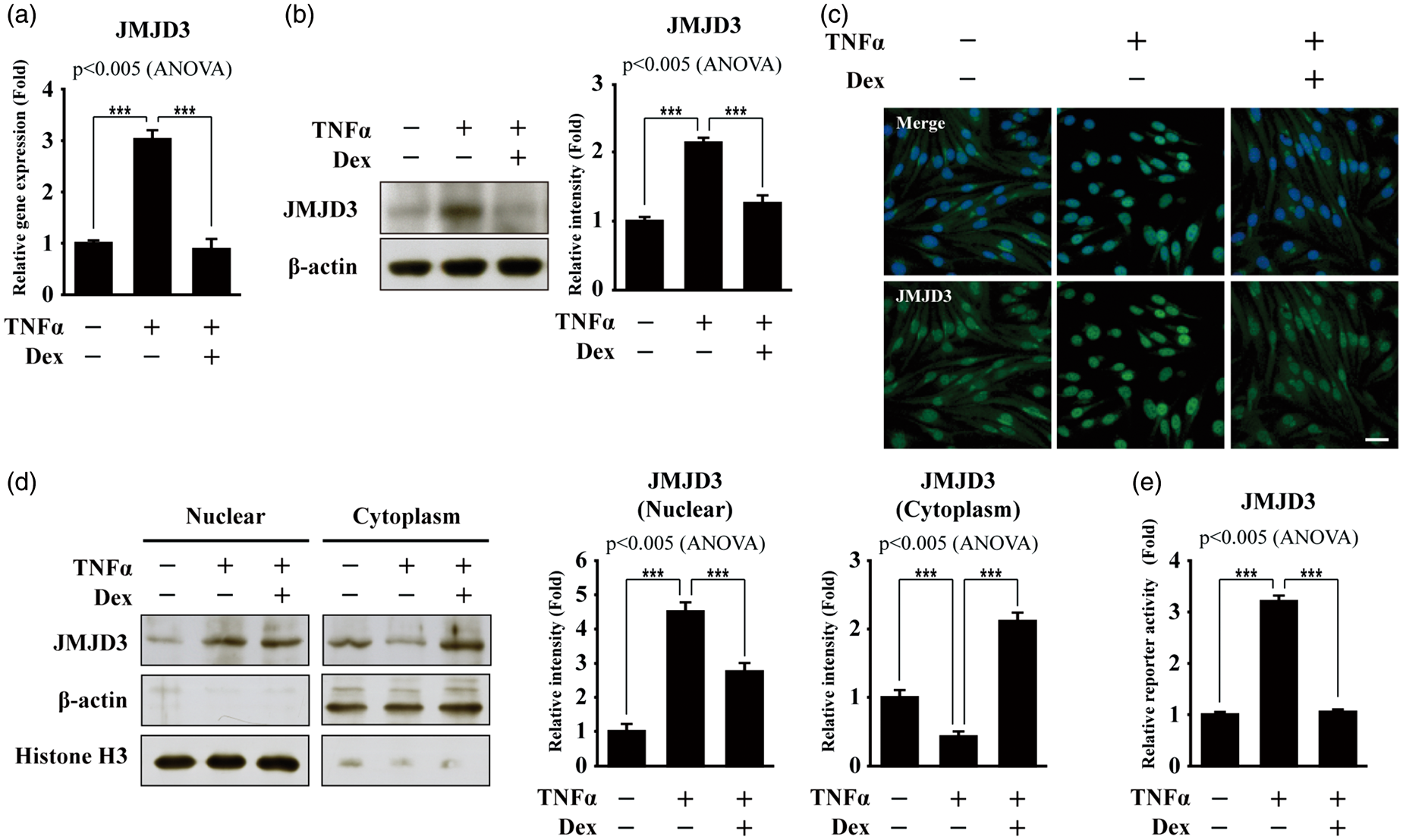
Negative GRE is required for dexamethasone-induced suppression of JMJD3 gene activation in bEnd.3 cells subjected to TNFα treatment
To identify the critical cis-element(s) in the upstream region of the JMJD3 gene, various reporter constructs were generated using the serially deleted upstream region of the JMJD3 gene. Interestingly, two putative nGREs (negative glucocorticoid response elements), which are involved in glucocorticoids-mediated negative gene expression, were found (Supplementary Figure S2).45 When the upstream region (−1494 to −1333 bp) containing nGRE1 was deleted, the Dex-induced suppression of JMJD3 promoter activity disappeared in the bEnd.3 cells subjected to TNFα treatment (Figure 2(a)). However, deletion of the upstream region (−1334–−1193 bp) containing nGRE2 and the NF-κB binding site produced no change in the JMJD3 gene promoter activity (Figure 2(a)). In addition, each nGRE was mutated and a reporter assay was performed. While the mutation of nGRE2 had no significant effect on the suppression of JMJD3 gene reporter activity following Dex treatment, the mutation of nGRE1 significantly attenuated the Dex-induced suppression of the JMJD3 gene reporter activity (Figure 2(b)). It was also confirmed that Dex induces the suppression of other known genes containing nGREs, such as IL-24 and Akt, in the bEnd.3 cells subjected to TNFα treatment (Supplementary Figure S3).
45
Negative GRE (nGRE) is required for dexamethasone-induced suppression of JMJD3 gene activation in bEnd.3 cells subjected to TNFα treatment. (a) Deletion of regulatory region containing nGRE abolishes Dex-induced suppression of reporter activity of JMJD3 gene in bEnd.3 cells subjected to TNFα treatment. Cells were transiently transfected with reporter vector containing serially deleted upstream region of JMJD3 gene in conjunction with control renilla luciferase expression vector (n = 3). Reporter activity is represented as fold activation relative to renilla luciferase activity. (b) Mutation of nGRE1 significantly attenuates Dex-induced suppression of reporter activity of JMJD3 gene in bEnd.3 cells subjected to TNFα treatment. Cells were transiently transfected with wild or mutant type JMJD3 gene-driven firefly luciferase reporter vector in conjunction with control renilla luciferase expression vector (n = 3). Reporter activity is represented as fold activation relative to renilla luciferase activity. All data represent mean ± S.E.M. Significance value was ***P ≤ 0.005.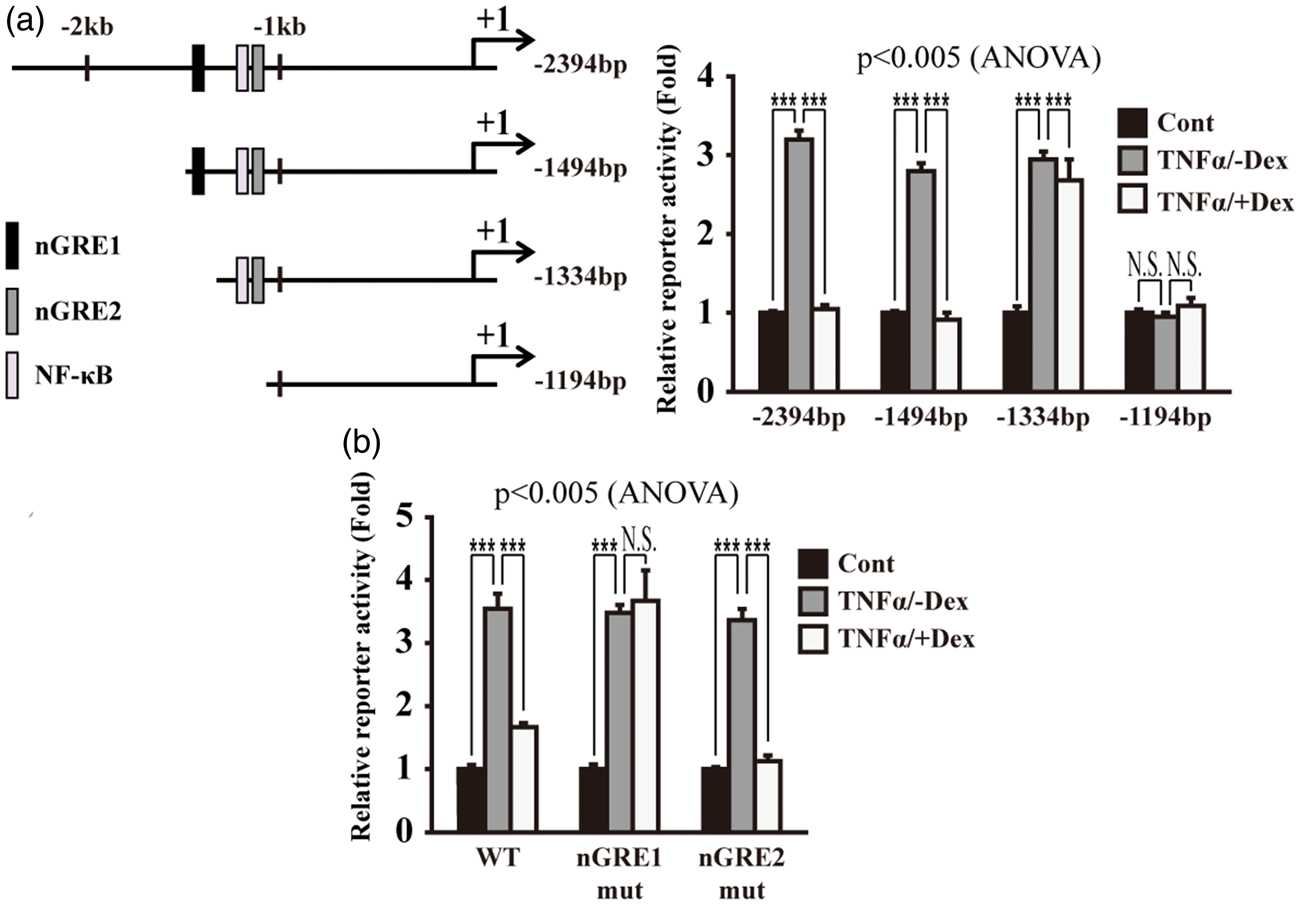
GRα-mediated dexamethasone-induced suppression of JMJD3 gene activation in bEnd.3 cells subjected to TNFα treatment
Given that Dex regulates the target gene expression through GRα (glucocorticoid receptor α) activation, we studied the involvement of GRα in the Dex-induced suppression of JMJD3 gene activation. The GRα expression was slightly down-regulated or did not change following Dex treatment in the TNFα treated-bEnd.3 cells at the RNA and protein level, respectively (Figure 3(a)). We also found that Dex induces the phosphorylation of GRα at serine 211 (S211) in the bEnd.3 cells subjected to TNFα treatment (Figure 3(b)). To confirm the GRα-dependent suppression of JMJD3 expression, the GRα was depleted using siRNA and the expression level of JMJD3 was determined. When compared with the control siRNA-transfected cells, the GRα depletion significantly abolished the Dex-induced suppression of JMJD3 gene activation in the bEnd.3 cells subjected to TNFα treatment (Figure 3(c)). Mifepristone (also known as RU486), which binds to the N-terminal of GR and mediates N-CoR co-repressor interaction, was also utilized as a GR antagonist.
46
Mifepristone treatment significantly abolished the Dex-induced suppression of JMJD3 gene activation in the bEnd.3 cells subjected to TNFα treatment (Figure 3(d)). We also found GRα-dependent expression of other known genes containing nGREs in the bEnd.3 cells subjected to TNFα treatment (Supplementary Figure S4).
Dexamethasone activates glucocorticoid receptor α in bEnd.3 cells subjected to TNFα treatment. (a) Dex slightly decreases GRα expression at RNA level in bEnd.3 cells subjected to TNFα treatment. However, expression level of GRα is not changed at protein level. Transcripts of GRα and GAPDH were determined by quantitative PCR (n = 3). Lysates were immunoblotted with anti-GRα and anti-β-actin antibodies, respectively (n = 3). Western blots were analyzed quantitatively. (b) Dex induces phosphorylation of GRα (S211) in bEnd.3 cells subjected to TNFα treatment. Lysates were immunoblotted with anti-GRα, anti-phospho GRα (S211), and anti-β-actin antibodies, respectively (n = 3). Western blots were analyzed quantitatively. (c) Depletion of GRα using siRNA attenuates Dex-induced suppression of JMJD3 gene activation in bEnd.3 cells subjected to TNFα treatment. After control (siCont) or GRα siRNA (siGRα) were transfected, transcripts of GRα, JMJD3, and GAPDH were determined by quantitative PCR (n = 3). (d) Mifepristone (Mif) attenuates Dex-induced suppression of JMJD3 gene activation in bEnd.3 cells subjected to TNFα treatment. Transcripts of JMJD3 and GAPDH were determined by quantitative PCR (n = 3). All data represent mean ± S.E.M. Significance values were *P ≤ 0.05 and ***P ≤ 0.005.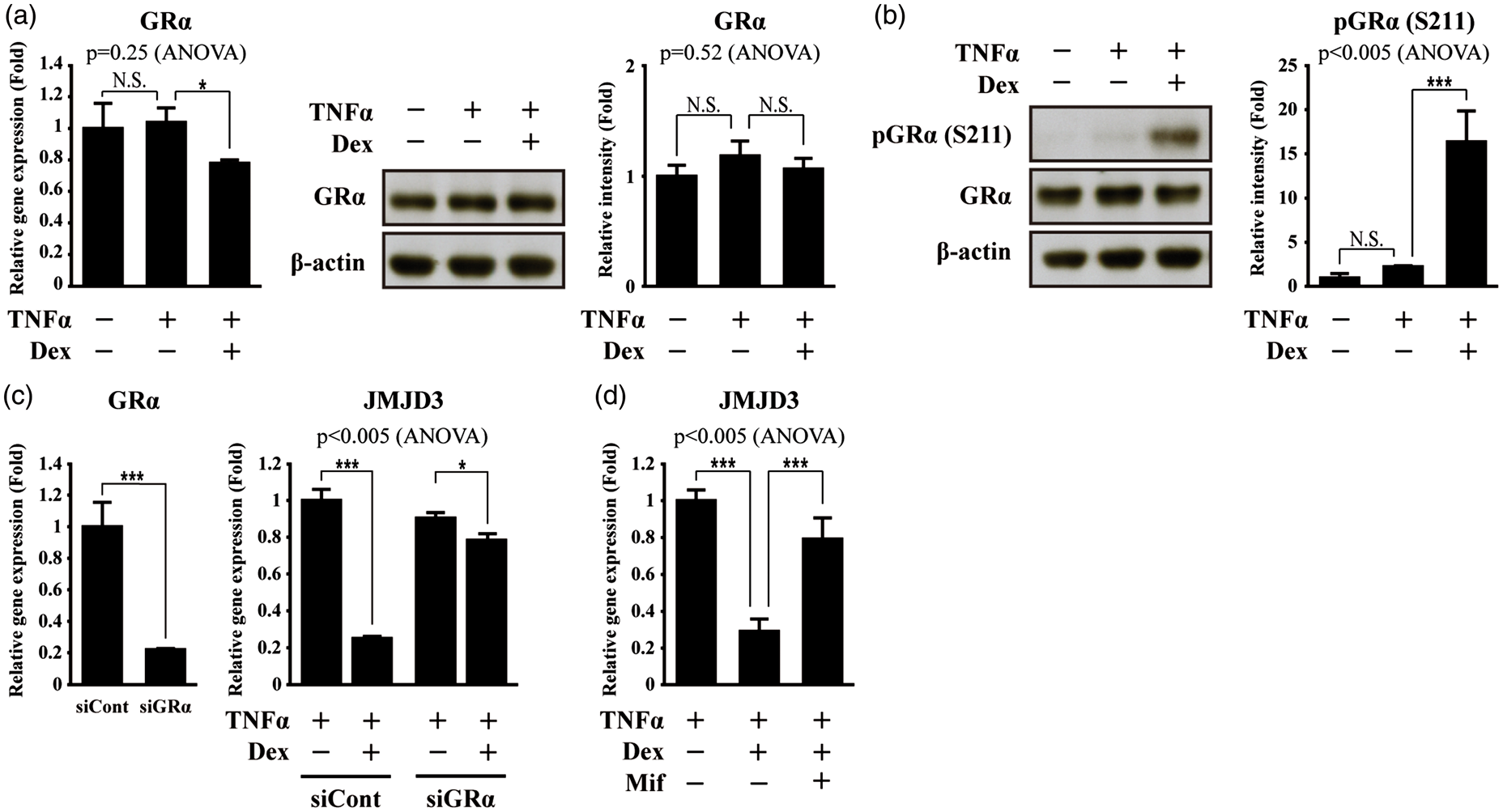
Dexamethasone induces recruitment of GRα and N-CoR to nGRE of JMJD3 gene in bEnd.3 cells subjected to TNFα treatment
It has been previously shown that GRα is recruited to nGREs with co-repressors, such as N-CoR.45,47,48 Thus, we investigated the interaction between GRα and N-CoR in the bEnd.3 cells. While GRα did not interact with N-CoR in the bEnd.3 cells with or without TNFα treatment, Dex did induce GRα interaction with N-CoR (Figure 4(a)). We further examined the recruitment of GRα and N-CoR to the nGRE in the JMJD3 gene using a chromatin immunoprecipitation (ChIP) assay. Dex induced the recruitment of GRα and N-CoR to the nGRE of the JMJD3 gene in the TNFα-treated bEnd.3 cells (Figure 4(b)). However, Mifepristone treatment abolished the Dex-induced recruitment of GRα and N-CoR (Figure 4(c)). Consistently, Mifepristone also decreased the recruitment of GRα and N-CoR to the nGREs in the upstream region of the IL-24 and Akt genes (Supplementary Figure S5).
Dexamethasone induces recruitment of GRα and N-CoR to nGRE in JMJD3 gene in bEnd.3 cells subjected to TNFα treatment. (a) Dex induces GRα interaction with N-CoR in bEnd.3 cells subjected to TNFα treatment. Lysates were immunoprecipitated with anti-GRα antibody, then immunoblotted with anti-N-CoR antibody (n = 3). Immunoprecipitation with normal IgG was used for negative control. (b) Dex induces recruitment of GRα and N-CoR to nGRE in JMJD3 gene in bEnd.3 cells subjected to TNFα treatment. Chromatin immunoprecipitation (ChIP) assay was performed using anti-GRα and anti-N-CoR antibodies, respectively (n = 3). Occupancy of each protein was quantified using real-time PCR in gene promoter region encompassing nGRE1. ChIP using normal IgG was performed as negative control. (c) Mifepristone attenuates Dex-induced recruitment of GRα and N-CoR to nGRE in JMJD3 gene in bEnd.3 cells subjected to TNFα treatment. ChIP assay was performed using anti-GRα and anti-N-CoR antibodies, respectively (n = 3). (d) Depletion of N-CoR using siRNA attenuates Dex-induced suppression of JMJD3 gene activation in bEnd.3 cells subjected to TNFα treatment. After control (siCont) or N-CoR siRNA (siN-CoR) were transfected, transcripts of N-CoR, JMJD3, and GAPDH were determined by quantitative PCR (n = 3). All data represent mean ± S.E.M. Significance value was ***P ≤ 0.005.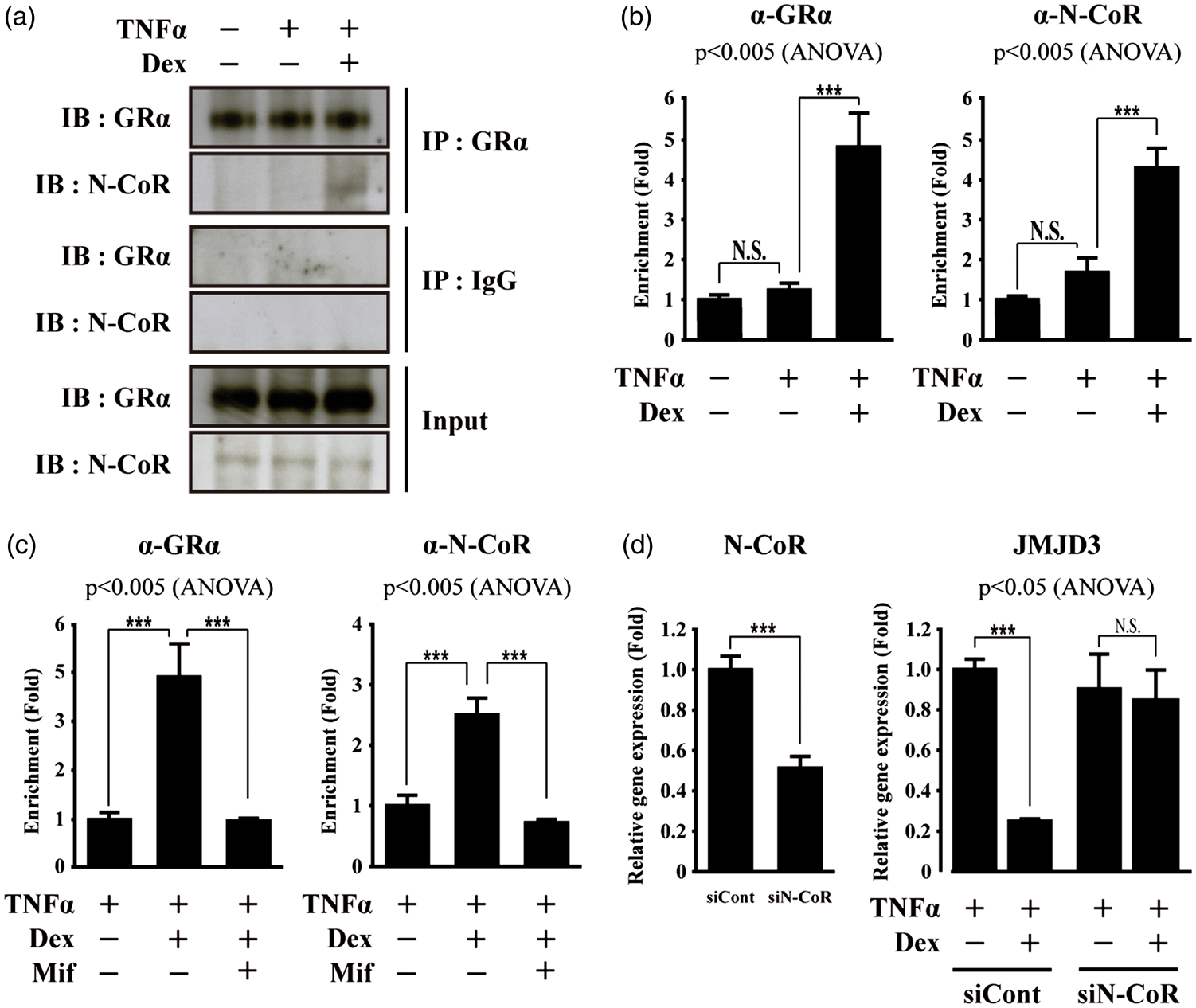
To confirm the involvement of N-CoR in the Dex-induced suppression of JMJD3 gene activation, the N-CoR transcripts were depleted using siRNA. Similar to GRα depletion, N-CoR depletion significantly eliminated the Dex-induced suppression of JMJD3 and other known gene activation in the bEnd.3 cells subjected to TNFα treatment (Figure 4(d) and Supplementary Figure S6). Collectively, our findings suggest that Dex induces the recruitment of GRα and N-CoR to the nGRE in the JMJD3 gene, thereby suppressing the JMJD3 gene activation in the bEnd.3 cells subjected to TNFα treatment.
Dexamethasone suppresses target genes of JMJD3 in bEnd.3 cells subjected to TNFα treatment
We recently reported that JMJD3 directly activates MMP-2, MMP-3, and MMP-9 gene expression in OGD/reperfusion-treated bEnd.3 cells.
38
Therefore, we wanted to investigate whether Dex treatment down-regulates MMP-2, MMP-3, and MMP-9 gene expression though the suppression of JMJD3 gene expression. Dex treatment abolished the up-regulation of MMP-2, MMP-3, and MMP-9 expression in the bEnd.3 and hCMEC/D3 cells subjected to TNFα treatment (Figure 5(a), Supplementary Figure S1). However, Mifepristone treatment or GRα depletion suppressed the Dex-induced down-regulation of MMP-2, MMP-3, and MMP-9 gene expression (Figure 5(a) and (b), Supplementary Figure S1). Consistently, zymography showed the Dex-induced suppression of MMP-2, MMP-9, and MMP-3 activities (Figure 5(c)). The recruitment of JMJD3 to the MMP-2, MMP-3, and MMP-9 gene promoters was investigated using a ChIP assay. While the recruitment of JMJD3 to the MMP-2, MMP-3, and MMP-9 gene promoters increased in the bEnd.3 cells subjected to TNFα treatment, Dex treatment produced a significant reduction in the recruitment of JMJD3 to the MMP-2, MMP-3, and MMP-9 gene promoters as well as maintenance of the H3K27me3 level in the bEnd.3 cells subjected to TNFα treatment (Figure 5(d)). However, Mifepristone treatment showed the opposite effect of Dex (Figure 5(e)).
Dexamethasone suppresses gene activation of matrix metalloproteinases in bEnd.3 cells subjected to TNFα treatment. Dex suppresses up-regulation of MMP-2, MMP-3, and MMP-9 genes in bEnd.3 cells subjected to TNFα treatment. However, Mifepristone treatment or depletion of GRα attenuates Dex-induced suppression of MMP-2, MMP-3, and MMP-9 gene activation by TNFα treatment. (a) Transcripts of MMP-2, MMP-3, MMP-9, and GAPDH were determined by quantitative PCR (n = 3). Lysates were immunoblotted with anti-MMP-2, anti-MMP-3, anti-MMP-9, and anti-β-actin antibodies, respectively (n = 3). Western blots were analyzed quantitatively. (b) After control (siCont) or GRα siRNA (siGRα) were transfected, MMP-2, MMP-3, MMP-9, and GAPDH were determined by quantitative PCR (n = 3). Lysates were immunoblotted with anti-MMP-2, anti-MMP-3, anti-MMP-9, and anti-β-actin antibodies, respectively (n = 3). Western blots were analyzed quantitatively. (c) Zymography was performed using equal amounts of lysates (n = 3). Zymographic images were analyzed quantitatively. (d, e) Dex reduces occupancy of JMJD3 in MMP-2, MMP-3, and MMP-9 gene promoters in bEnd.3 cells subjected to TNFα treatment. However, Mifepristone inhibits Dex-induced reduction of JMJD3 recruitment to MMP-2, MMP-3, and MMP-9 gene promoters. ChIP assay was performed using anti-JMJD3, anti-H3K27me3, and anti-H3K27me1 antibodies, respectively (n = 3). Occupancy of each protein was quantified using real-time PCR in gene promoter region encompassing NF-κB binding site. ChIP using normal IgG was performed as negative control. All data represent mean ± S.E.M. Significance values were *P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.005.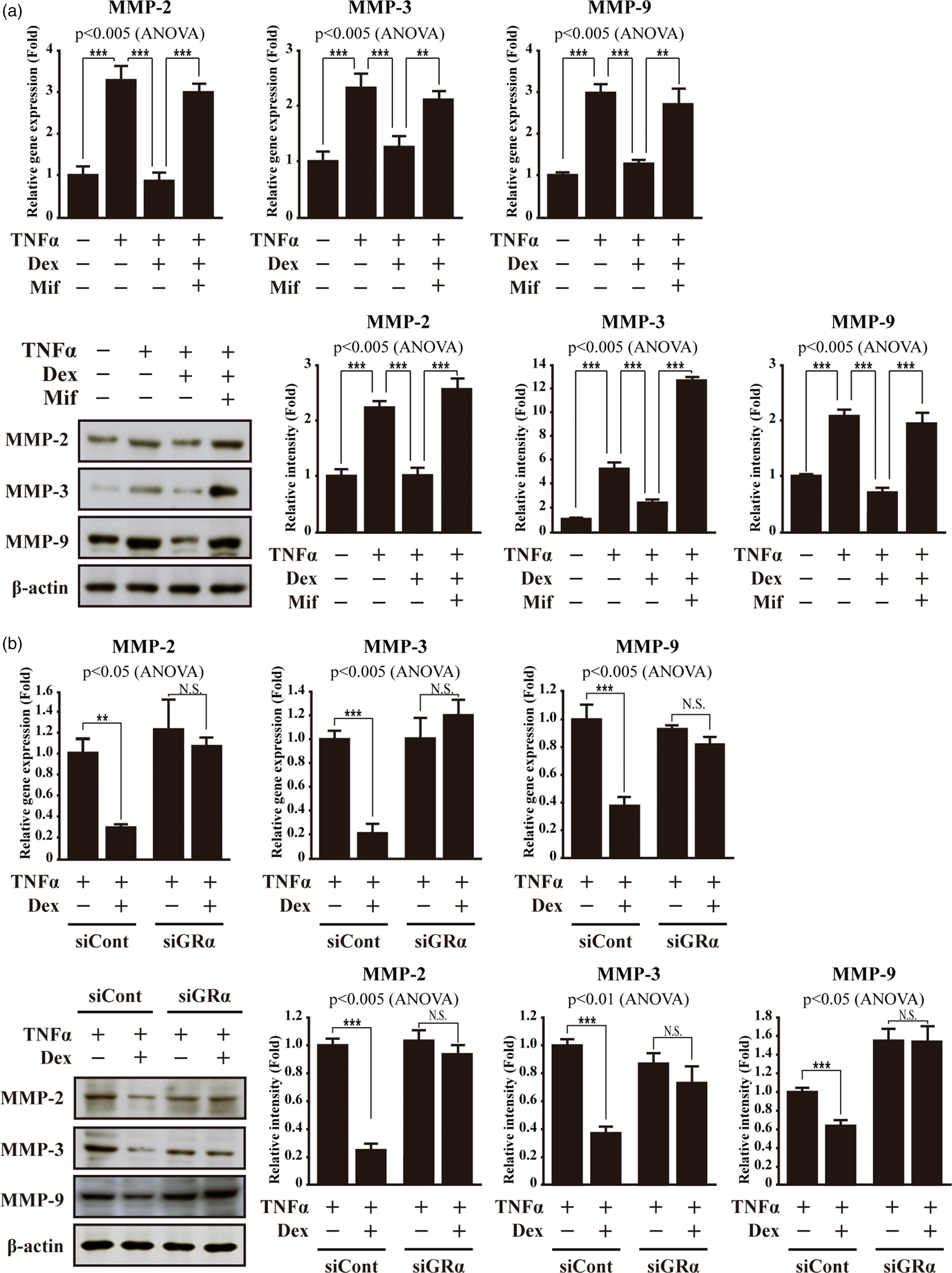
Dexamethasone maintains integrity of tight junction in bEnd.3 cells subjected to TNFα treatment
Since MMP-2, MMP-3, and MMP-9 all have a detrimental effect on the integrity of the BBB via the degradation of tight junction proteins,49–56 we next investigated the effect of Dex treatment on the expression of tight junction proteins. The levels of tight junction proteins, such as claudin 5 and occludin, were examined using immunocytochemistry and Western blot analysis. The levels of claudin 5 and occludin were lower in the bEnd.3 or hCMEC/D3 cells subjected to TNFα treatment (Figure 6(a) and (b), Supplementary Figure S7(a)). However, Dex attenuated the decreased levels of claudin 5 and occludin. Mifepristone treatment abolished the effects of Dex on the tight junction protein (Figure 6(a) and (b), Supplementary Figure S7(a)). We also examined the RNA levels of claudin 5 and occludin. Dex treatment increased the expression of claudin 5 and occludin genes in the TNFα-treated bEnd.3 cells when compared with the TNFα-treated control cells (Figure 6(c)). However, Mifepristone abolished the effect of Dex on the claudin 5 and occludin gene expression (Figure 6(c)). Consistently, a similar gene expression pattern was observed in the hCMEC/D3 cells (Supplementary Figure S7(b)). We also measured trans-endothelial electrical resistance (TEER) to monitor the integrity of the brain microvascular endothelial cells. While TNFα treatment induced a decrease in the TEER when compared with the untreated control, Dex treatment increased the TEER of the bEnd.3 and hCMEC/D3 cell subjected to TNFα treatment (Figure 6(d) and Supplementary Figure S7(c)). Mifepristone treatment or GR depletion abolished the Dex-induced increase in the TEER.
Dexamethasone maintains integrity of tight junction in bEnd.3 cells subjected to TNFα treatment. (a) Representative photomicrographs of claudin 5 and occludin expression in bEnd.3 cells. Dex attenuates decrease of claudin 5 and occludin expression in bEnd.3 cells subjected to TNFα treatment. However, Mifepristone inhibits protective role of Dex in claudin 5 and occludin expression. Cells were immunostained with anti-claudin 5 and anti-occludin antibodies, respectively (n = 3). Nuclei were identified using DAPI staining. Scale bar, 25 µm. (b) Lysates were immunoblotted with anti-claudin 5, anti-occludin, and anti-β-actin antibodies, respectively (n = 3). Western blots were analyzed quantitatively. (c) Transcripts of claudin 5, occludin, and GAPDH were determined by quantitative PCR (n = 3). (d) Dex increases trans-endothelial electrical resistance (TEER) in bEnd.3 cells subjected to TNFα treatment. However, Mifepristone treatment or depletion of GRα attenuates Dex-induced increase in TEER in bEnd.3 cells subjected to TNFα treatment. TEER was determined using voltohmmeter (n = 3). All data represent mean ± SEM. Significance values were *P ≤ 0.05, **P ≤ 0.01, and ***P ≤ 0.005.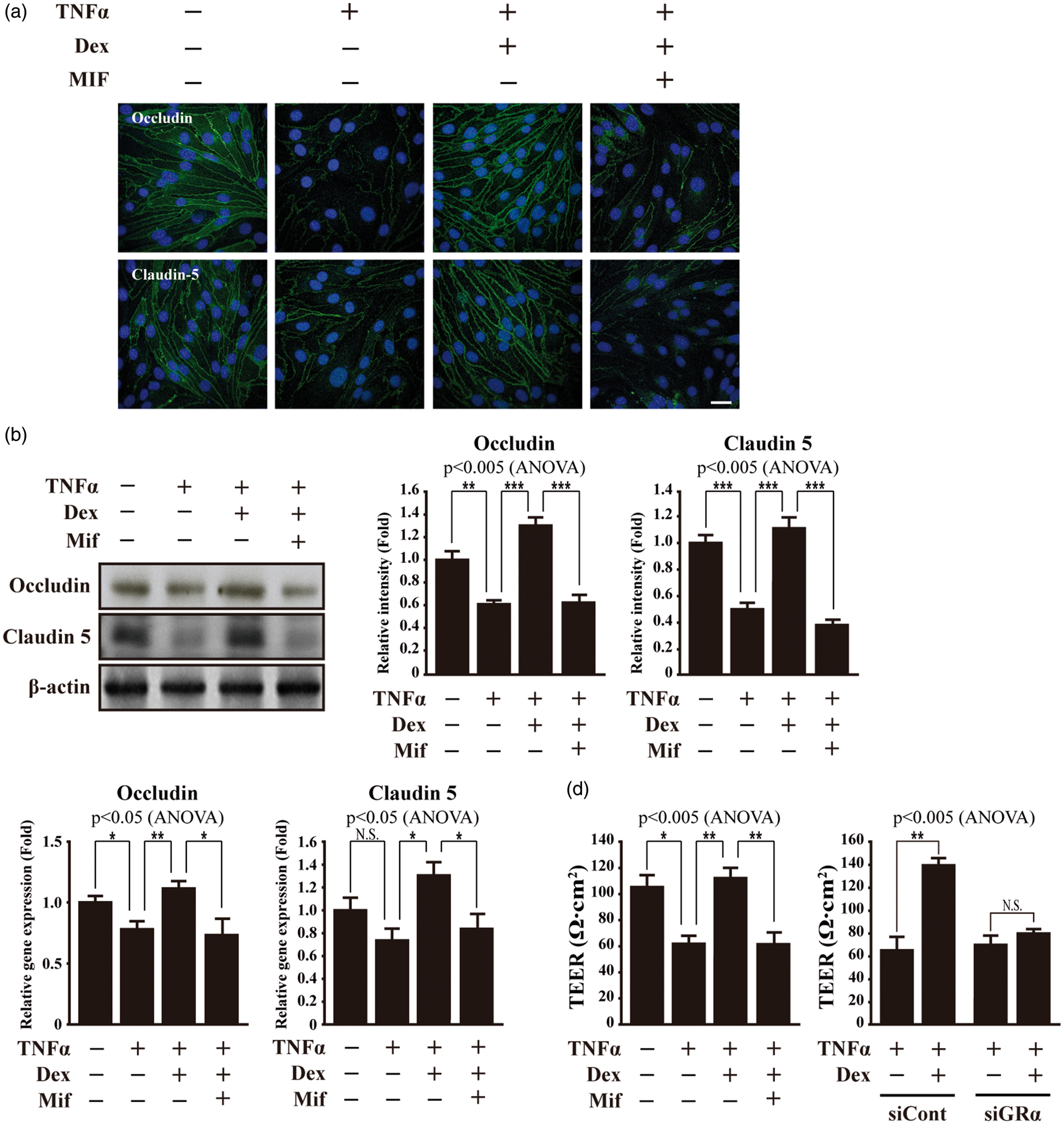
Discussion
We previously demonstrated that histone H3K27 demethylase JMJD3 is up-regulated in injured blood vessels after spinal cord injury.31,38 We also showed that JMJD3 is required for MMP-2, MMP-3, and MMP-9 gene activation in bEnd.3 endothelial cells subjected to OGD/reperfusion injury, indicating that the regulation of JMJD3 could be a new strategy to protect against BBB disruption after CNS injury. 38 In the current study, we found that TNFα treatment, which disrupts BBB, up-regulates JMJD3 expression in brain microvascular endothelial cells. Consistently, the functional NF-κB binding site is present in the upstream region of the JMJD3 gene.38,57 TNFα treatment also induced nuclear localization of JMJD3 in the brain microvascular endothelial cells. In fact, it has been shown that JMJD3 contains 2 classical nuclear localization signals in the N-terminus and its nucleocytoplasmic shuttling seems to be dynamically regulated. 58
We also demonstrated that dexamethasone suppresses the up-regulation of JMJD3 expression in brain microvascular endothelial cells subjected to TNFα treatment. Negative regulation of JMJD3 gene expression was achieved by the recruitment of GRα and N-CoR to the nGRE in the upstream region of the JMJD3 gene.45,59 It is previously known that nGRE differs from classic GRE in sequence and is involved in glucocorticoid-mediated anti-inflammatory effects.45,47,48 This study further supports the anti-inflammation function of glucocorticoids, as JMJD3 is a well-known epigenetic regulator in inflammatory gene activation.57,60–62
Glucocorticoids have beneficial effects against CNS damage via several mechanisms, such as reducing excessive inflammation, edema, and BBB/BSCB disruption.63,64 In particular, glucocorticoids induce improved tight junctions by increasing the levels of tight junction proteins, including claudin 5, occludin, ZO-1, and VE-cadherin, in vascular endothelial cells.19,22–27 In present study, we further suggest that dexamethasone improves the integrity of tight junctions by decreasing MMP-2, MMP-3, and MMP-9 gene expression through the down-regulation of JMJD3, which is a transcriptional activator of MMP-2, MMP-3, and MMP-9 genes, in brain microvascular endothelial cells. It is already known that MMPs play a critical role in the integrity of the tight junctions in injured cerebral ischemia.65,66 After cerebral ischemia, MMP-2 mediates occludin degradation and claudin 5 redistribution, which cause BBB disruption.54,56,67 Furthermore, studies of MMP-3 KO mice have shown that up-regulated MMP-3 in blood vessels contributes to the degradation of tight junction proteins, such as claudin 5 and occludin, after LPS administration or CNS injury.50,51 MMP-9 also plays an important role in the integrity of tight junctions via the degradation of occludin, ZO-1, and claudin 5, resulting in BBB disruption.32,56
We confirmed previous reports that glucocorticoids up-regulate the expression of the claudin 5 and occludin genes in microvascular endothelial cells. For example, hydrocortisone induces occludin gene activation through an imperfect GRE site in the gene promoter.24,68 Claudin 5 is also up-regulated by glucocorticoids in brain microvascular endothelial cells.24,69 While GRE sites are present in the promoter of the claudin 5 gene, it is not yet clear whether these sites are functional. 69 Interestingly, dexamethasone was found to activate the expression of occludin and claudin 5 genes via binding p54/NONO to the occludin enhancer element (OEE) in the gene promoters in retinal endothelial cells.70,71
Collectively, dexamethasone induced the down-regulation of JMJD3 activation via the nGRE in the upstream region of the gene, thereby suppressing the up-regulation of MMP-2, MMP-3, and MMP-9 gene. Dexamethasone also activated the expression of the claudin 5 and occludin genes. These events further resulted in the maintenance of the integrity of the tight junctions in the brain microvascular endothelial cells subjected to TNFα treatment, indicating that glucocorticoids may protect the BBB disruption via transcriptional and post-translational regulation of tight junction proteins.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2009-0093822, 2013R1A1A2008384) to BJ.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
WN, JYS performed experiments, analyzed data. BJ, JYS and SJ planned experiments, wrote manuscript. JYL, WK, and TYY analyzed data.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.