
Abstract
Brain capillary endothelium mediates the exchange of nutrients between blood and brain parenchyma. This barrier function of the brain capillaries also limits passage of pharmaceuticals from blood to brain, which hinders treatment of several neurological disorders. Receptor-mediated transport has been suggested as a potential pharmaceutical delivery route across the brain endothelium, e.g. reports have shown that the transferrin receptor (TfR) facilitates transcytosis of TfR antibodies, but it is not known whether this recycling receptor itself traffics from apical to basal membrane in the process. Here, we elucidate the endosomal trafficking of the retrograde transported cation-independent mannose-6-phosphate receptor (MPR300) in primary cultures of brain endothelial cells (BECs) of porcine and bovine origin. Receptor expression and localisation of MPR300 in the endo-lysosomal system and trafficking of internalised receptor are analysed. We also demonstrate that MPR300 can undergo bidirectional apical–basal trafficking in primary BECs in co-culture with astrocytes. This is, to our knowledge, the first detailed study of retrograde transported receptor trafficking in BECs, and the study demonstrates that MPR300 can be transported from the luminal to abluminal membrane and reverse. Such trafficking of MPR300 suggests that retrograde transported receptors in general may provide a mechanism for transport of pharmaceuticals into the brain.
Keywords
Introduction
Polarized brain endothelial cells (BECs) that form brain capillaries are connected by the most effective tight junctions among all endothelial cells found in the mammalian organism.1–4 BECs interact with pericytes and the end-feet of astrocytes, both of which are important for blood–brain barrier (BBB) development and maintenance.1,2,5 These cells, together with associated basement membranes, form the neurovascular unit.1,2,5 The BBB regulates the transport of nutrients, metabolites and cells between blood and brain parenchyma.1,2 While this helps to protect the brain from xenobiotics, it also hinders entry of a number of pharmaceuticals (including large molecule drugs) to the brain parenchyma.1,2,5
Transport of receptor ligands between the luminal and the abluminal membranes, known as receptor-mediated transcytosis (RMT), is one of the natural routes across the BBB. RMT has the potential to deliver pharmaceuticals and nanoparticles to the brain (for review, see earlier studies2,6,7). Hence, there has been intense interest in potential transcytosis routes across the BBB. However, it is not clear whether the receptors are themselves trafficked from apical to basal cell membrane (see supplementary text for discussion of terminology of this process).
Endocytic receptors can be divided into recycling receptors and retrograde transported receptors, depending on their fate after endocytosis; recycling receptors are recycled to the plasma membranes from endosomes, whereas retrograde transported receptors are transported in a retrograde manner from endosomes towards the trans-Golgi network (TGN). A number of recycling receptors (e.g. transferrin receptor (TfR) and lipoprotein receptor-related protein 1) have been shown to be involved in RMT across the BECs.6–11 Strategies using antibodies against TfR and antibodies/ligand-coated vesicles have been used to deliver drugs to the brain parenchyma with success, although the capacity appears to be low.
The cation-independent mannose-6-phosphate receptor (MPR300; 300 kDa) together with cation-dependent mannose-6-phosphate receptor (MPR46, 46 kDa) are canonical retrograde transported receptors with a high degree of overlapping functions.12–18 Their major function is delivery of the acidic hydrolases from the TGN to the lysosomes. MPR300 is also involved in other functions,16,19,20 e.g. development, angiogenesis and biogenesis of secretory granules in certain types of cells.19,21–23 The receptor can be found in the TGN, vesicular compartments and in small amounts at the plasma membrane in steady-state.19,24,25 MPR300 binds its endogenous ligands in the TGN and transports them to the endosomal compartment. This is mediated by cytosolic adaptor proteins such as AP-1, GGAs and clathrin.26–30 Upon acidification of endosomal pH, the ligand is released in late endosomes, and the receptor is trafficked in a retrograde manner back to the TGN where it can bind new ligands.15,17,19,31–34 The receptor retrieval involves Rab9 protein as well as AP-1 and PACS.35–40 Part of the receptor fraction can undergo retrograde trafficking from maturing endosomes, mediated by the Rab29 and retromer complex.33,41–46 MPR300 is also involved in endocytosis of secreted acidic hydrolases and other ligands.19,22,47,48 Receptor endocytosis to early endosomes (EE)/sorting endosomes (SE) depends on scaffolding and cytoplasmic adaptor proteins, primarily clathrin, AP-2, epsinR and RFN126.
There is little information regarding function and trafficking of MPR300 in BECs. MPR300 is a target for enzyme replacement therapy for lysosomal storage disorders,49–52 a group of disorders caused by malfunction of lysosomes, which can be treated by external administration of lysosomal enzymes. 52 As such, MPR300 has been studied in relation to delivery of lysosomal enzymes to the brain. Urayama et al. 49 reported that MPR300 is downregulated in the mouse BBB during development. A study using monocultured primary porcine BECs (pBECs) as an in vitro BBB model indicated some involvement of MPR300 in delivery of arylsulfatase A across the BBB model. 50 However, little is known regarding the intracellular fate the receptor.
In the present study, we compared expression of MPR300 in three in vitro BBB models: mono-culture (MC), astrocyte-induced monoculture and non-contact co-culture (NCC) with primary rat astrocytes. We also investigated the receptor internalisation pathway in primary BECs from adult mammals in the NCC BBB model. Finally, we demonstrate that MPR300 is trafficked in both directions across porcine BECs.
Materials and methods
Purification of brain capillaries
Porcine brain capillaries were purified based on established protocols.53,54 In brief, grey matter from six-month-old animals was isolated, and homogenised using a 40-mL Dounce tissue grinder (Kimble Chase). The homogenate was filtered through 140 μm mesh filter (Merck Millipore, NY4H04700). The filtrate was enzymatically digested for 1 h with 500 μg·ml−1 Collagenase type II (Thermo Fisher Scientific, 17101-015), 2.5% Trypsin-EDTA (1:10 dilution; Thermo Fisher Scientific, 15090-046) and 50 μg·ml−1 DNAse I (Sigma-Aldrich, D4513). Digested capillaries were collected by centrifugation at 240 rcf, 4℃ for 5 min. Capillaries from bovine brains (source Mogens Nielsen Kreaturslagteri A/S, Herlufmagle, Denmark) were isolated in a similar way to porcine capillaries (for details, see Helms and Brodin 55 ).
Cell culture
pBECs were grown until 90% confluency according to an established protocol53,54,56 in a T75 cell culture flask pre-coated with a mixture of collagen IV (Sigma-Aldrich, C5533) and fibronectin (Sigma-Aldrich, F1141), in 10 mL pBEC-conditioned medium (CM) (Table S1) supplemented with 4 μg·mL−1 puromycin (Sigma-Aldrich, P8833) to kill contaminating pericytes.
Primary rat astrocytes were grown as previously described 57 in poly-L-lysine (Sigma-Aldrich, P1524) coated 12-well plate, in 1 mL per well of Astro-medium (Table S1). Once the astrocytes reached 100% confluency, the medium was changed every three days. Used medium was collected, filtered through 0.22 μm syringe filter and stored at −20℃ to preserve growth factor activity (astrocyte-conditioned medium (ACM) needed in the BBB model).
Bovine brain endothelial cells (bBECs) were grown as previously described. 55 In brief, T75 flasks were coated with collagen type IV and fibronectin. Bovine capillary suspensions were seeded in 10 mL growth medium (GM+) (Table S1) containing 4 µg·ml−1 puromycin (37℃, 10% CO2). The cells were grown for five days with change of medium on day 3 to GM + without puromycin.
Immortalised mouse embryonic fibroblast (MEF) cells were grown in DMEM medium supplemented with 10% FBS and 1% P/S. For immunostaining, cells were plated on #1.5 coverslips pre-coated with poly-L-lysine (5 μg/mL; 2 h at 37℃). MEF MPR300 KO, MEF transfected with human MPR300 17 and wild-type MEF were used as a control for antibody used for detection of MPR300.
Culture of in vitro BBB models
The porcine in vitro BBB model was based on a number of previous studies;53,54,56 24 h before seeding pBECs on inserts, the medium in astrocyte cultures was refreshed: 1.5 mL of fresh Astro-medium was added to each well with cells. 1.1 × 105 cells in 500 μL of pBEC-CM were seeded per Transwell (Corning, 3401). For NCC, Transwells were transferred to wells with primary rat astrocytes. After 24 h, medium was changed to 1.5 mL of serum-free pBEC-CM in the wells and 500 μL of pBEC-CM in Transwells. The following differentiation factors were added to wells and Transwells: 250 μM cAMP (Biolog, C010), 17.5 μM 4-(3-Butoxy-4-methoxybenzyl)imidazolidin-2-one (RO; Sigma-Aldrich, B8279) and 550 nM Hydrocortisone (Sigma-Aldrich, H4001). Cells were incubated until establishment of high transendothelial electrical resistance (TEER) values (usually three to four days) before use in experiments. Every 24 h, TEER measurement was performed using EndOhm-12 and EVOM measurement device (World Precision Instruments), and a new aliquot of differentiation factors was added. On the second day, complete medium refreshment took place. For MC, cells were grown as described above except that a 12-well plate without astrocytes was used for culture. MCs induced with ACM (MC+ACM) were grown as described above except that a 12-well plate without astrocytes was used for culture, and medium in the well during the first 24 h was a 1:1 mixture of pBEC-CM and ACM.
The bovine in vitro BBB model was based on a previously published study. 55 For co-culture at day 3 of cell growth, Transwell filter inserts (area = 1.13 cm2) were coated with collagen type IV and fibronectin, and primary rat astrocytes were seeded on the underside of the filter inserts or on the bottom of the well in 12-well plate, 1.2· × 105 cells/filter insert or well (37℃, 10% CO2), for contact co-culture (CCC) and NCC, respectively. At day 5 after seeding, when the endothelial cells had reached a confluence of approximately 60%–80% in the flasks, the medium in the Transwell plates with the filter inserts containing astrocytes was changed to Growth Medium – (GM-) (Table S1), and the endothelial cells were passaged from the flasks onto the filter inserts (100,000 cells/filter insert). The endothelial cells were then grown for an additional six days on the filter inserts with medium change from GM- to Differentiation Medium (DM) (Table S1) at day 3.
Western blot
Six Transwell inserts with a tight layer (high TEER) of pBEC or bBECs were washed with phosphate-buffered saline (PBS) (Sigma-Aldrich, D8537) and lysed in ExB lysis buffer (150 mM NaCl, 20 mM MgCl2, 20 mM CaCl2, 100 mM HEPES, 1% TritonX-100, supplemented with Complete Protease Inhibitor; Roche Diagnostics, 10765800). As a positive control, porcine primary pericytes were used; 2.5 μg of each protein sample was loaded on a 4%–12% polyacrylamide gel (NuPage, Thermo Fisher Scientific). Protein was transferred to nitrocellulose and blocked in 5% milk in TTBS solution (1 M Trisma-Base, 150 mM NaCl, 0.1% Tween 20), followed by incubation with primary antibodies overnight at 4℃: Rabbit polyclonal anti-MPR300 (Abcam, ab32815; 1:500), rabbit monoclonal anti-MPR46 (Abcam, ab134153; 1:1000), mouse monoclonal anti-bovine MPR300 (DSHB, 86f7; 1:1000), mouse monoclonal anti-bovine MPR46 (DSHB, 22d4; 1:1000) and mouse anti-β-actin (Sigma-Aldrich, A5441; 1:1000). Secondary antibodies were incubated for 1 h at room temperature: goat anti-rabbit-HRP conjugated (1:2000; Cell Signalling, 7074) and horse anti-mouse-HRP conjugated (1:2000, Cell Signalling, 7076). Bands were visualised using Fuji LAS 4000.
Quantitative PCR
RNA was purified from cells grown in the different in vitro models using NucleoSpin®RNA (Macherey-Nagel, 740955.50). Nucleic acid concentration and purity were measured using NanoDrop ND-1000 (Thermo Scientific). Linear amplified cDNA was generated using Ovation PicoSL WTA System V2 (NuGENE, 3312-12) from 35 ng of total RNA. The cDNA was purified using the MinElute Reaction Cleanup Kit (Qiagen, 28204). Primers were designed to span exons. Design of primers was performed using Pearl Primer software 58 and the NCBI primer design tool (http://www.ncbi.nlm.nih.gov/tools/primer-blast) (Table S2). Primers were purchased from MWG-Biotech (Eurofins). The quantification of transcript was performed using real-time quantitative polymerase chain reaction (qPCR). Each cDNA sample was amplified in triplicate using LightCycler 480 SYBR Green I Master version 12 (Roche, 04707516001) in total reaction mixture of 10 μL, containing 5 μL 2 × Mastermix, 1 μL forward and reverse primers (5 μM), 10 ng cDNA template and nuclease-free H2O to 10 μL. The PCR was run on a LightCycler®96 (Roche), using LightCycler®480 Multiwell Plate 96. The programme was as follows: 95℃ for 5 min (pre-incubation), 45 × (95℃ for 10 s, 60℃ for 10 s, 75℃ for 15 s) (amplification), then 95℃ for 10 s, 65℃ for 1 min (melting curve) and finally 40℃ for 10 s (cooling). A non-template control was included for each gene in the individual qPCR runs. Primer efficiencies were tested by dilution series of cDNA template on cDNA generated from pericytes for each primer. Data analysis was performed in GraphPad Prism 6 software (GraphPad), using unpaired Student's t-test with Welch's correction to test significance of results.
Receptor internalisation
Transwell inserts with a tight layer of pBECs (TEER > 600 Ω cm2) were transferred to a new 12-well plate, washed twice with PBS, and fresh pBEC medium was added, 300 μL to the upper (luminal) compartment and 700 μL to the lower (abluminal) compartment. Cells were incubated for 45 min at 4℃ to arrest endocytotic events. Primary antibody against MPR300 was added to the upper or lower compartment, and incubated at 4℃ for 1.5 h to allow antibody to bind to cell surface receptors. Then, each Transwell insert was washed twice with cold PBS, followed by addition of pBEC medium to both sides of the monolayer (except for those inserts which represented t = 0 min, which were fixed immediately with 4% paraformaldehyde (PFA)). The rest of the samples were returned to the incubator to release cells from arrest and allow internalisation of receptor-bound antibodies. Polycarbonate supports were cut out from the Transwell inserts, washed two times with PBS and fixed with 4% PFA at time points 10, 20 and 40 min. After fixation, cells were used for immunostaining with markers for subcellular localisation.
Detection of receptor trafficking across polarized BECs
Bidirectional apical–basal trafficking detection assay was carried out as described previously. 59 Transwell inserts with tight layers of pBECs were transferred to a new 12-well plate, washed twice with PBS, and pBEC medium was added to both chambers (300 μL to Transwell and 700 μL to the well). After 30 min equilibration in the incubator, primary antibody against receptor (or blank), and compatible secondary antibody were added to opposite (apical and basal) compartments. Four combinations were prepared, described in detail in figure legend (Figure 5). Each of the four setups was cultured for 45 min, washed with PBS and fixed with 4% PFA. After fixation, Transwells were used for immunostaining with subcellular localisation markers.
Immunostaining, imaging and image processing
Fixed cells were permeabilised with 0.1% TritonX-100 in PBS and blocked with 2% bovine-serum albumin (BSA) (Sigma-Aldrich, A7906) in PBS. Next, Transwell membranes were incubated with primary antibody solution in 2% BSA in PBS for 1 h, washed and then further incubated for 1 h with secondary antibodies. The antibodies used in this study are listed in Table S3. Finally, 0.5 μg·mL−1 Hoescht 32528 (Sigma-Aldrich, B1155) in ddH2O was used to stain nuclei and Transwell membranes mounted in mounting medium (Dako, S3023) under #1.5 coverslips. Confocal imaging was done using Olympus IX-83 fluorescent microscope with Andor confocal spinning unit and Andor iXon Ultra 897 camera, using Olympus cellSens software (Olympus). Images of cells were taken using Olympus UPSALO O, 100× magnification, 1.40 NA, oil objective lens; images of capillaries were taken using Olympus UPSALO W, 60× magnification, 1.20 NA water objective lens. Images were processed using Fiji software; 60 for each channel, independent brightness and contrast adjustments were applied. For co-localisation measurement, Pearson's correlation coefficients (PCCs) were calculated using co-loc plug-in in Olympus cellSens software. For each measurement, three cells from individual experiments were analysed.
Ethical information
Bovine and porcine brains were obtained as a by product from the Danish food industry. Danish Slaughterhouses are under currently supervision and observation from the Danish Ministry of Environment and Food. Rats used for isolation of astrocytes were bred and group housed in the local animal facility at an ambient temperature of 22℃–23℃ and on a 12/12 h dark/light cycle (lights on 7 a.m.) under inspection of veterinarian and according to Danish regulation for lab animals. The rats were euthanized before they were sacrificed in accordance with international guidelines on the ethical use of animals (European Communities Council Directive of 24 November 1986; 86/609/EEC) and Danish guidelines. No in vivo experiments on animal nor human material were used in these experiments.
Results
In vitro BBB models
For studying expression and trafficking of MPRs, we used porcine and bovine in vitro BBB models, which in several previous publications have been shown to have similar to in vivo properties with respect to tightness and polarity.53–55,61 Primary BECs were grown in Transwell system as MCs or in co-culture with primary rat astrocytes. For qPCR experiments MC, MC+ACM and co-culture models were used. The rest of the experiments were conducted using co-culture models. The integrity of the endothelial monolayers grown in co-culture was assessed by measurements of the TEER. The porcine co-cultures had a TEER of 924 ± 166 Ω·cm2 (mean ± SD, n = 133) with 1307 ± 136 Ω·cm2 being the highest value measured on the day of experiment (Figure 1(a)). Immunofluorescence staining for tight junction proteins was also performed on the BECs (Figure 1(b)); the cells showed clear junctional staining for p120, claudin-5, ZO-1 and occludin. These results confirmed the quality of the model and its suitability for study of receptor expression and trafficking in the brain mammalian BECs. The bovine models had an average TEER value of 1662 ± 452 Ω·cm2 (mean ± SD, n = 5); expression of Claudin-5 was described previously.
62
BBB in vitro model. (a) Graph showing TEER values measured from pBECs grown in non-contact co-culture with primary rat astrocytes. Measurement was done at 24 h intervals following incubation with differentiation factors using EndOhm 12 device. Data corrected for values of TEER measured on empty Transwell, n = 11, mean ± SD and (b) immunostaining of pBECs grown in non-contact co-culture with primary rat astrocytes for proteins associated with TJs. Cells fixed and immunostained on third day of culture; nucleus (blue), TJ markers (red and green), scale bar 10 μm.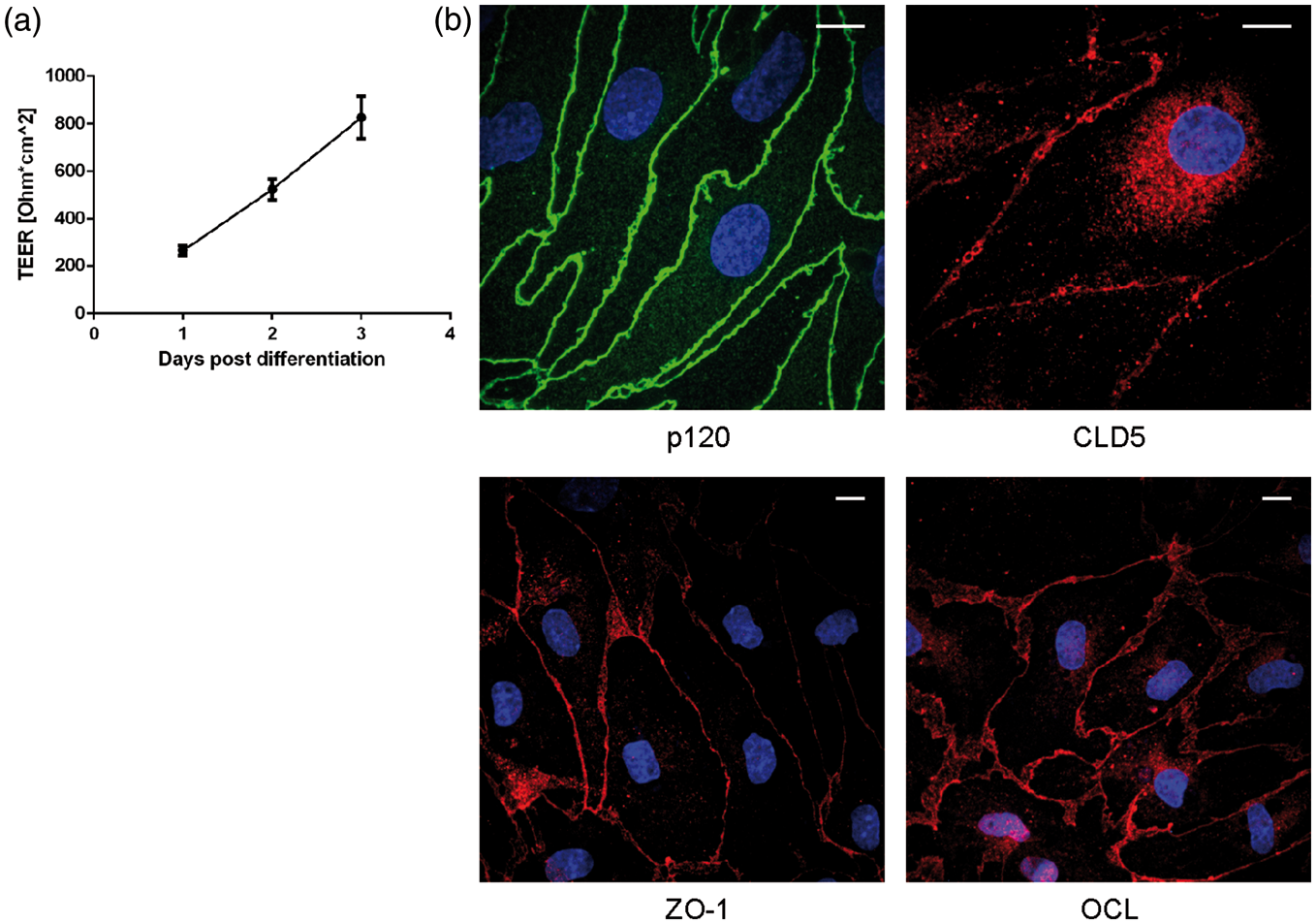
Expression of MPR300 in mammalian BECs and capillaries
First, we examined expression levels of MPR300 in BECs in the porcine and bovine models. Porcine endothelial cells were grown as MC, mono-culture in the presence of astrocyte-conditioned medium (MC+ACM) or in NCC with primary rat astrocytes, and for the bovine model as a MC, NCC and CCC with primary rat astrocytes. When the barrier was well established (based on TEER measurement), cells were washed with PBS, lysed and mRNA was purified. Expression of the receptors was measured using qPCR and normalised against the mRNA expression of β-actin (Figure 2(a)). In the porcine models, the expression level of MPR300 was highest in the MC with ACM (0.121 ± 0.006 and 0.099 ± 0.002, respectively; Figure 2(a)). In the NCC culture condition, expression of MPR300 (0.086 ± 0.001) was less than for MC+ACM but still higher than in MC (0.048 ± 0.006; Figure 2(a)). In the bovine model, the MC condition showed the highest level of MPR300 mRNA (0.018 ± 0.007), which was lower in NCC and CCC conditions (0.013 ± 0.002 and 0.012 ± 0.002, respectively); however, none of these differences were significant (Figure 2(a)). The differences in the receptor expression between the various model configurations were not significant. For the following experiments, the NCC variant of the models was used as the most convenient for performing transcytosis and internalisation experiments in the presence of astrocyte factors.
Expression of MPR300 in mammalian BBB in vitro model and fresh capillaries. (a) qPCR data showing relative level of expression of MPR300 in pBEC and bBEC. All data were normalised to expression of β-actin; (b) Western blot showing detection of MPR300 in pBEC, bBEC, porcine pericytes and mice embryonic fibroblasts (knock-out for MPR300, MPR300-transfected MPR300 knock-outs and wild type); (c) Immunostaining of mammalian BECs against proteins associated with TJs and MPR300. Detection of MPR300 (red) in pBEC and bBEC; (d) Immunostaining of freshly isolated brain capillaries showing detection of MPR300 (red) co-stained with tight junction associated protein p120 (green) in porcine, and MPR300 (green) co-stained with tight junction protein CLD5 (red) in bovine and (e) Immunostaining of mice embryonic fibroblasts (knock-out for MPR300, MPR300 transfected MPR300 knock-outs) against MPR300 (green). In all pictures, nucleus showed in blue. Scale bars 10 μm. For qPCR, the mean ± SD of six inserts for porcine and three for bovine are shown; *p < 0.01, **p < 0.0001, ns: not significant with p > 0.05; unpaired Student's t-test with Welch's correction.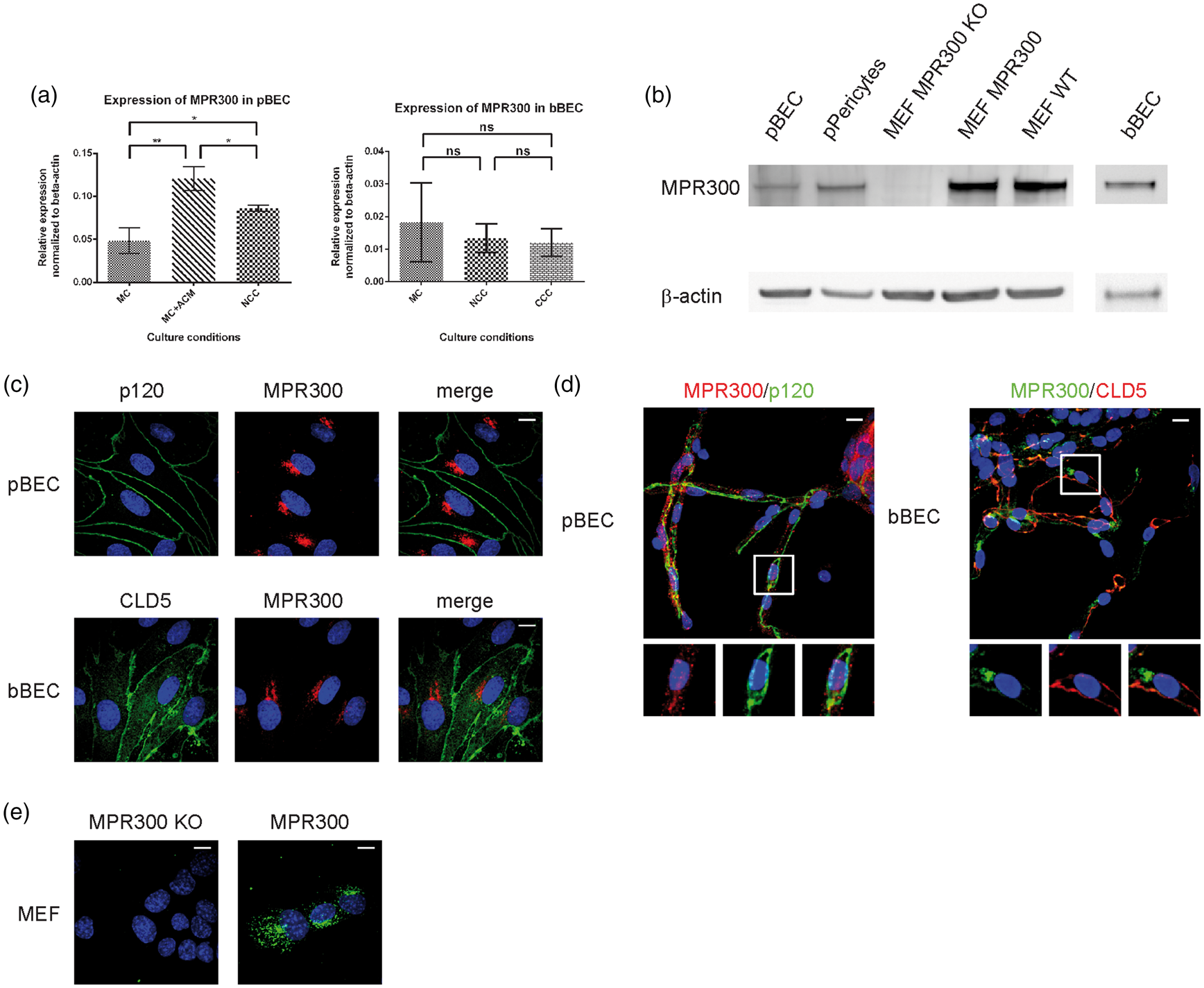
In order to investigate whether MPR300 was present at the protein level, we performed Western blot analysis on cell lysates (Figure 2(b)). Bands corresponding to MPR300 in lysates from pBEC and bBEC were detected; porcine pericytes were used as positive control. Mice embryonic fibroblast MPR300 knock-out cells (MEF MPR300 KO) and MEF MPR300 KO transfected with MPR300 were used as a negative and positive control for anti-MPR300 (Figure 2(b)). These results show presence of the receptor in BECs derived from these adult mammals. Expression of the receptor at the protein level in both porcine and bovine BECs was also demonstrated by immunocytochemical studies. MPR300 staining showed a punctate localisation in the cytoplasm and in the perinuclear region (Figure 2(c)), consistent with its trafficking pathway14,19,20,22 between TGN and endosomal compartments. Antibody specificity for immunofluorescense studies was checked using MEF MPR300 KO and MEF MPR300 KO transfected with MPR300 (Figure 2(e)).
Finally, MPR300 detection in cultures was compared to capillaries freshly purified from porcine and bovine brains. Capillaries were co-stained with junctional markers p120 or claudin-5 (CLD5), which clearly labels the cell outlines (Figure 2(d)). Signal from MPR300 was present as bright punctate spots mainly around the nucleus.
These results show that MPR300 is present in adult mammalian brain capillaries and their expression is not an artefact of cell culture used for the other experiments in this study.
Subcellular localisation of MPR300 in mammalian BECs
Endothelial cells grown under NCC culture conditions were co-stained for MPR300 and markers of different cellular compartments, in order to establish which subcellular vesicle compartments contained MPR300. In porcine cells, we co-stained MPR300 (Figure 3) with EEA1 (EE marker), clathrin heavy chain (CHC, marker for clathrin-mediated trafficking), caveolin 1 (Cav1, marker for non-clathrin-mediated endocytosis pathway(s)), VTI1a (for TGN) and Vps35 (as a marker for the retromer complex). There is strong co-localisation of the receptor with CHC and EEA1 indicating that MPR300 from the plasma membrane is internalised into EE/SE in a process involving clathrin (Figure 3). The co-localisation with CHC could also reflect TGN staining, as clathrin-coated pits are also involved in the membrane-budding at TGN exit. We observed only a weak signal for co-localisation of the receptor with Cav1. Although a caveolin-mediated and therefore clathrin-independent pathway could be a factor in the process of receptor internalisation, its contribution appears to be minor (Figure 3). Finally, we saw strong co-localisation of receptor signal with VTI1a in the perinuclear region as well as strong punctate co-localisation with Vps35 (Figure 3), marking TGN and retromer binding vesicles, respectively.
Co-localisation of MPR300 with makers for internalisation and trafficking pathways in pBEC. Immunostaining of pBEC for MPR300 (red) and markers of internalisation and trafficking pathways (green), nucleus (blue); scale bar 10 μm. Z-stack shown in maximum projection over Z, marked region is shown in inset and as XZ projection with luminal and abluminal (Transwell) sides in top-bottom orientation.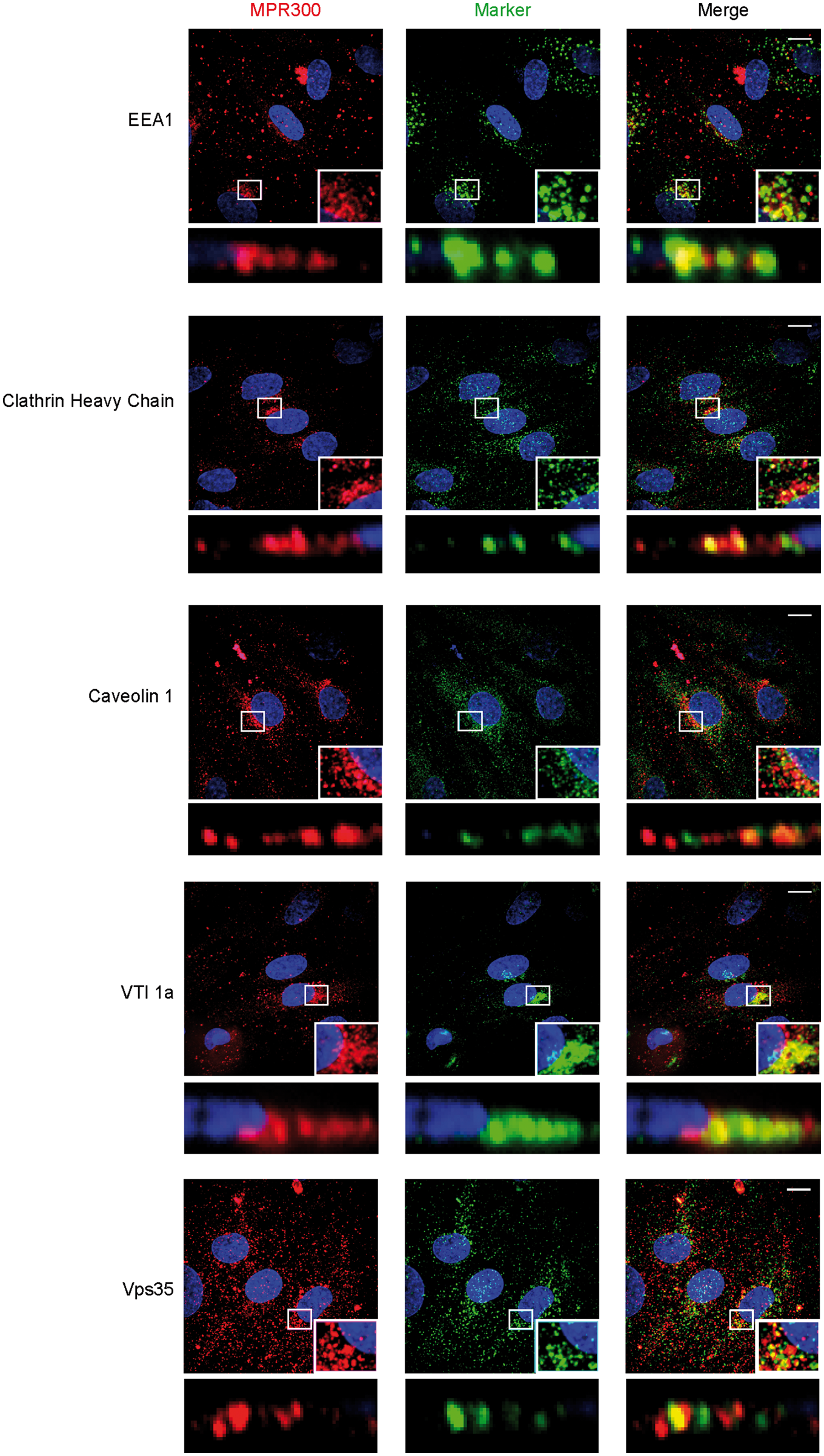
In bovine BECs, we performed co-localisation studies of MPR300 (Figure S1) with EEA1, Rab7 (late endosome marker), Lamp1 (lysosome marker) and Vps35. We observed strong punctate co-localisation of MPR300 signal with EEA1 and Vps35 signal, as well as weak co-localisation with Rab7 (Figure S1). We also observed very weak co-localisation of the receptor signal with Lamp1 signal (Figure S1). Normally, MPR300 does not go to the lysosomes; however, a small fraction of receptor in this compartment could correspond to receptor undergoing degradation.
Taken together, these results show that MPR300 is localised in vesicles that are involved in trafficking in the endo-lysosomal system in BECs.
Because the results from porcine and bovine models were similar, further experiments were performed in the porcine models.
Internalisation of MPR300 in mammalian BECs
Next, we investigated the internalisation MPR300 from apical and basal sides of the cells by saturation of surface localised receptor on either apical or basal side with primary antibodies (at 4℃). Subsequently, cells were fixed for time 0 min, or after 10, 20 and 40 min internalisation. All samples were immunostained for receptor and intracellular markers. Results from internalisation of MPR300 are shown in Figure 4.
Internalisation of MPR300 in pBEC at luminal and abluminal side. Immunostaining of pBECs for MPR300 (red) and markers for internalisation pathways (green); nucleus (blue; in all pictures); scale bar 10 μm; Z-stack of pBECs presented as maximum projection over Z; marked region is shown in inset and as XZ projection with luminal and abluminal (Transwell) sides in top-bottom orientation.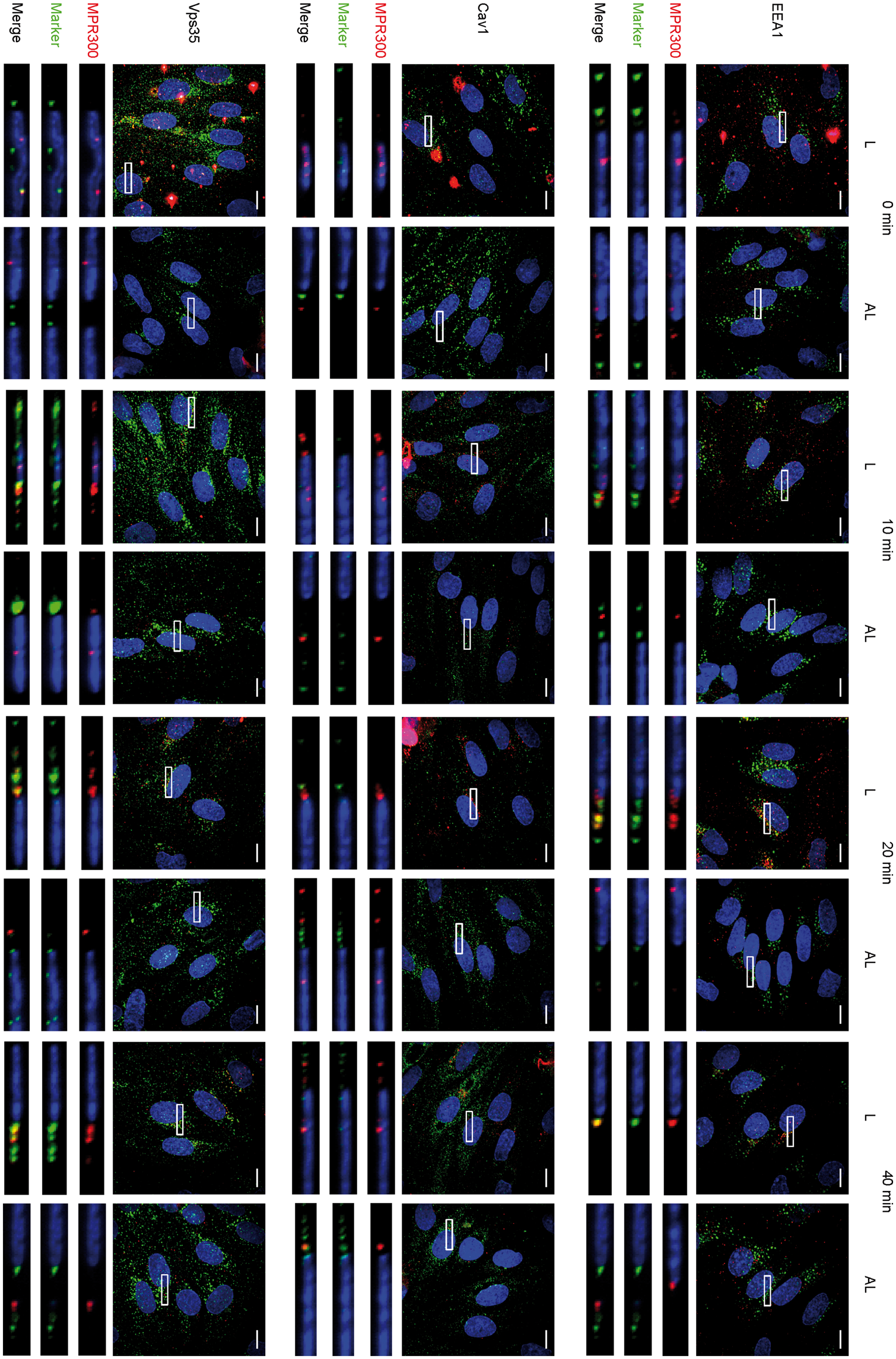
For MPR300, we detected signal in spots scattered across the whole cell surface at time 0 min; at later time points, those spots begin concentrating around the perinuclear region. For the apical side, this peaks at 20 min and becomes less pronounced at 40 min. For basal detection, concentration near the perinuclear region is most intense at 40 min (Figure 4). This indicates that internalisation of the receptor occurs from both sides of the cell, although speed or trafficking routes appear to be different.
Pearson's correlation coefficient factor values for co-localisation of MPR300 and MPR46 with subcellular markers in internalisation experiments.
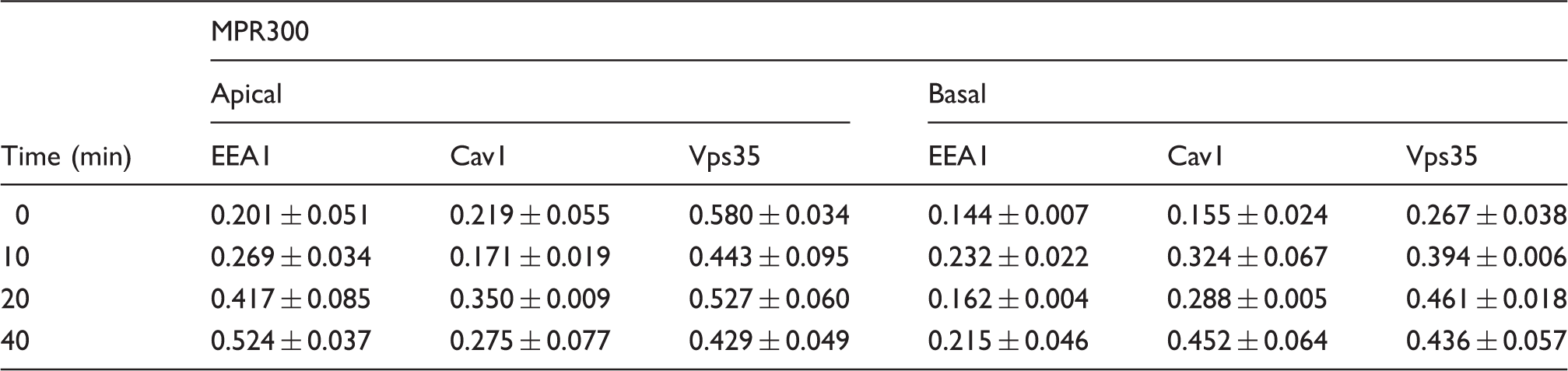
Values show mean ± SEM of measurement on three individual cells for each staining.
PCCs of MPR300 with EEA1 and Cav1 were low at 0 min. Co-localisation with Cav1 varied over time with increase at 20 min (r = 0.350 ± 0.009) for apical side internalisation, and at 40 min (r = 0.452 ± 0.064) for basal side internalisation. Co-localisation with EEA1 steadily increased over time for apical side internalisation, but varied, staying low for basal side internalisation.
Taken together, MPR300 is internalised from both apical and basal sides by BECs, and probably enters the canonical, retrograde pathway. Co-localisation with different markers showed that the internalisation process could differ depending on the membrane side from which it takes place. However, further investigation is needed to map the exact consecutive routes MPR300 follows from the apical or basal side.
Bidirectional apical to basolateral polarised receptor trafficking of MPR300 in mammalian BECs
Next, we examined MPR300 trafficking from the apical to the basal membrane and vice versa. Physiological ligands for MR300 are uncoupled in the low pH of endosomes then forwarded to lysosomes and are therefore not useful to study the retrograde trafficking route of these receptors. Hence, we used primary antibodies against the receptors, added to either apical or basal side of the cells, with labelled secondary antibody added to the opposite chamber (Figure 5(a)). With this procedure, receptors localised to the plasma membrane on one side of the cell can be bound by the antibody, with only those receptor bound antibodies transported to the opposite membrane being able to be bound by labelled secondary antibody added to the opposite chamber. After 45 min, cells were fixed and immunostained against MPR300, EEA1 and Vps35. Results from these experiments are presented in Figures 5(b) and S2.
Detection of MPR300 trafficked across cells in pBECs. (a) Scheme showing concept of experiment and experimental set-ups used in study. Four combinations were prepared (Transwell/well): (I) Rabbit anti-MPR300 (1:100)/Donkey anti-rabbit IgG conjugated to AlexaFluor 488 (1:200); (II) Donkey anti-rabbit IgG conjugated to AlexaFluor 488/Rabbit anti-MPR300; (III) Non-treated (NT)/donkey anti-rabbit IgG conjugated to AlexaFluor 488 and (IV) Donkey anti-rabbit IgG conjugated to AlexaFluor 488/non-treated (NT) and (b) Immunostaining of pBECs for trafficked across cell plasma membranes MPR300 (green) and Vps35 (red); nucleus (blue; in all pictures); scale bar 10 μm; Z-stack of pBECs presented as maximum projection over Z; marked region presented in inset for separate channels, from left: receptor staining, marker staining, merged.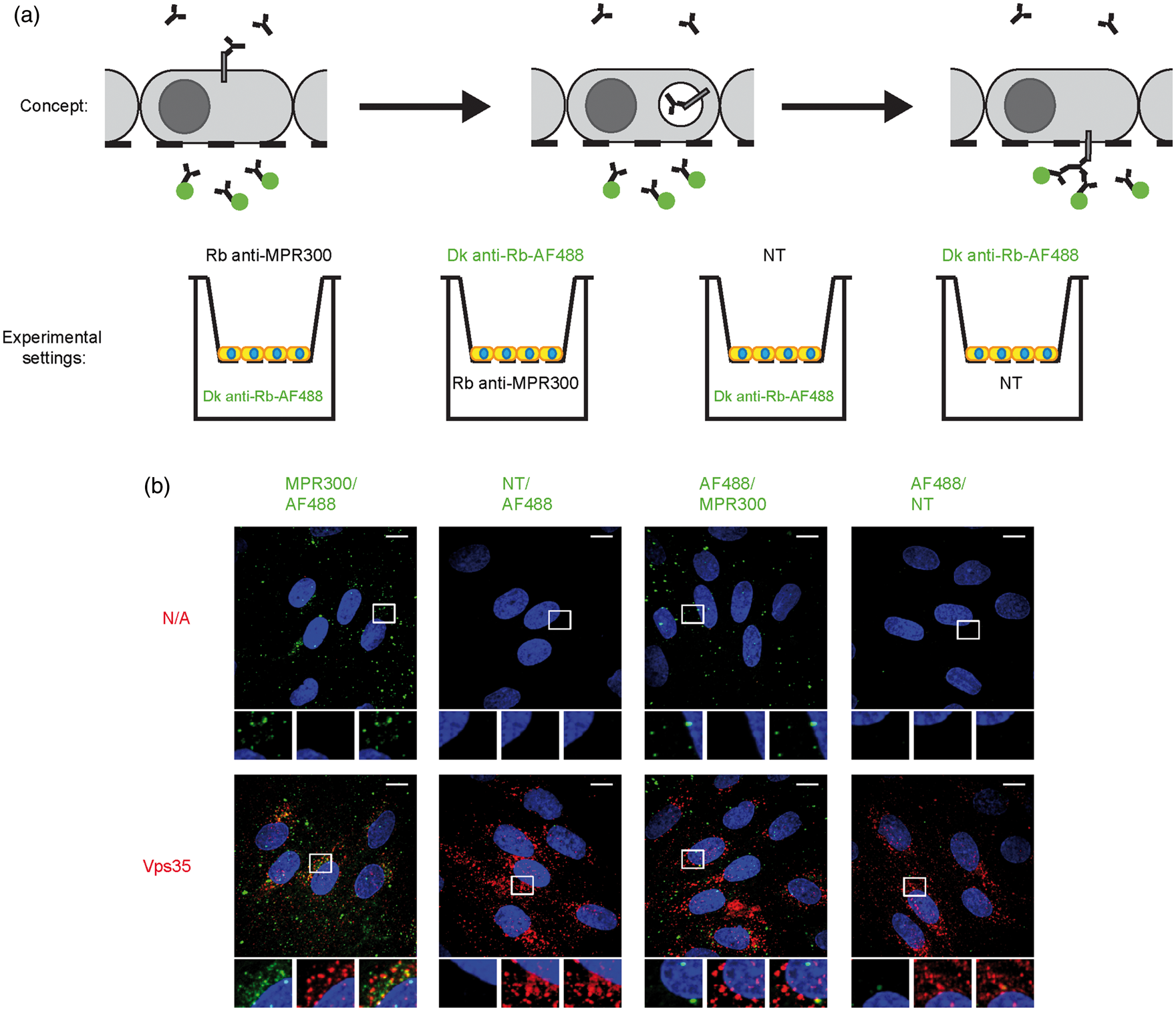
For MPR300, we observed a punctate signal from receptor trafficked across the cell (green) following both apical and basal application of primary antibody (Figure 5(b)) indicating that MPR300 is transported across cell from the apical membrane to the basal side (and the other way). At the same time, we observed more spots when primary antibody was applied to the apical side of the cell layer than to basal. Co-staining with MPR300 after cell permeabilisation (red) showed a few strong spots where both signals co-localised. This confirms that the detected signal was coming from MPR300-positive vesicles (Figure S2). We also observed a few weak spots for co-localisation with EEA1, again mostly present in the apical loading experiments (Figure S2). This agrees with our internalisation studies, where at later time points the amount of receptor co-localising with EEA1 also drops after the peak at 20 min. Finally, we observed co-localisation with Vps35, which is known to be involved in trafficking of the MPR300 (Figure 5(b)).
Discussion
RMT is currently being intensively studied as an approach to deliver pharmaceuticals to the central nervous system (CNS).6,7 The recycling TfR is the most studied receptor system for this purpose. It has been reported by several groups to deliver antibodies and particles to the brain.63–72 However, the yield is low, and targeting TfR for brain delivery may have health issues. 73 Studying other types of receptors will extend the number and variety of possible targets and could lead to better pharmaceutical delivery strategies. In the current study, we investigated the expression and transcytosis potential of cation-independent mannose-6-phosphate receptor in primary cultures of brain capillary endothelial cells from adult mammals. Besides being an endocytic receptor, MPR300 (in contrast to TfR) is a retrograde transported receptor that mainly shuttles between the endosomal compartments and the TGN.22,74,75 MPR300 delivers acidic hydrolases to lysosomes and is used as a target for treatment of lysosomal storage disorders throughout the body including the CNS.49–52,76,77 Zhou et al. 77 studied delivery of antibodies against insulin receptor conjugated with MPR300 ligand, idurate-2-sulfate. They observed uptake of the antibodies to a number of tissues, but not to the brain of adult mouse, and concluded that MPR300 is absent from the plasma membrane of BECs. Urayama et al. 49 reported that phosphorylated-glucuronidase (P-GUS), another ligand of MPR300, can enter the brain of neonatal rats. Brain uptake declined as a function of age, and at week 7, no uptake of P-GUS was observed. The authors concluded that MPR300 is down-regulated during development, an observation supported by other reports.78–82 We were able to show mRNA and protein expression of MPR300 in cultured BECs from adult pigs and cows (Figure 2). The presence of the receptors in the native endothelium was confirmed by immunostaining of freshly isolated capillaries from porcine and bovine brains (Figure 2). Matthes et al. 50 previously showed MPR300-mediated delivery of arylsulfatase A across monolayers of primary porcine endothelial cells grown in vitro. However, the observed transport appeared to be non-saturable, and involvement of MPR300-dependent pathways was not demonstrated unequivocally. In their study, a MC model was used, which in many studies show less pronounced apical–basal polarisation, and a less complete BBB phenotype compared to co-cultured models. Co-culture models may thus more closely resemble the in vivo state of BECs. Acidic hydrolases, the natural ligands for MPR300, are trafficked into late endosomes where they are released from the receptor. The acidic hydrolases are then further transported to the lysosomes, whereas MPR300 is directed in the retrograde direction of the TGN. This uncoupling can prevent efficient transcytosis of these enzymes across endothelial cell barriers. Conjugation of these acidic hydrolases to antibodies or to other pH-independent MPR300 tags would direct them to the retrograde pathway along with MPR300 and could be a better strategy for treatment of lysosomal storage disorders in the CNS.
Treatment of eight-week-old mice with epinephrine has been shown to increase brain delivery of the MPR300 ligand P-GUS. 76 The team concluded that epinephrine might modify endocytic properties of the cells and stimulate plasma membrane trafficking of intracellular pools of receptor. 76 Similar effects were observed for other cell types upon stimulation with hormones or phorbol esters.83–87 The authors further demonstrated that co-administration of P-GUS with increasing doses of mannose-6-phosphate caused an inhibition of P-GUS uptake, suggesting that the uptake was MPR-dependent.
Here, we investigated subcellular localisation and trafficking of MPR300, and our observations are summarised in Figure 6. We demonstrated co-localisation of the receptor with EEA1 suggesting that endocytosed receptor is trafficked to EE (Figures 3 and S1). Co-localisation with CHC and Cav1 suggests that both clathrin and caveolae pathways are involved in the MPR300 internalisation. Co-localisation with VTI1a (TGN marker) and Vps35 (retromer complex marker) was, as expected, also observed, since both TGN and retromer are known to be involved in trafficking of the receptor.22,26,27,33,41–46 In TGN, MPR300 normally enters a shuttle route between TGN and endosomes, and in the endosomes minor amounts of the receptor will eventually be incorporated into intraluminal vesicles and end up in lysosomes for degradation. Accordingly, we also observed weak co-localisation with Rab7 (late endosomal marker) and Lamp1 (lysosomal marker). We also examined internalisation of MPR300 by BECs, showing that the receptor is internalised by these cells from both apical and basal membranes (Figure 4), and probably follows the retrograde pathway to TGN. Co-staining with different markers and evaluation of co-localisation indicated that the kinetics and pathways involved in this process could differ between plasma membranes from which internalisation takes place. We could detect only low co-localisation with EEA1 or Cav1 at initial time points, suggesting that these compartments are not strongly involved in internalisation of the receptors. It also cannot be excluded that internalisation occurs very fast and receptor has travelled through these compartments by 10 min. To our surprise, we observed high co-localisation with Vps35, part of the retromer complex, already at 0 min. The retromer complex is the main mediator of retrograde trafficking in the endo-lysosomal system.20,32,44 It has also been reported that in the polarised epithelial cell line MDCK, the Vps35 (protein forming retromer core, together with Vps29 and Vps26) can mediate polarized trafficking of receptors between apical–basal membranes, not only retrograde trafficking.
88
Although there are no reports demonstrating that Vps35 is involved in internalisation, our data suggest that Vps35 is involved in trafficking events of MPR300 shortly after it has been internalised. This is in line with previous observations showing a role for Vps35 in retrieval of protein from EE,89–91 and it may explain the fast RMT that has also been reported in BEC.9,10 Although co-localisation between MPR300 and Vps35 at the plasma-membrane is controversial, Vps35 was shown to interact with Vps26b at the plasma membrane in mouse brain cells.92,93 Studies in epithelial cells have shown that Vps35 is involved in transcytosis and even though it is well accepted that the same cytosolic machinery is involved in similar trafficking events in different polarized cells (e.g. apical/basolateral in epithelia vs. axonal/somadendritic in neurons), we cannot conclude that the high PCC we observed between Vps35 and MPR300 in our internalisation experiment is a snapshot of transcytotic vesicles or whether it reflects vesicles undergoing retrograde transport to TGN. The exact order of events and compartments involved in internalisation requires further more detailed studies.
General overview of compartments and pathways involved in MPR300 trafficking in BEC. Schematic representation of hypothesized MPR300 trafficking routes in BEC, and compartments involved in this process. MPR300 is internalized from clathrin-coated pits (CHC) to EE/SE from where it can be retrograde transported to either late endosomes (LE) or trans-Golgi network (TGN). From LE, MPR300 can enter a shuttle loop between endosomes and TGN. Small fractions of MPR300 are constitutively incorporated into intraluminal vesicles in LE (shown as small vesicles inside LE) and then transported to lysosomes for degradation. A minor fraction of MPR300 is also likely to be internalized through caveolae, as some co-localisation with Cav1 is observed. The observed trafficking across the cells (TC) of MPR300 can theoretically occur via EE/SE, LE or caveolae. Markers used in the study to identify sub-cellular compartments and endocytosis pathways are shown in italics.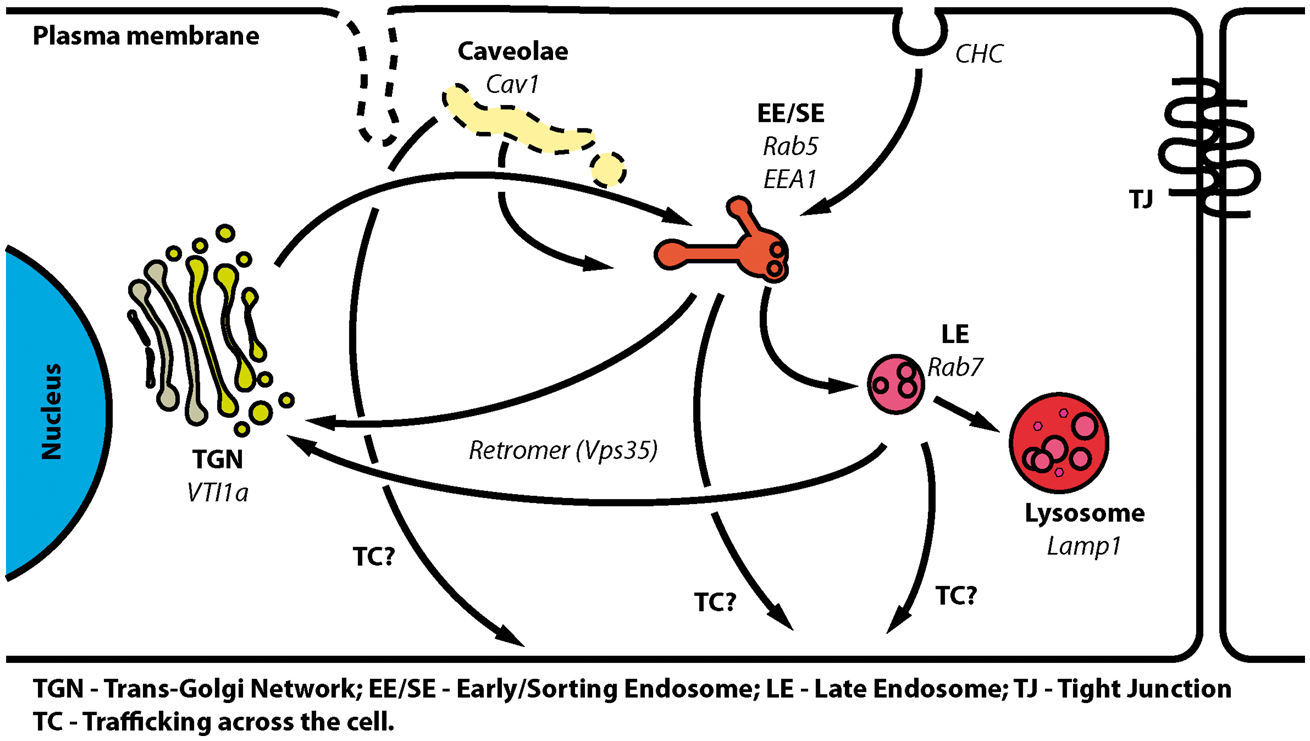
We observed co-localisation of MPR300 with Vps35 in our earlier antibody-based fluorescent assay designed to investigate polarized trafficking of receptors across cell 59 (Figure 5). At the same time, we could not detect co-localisation with EEA1, suggesting that shortly after clathrin-mediated endocytosis, receptors that eventually show polarized plasma membrane-to-membrane trafficking will be guided to special Vps35-positive vesicles and thereby escape the early-to-late endosome pathway. This would also explain the minimal co-localisation with the late endosome marker Rab7 in the bovine BEC. However, this study shows localisation of the accumulated, trafficked across cell, MPR300 after 45 min, while other reports showed RMT through BECs within 10–20 min.9,10 Further experiments are needed to elucidate the time-course of MPR300 trafficking across BECs.
In summary, we have shown that MPR300 is expressed in brain capillary endothelial cells of adult mammals, using primary porcine and bovine BBB co-culture models. The receptor was rapidly internalised, probably via clathrin-coated pits, and was trafficked through EEA1-positive and Vps35-positive vesicular compartments. Finally, using a novel antibody-based approach, we were able to show direct plasma membrane-to-membrane trafficking of MPR300 in BECs, which could be mediated by Vps35-positive vesicles. This shows that retrograde transported receptors should be considered as putative drug delivery routes across the BBB.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by The Lundbeck Foundation, grant number R155-2013-14113.
Acknowledgements
The authors would like to acknowledge Niels M. Kristiansen, Charlotte Goldeman Andersen and Natasia Pretzner for technical assistance.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors' contributions
PS, MNSH, BB and MSN were all involved in the experiments with bovine BEC. PS, KBJ, LBT, TLA, TM, NJA and MSN were involved in experiments concerning porcine BEC. PS, MNSH and KLH did qPCR experiments. MSN and PS conceived the idea for the project and wrote most of the manuscript.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.