
Abstract
The blood—brain barrier (BBB) is composed of uniquely differentiated brain microvascular endothelial cells (BMEC). Often, it is of interest to replicate these attributes in the form of an in vitro model, and such models are widely used in the research community. However, the BMEC used to create in vitro BBB models de-differentiate in culture and lose many specialized characteristics. These changes are poorly understood at a molecular level, and little is known regarding the consequences of removing BMEC from their local in vivo microenvironment. To address these issues, suppression subtractive hybridization (SSH) was used to identify 25 gene transcripts that were differentially expressed between in vivo and in vitro BMEC. Genes affected included those involved in angiogenesis, transport and neurogenesis, and real-time quantitative polymerase chain reaction (qPCR) verified transcripts were primarily and significantly downregulated. Since this quantitative gene panel represented those BMEC characteristics lost upon culture, we used it to assess how culture manipulation, specifically BMEC purification and barrier induction by hydrocortisone, influenced the quality of in vitro models. Puromycin purification of BMEC elicited minimal differences compared with untreated BMEC, as assessed by qPCR. In contrast, qPCR-based gene panel analysis after induction with hydrocortisone indicated a modest shift of 10 of the 23 genes toward a more ‘in vivo-like’ gene expression profile, which correlated with improved barrier phenotype. Genomic analysis of BMEC de-differentiation in culture has thus yielded a functionally diverse set of genes useful for comparing the in vitro and in vivo BBB.
Introduction
The microvessels within the brain are made up of specialized endothelial cells (EC) that display distinct attributes as compared with the EC found throughout the majority of the body. A single layer of tightly adjoined brain microvascular EC (BMEC) forms the lumen of capillaries and effectively blocks the free diffusion of solutes between the blood and the brain. The impermeable nature of brain microvessels has resulted in them being called the ‘blood—brain barrier (BBB)’. Although the BBB possesses barrier properties, it is also endowed with selective transport systems, which provide the brain with necessary nutrients. In addition, BMEC interact intimately with other brain cells of the neurovascular unit, and hence can act as mediators between blood and brain. As a result of these functional attributes, the BBB plays an important role in a number of neurological diseases and hampers drug delivery efforts by preventing access to the brain.
The study of these and other aspects of the BBB can be facilitated by in vitro models. Such models often consist of a monolayer of BMEC grown on a porous membrane, effectively dividing a culture chamber into two compartments representing the blood and the brain. However, data generated from in vitro systems have to be carefully interpreted since cultured BMEC tend to lose many of their in vivo characteristics, and it is unclear to what extent the de-differentiation is occurring. A good deal of this de-differentiation has been attributed to the removal of BMEC from their microenvironment within the brain. In addition to the barrier-forming BMEC, astrocytes, neurons, and pericytes are intimately associated with the microvessels and are responsible for inducing various aspects of the BBB phenotype. As a result, astrocytes and their conditioned medium have been shown to increase γ-glutamyl transpeptidase activity, tight junction complexity, and barrier properties in cultured BMEC (Rubin et al, 1991). Neurons co-cultured with BMEC increased γ-glutamyl transpeptidase activity and decreased the flux of dopamine through a BMEC monolayer (Tontsch and Bauer, 1991). Few data exist that describe the exact influence of pericytes on BMEC, but they can improve barrier properties. In addition, soluble factors have been shown to improve in vitro BBB model properties. Cyclic adenosine monophosphate (Rubin et al, 1991) and glucocorticoids, such as hydrocortisone (HC) (Calabria et al, 2006; Hoheisel et al, 1998) and dexamethasone (Grabb and Gilbert, 1995), have been successful in partially reinstating barrier properties for in vitro BMEC. Another important component of the in vivo microenvironment is blood flow and associated shear stress. Recent genomic studies employing a dynamic in vitro BBB have showed that flow regulates glucose metabolism and the oxidative environment of the BMEC (Desai et al, 2002; Marroni et al, 2003). Unfortunately, the BBB properties of in vitro models continue to fall short of their in vivo counterpart despite the aforementioned enhancements.
The loss of in vivo BBB characteristics is often assessed by monitoring properties such as transendothelial electrical resistance, small molecule permeability, enzyme activity, and transporter expression. Transendothelial electrical resistance is a useful technique to gauge the barrier properties of in vitro BBB models. The transendothelial electrical resistance in cultured BMEC monocultures is often 100 Ω cm2 or lower (Calabria et al, 2006; Rubin et al, 1991), well below the value of nearly 1,500 Ω cm2 observed in the brain (Butt et al, 1990). Transcellular permeability to small molecule tracers, such as fluorescein and sucrose, can also yield valuable information regarding barrier integrity. Although barrier properties are an often-used measure to assess the characteristics of in vitro BBB models, several morphological and biochemical criteria have also been employed. The quality of the tight junctions has been evaluated by monitoring the expression levels and localization of various junctional proteins, such as zonula occluden, occludin, and claudin-5 (Calabria et al, 2006; Forster et al, 2005). Enzymatic activities such as those of γ-glutamyl transpeptidase and alkaline phosphatase are also sometimes evaluated. Finally, the expression levels of BBB-resident transporters such as glucose transporter 1 (GLUT-1) (Boado et al, 1994) and the efflux transporter p-glycoprotein (P-gp; MDR1A) have been investigated (Gaillard et al, 2000). Taken together, the effects of culture manipulation and refinement on in vitro BBB model quality are often evaluated using a few discrete phenotypic and molecular characteristics, but from these measurements alone it is difficult to determine the magnitude of departure from the in vivo situation.
Thus, we have chosen to use a global profiling technique to measure simultaneously multiple BBB attributes in a single experiment to answer questions regarding the extent of BMEC de-differentiation in culture. Although many studies have used genomics and other global profiling techniques to investigate how the introduction of inductive factors, inductive cells, and flow can affect BMEC characteristics in side-by-side culture comparisons (Calabria and Shusta, 2006; Marroni et al, 2003), little has been done to link the findings to the in vivo transcriptome and determine how well these changes actually reconstitute the in vivo BBB. To address this shortcoming, we used suppression subtractive hybridization (SSH) to, for the first time, directly compare various in vitro situations directly to the in vivo BBB. We identified a host of BMEC genes, whose expression levels were dramatically changed when comparing the in vivo and in vitro BBB, many of which had not previously been shown to be altered upon in vitro culture. The resulting gene panel, representing the differences between in vivo and in vitro gene expression, was then utilized to evaluate effects of puromycin purification on BMEC. The expression profiles indicate that puromycin treatment is a useful method to obtain purified BMEC in culture without dramatically altering gene expression. Finally, HC induction of BMEC was investigated using the gene panel, and this treatment was successful in partially restoring barrier properties and the in vivo gene expression profile.
Materials and methods
Isolation of Rat Brain Microvessels for mRNA Recovery
Brains were removed from male rats (220 to 250 g; Harlan, Indianapolis, IN, USA) and stored in Dulbecco's modified Eagle's medium (DMEM; Sigma, St Louis, MO, USA) on ice. The remainder of the isolation took place in a cold room at 4°C. The meninges were removed, and the cortices were dissected away from the surrounding tissue. The cortices were homogenized using a Dounce tissue grinder and homogenized material was filtered through a 210 μm nylon mesh to remove larger vessels. The flow-through was centrifuged in a 20% (w/v) dextran solution at 5,000g for 15 mins at 4°C in a swinging bucket rotor. The fatty top layer was discarded and the pellet was resuspended in DMEM. This solution, primarily containing microvessels and red blood cells, was filtered through a 44 μm nylon mesh. The microvessels retained on the nylon mesh were washed thoroughly with DMEM and were free of adjoining material when examined by microscopy. These microvessel preparations were then used for poly(A +) RNA isolation as described below.
Isolation and Culture of Rat Brain Microvascular Endothelial Cells
An enzymatic isolation strategy was performed, as described previously to isolate BMEC for in vitro culture (Calabria et al, 2006). Briefly, brain cortices were obtained from male rats (220 to 250 g; Harlan) and digested in a mixture of 0.7 mg/mL type 2 collagenase and 39 U/mL DNase I (Worthington Biochemical Corp., Lakewood, NJ, USA) in DMEM for 1.25 h at 37°C. A microvessel-enriched pellet was obtained by re-suspending the digested material in a 20% (w/v) bovine serum albumin (Sigma) and DMEM solution and centrifuged at for 8 min at 1,000g and 4°C in a swinging bucket rotor. This pellet was further digested in a solution of 1 mg/mL collagenase/dispase (Roche Applied Science, Indianapolis, IN, USA) and 39 U/mL DNase I in DMEM for 1 h at 37°C. Purified microvessels were then isolated using a continuous 33% Percoll (Amersham Biosciences, Piscataway, NJ, USA) gradient, and the cells were plated onto collagen IV/fibronectin-coated tissue culture plates or 0.4 μm polyester filters (Corning, Acton, MA, USA). Culture medium contained DMEM with 20% (v/v) bovine platelet-poor plasma-derived serum (Biomedical Technologies Inc., Stoughton, MA, USA), 1 ng/mL basic fibroblast growth factor (Roche Molecular Biochemicals, Indianapolis IN, USA), 1 μg/mL heparin (Sigma), 2 mmol/L
For HC induction experiments, BMEC were cultured on Transwell filters as described above for 3.5 DIV before switching to a serum-free medium containing a 1:1 ratio by volume of DMEM and Ham's F12 nutrient mixture, 2 mmol/L
Isolation of Poly(A +) RNA
Poly(A +) RNA was isolated from freshly isolated rat brain microvessels or from cultured rat BMEC using the MicroPoly(A)Purist™ mRNA purification kit (Ambion, Austin, TX, USA). The yield of poly(A +) RNA from microvessels was 150 ng/rat brain and from cultured BMEC, yield was 90 ng/cm2. The integrity of the mRNA samples used for the SSH procedure was confirmed by actin Northern blotting.
Immunocytochemistry and Quantitative Analysis of Culture Purities
Primary antibodies were incubated with BMEC at 20°C for 1 h. Antibodies used were rabbit anti-human von Willebrand factor (14 μg/mL; Dako, Carpinteria, CA, USA), rabbit anti-cow glial fibrillary acidic protein (GFAP) (11 μg/mL; Dako-Cytomation, Carpinteria, CA, USA), mouse anti-α actin (2 μg/mL; American Research Products, Belmont, MA, USA), and mouse anti-rat prolyl-4-hydroxylase-β (10 μg/mL; Acris Antibodies GmbH, Hiddenhausen, Germany). Secondary antibodies (Texas Red goat anti-rabbit IgG antibody, 2 μg/mL; Alexa Fluor goat anti-mouse IgG antibody, 2 μg/mL; Invitrogen Corp.) were incubated with primary antibody-labeled samples for 1 h at 20°C. The nuclear stain 4′,6-diamidino-2-phenylindole was added to the wells for 5 mins at 20°C at a concentration of 300 nmol/L (Invitrogen Corp.).
Photographs were acquired using an Olympus fluorescence microscope coupled with a camera (Diagnostic Instruments Inc., Sterling Heights, MI, USA) controlled by MetaVue software version 5.0r1 (Downingtown, PA, USA). Counting was carried out after overlaying von Willebrand factor, anti-αactin, and 4′,6-diamidino-2-phenylindole nuclear stain images from the same field in Adobe Photoshop Elements version 2.0. The percent culture purity was calculated by using the number of α-actin-positive cells, unstained cells, and the total number of cells (4′,6-diamidino-2-phenylindole stain) in each image. Unstained cells were designated as non-endothelial due to their lack of von Willebrand factor staining. Thus, % EC = (total cells–α-actin-positive cells—unstained cells)/total cells × 100%. At minimum, 2,000 cells were counted for each labeling condition. Owing to the high purity of puromycin-treated cultures, entire wells were evaluated to determine culture purity.
Suppression Subtractive Hybridization
Suppression subtractive hybridization was performed as described previously (Li et al, 2001; Shusta et al, 2002) using the polymerase chain reaction (PCR)-select cDNA subtraction kit according to the manufacturer's instructions (Clontech, Palo Alto, CA, USA). First, mRNA samples were treated by the TURBO DNA-free™ kit (Ambion) to remove genomic DNA. The brain microvessel and cultured BMEC mRNA levels were then normalized by spectrophotometry. The freshly isolated brain microvessel poly(A +) RNA was then used to produce 2.0 μg of tester cDNA, and the subtraction procedure was completed using driver cDNA from cultured BMEC mRNA (to determine gene transcripts downregulated in culture). The subtraction was also performed in the reverse direction with the cultured BMEC mRNA used to create 2.0 μg of tester cDNA, and the freshly isolated rat brain microvessel mRNA was used to produce driver cDNA (to determine gene transcripts upregulated in culture). The SSH-PCR products were cloned into the pCRII vector (Invitrogen Corp.), and the resultant library of cloned cDNA fragments was expanded in Escherichia coli XL1-Blue supercompetent cells (Stratagene, La Jolla, CA, USA). Differential Southern blot hybridization was employed in a 96-well format to identify positive clones as described previously (Li et al, 2001; Shusta et al, 2002). Briefly, bacterial clones were blotted using subtracted and unsubtracted probes to enable a comparison of relative hybridization intensity (data not shown). Clones that hybridized to tester probes, and not to driver probes, were defined as differentially expressed and selected for sequencing.
Isolated clones were sequenced using the standard M13 forward primer at the University of Wisconsin's Comprehensive Cancer Research Center (Madison, WI, USA). The BLAST program (NCBI and NIH) was utilized to determine gene identity using the nr database. The accession numbers and gene names for the corresponding matches to rat sequences are given in Table 3. For some genes, rather than report accession number matches to known, validated mRNA from other species, we instead reported accession numbers (XM_ prefix) for the homologous rat genes that, to date, were annotated as ‘predicted’.
Quantitative PCR primers

GAPDH, glyceraldehyde-3-phosphate dehydrogenase.
Quantitative analysis of cellular distribution in untreated BMEC cultures

The results are the mean ± s.d. of three culture wells from a 24-well culture plate.
Panel of genes differentially expressed upon BMEC culture
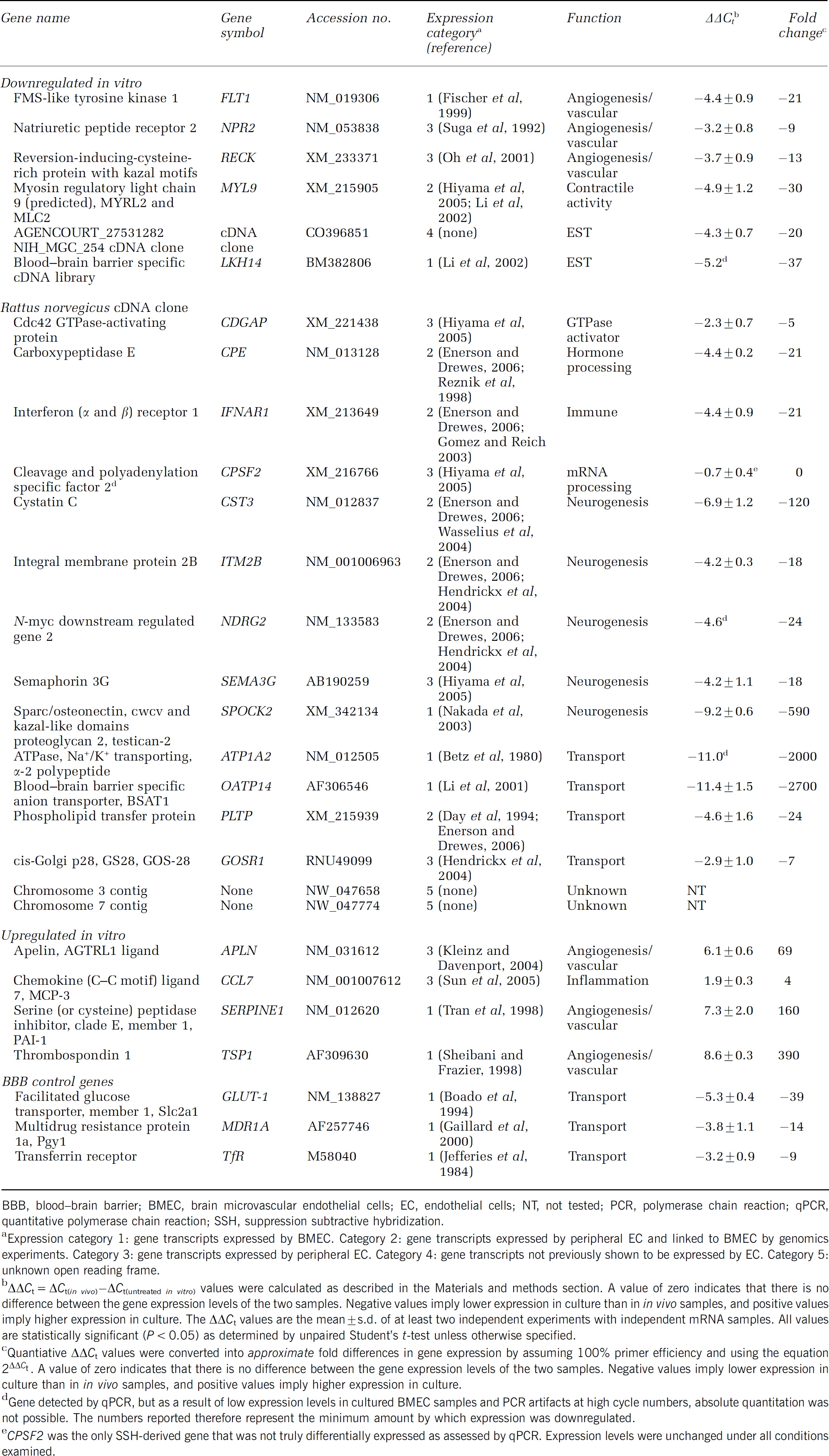
BBB, blood—brain barrier; BMEC, brain microvascular endothelial cells; EC, endothelial cells; NT, not tested; PCR, polymerase chain reaction; qPCR, quantitative polymerase chain reaction; SSH, suppression subtractive hybridization.
Expression category 1: gene transcripts expressed by BMEC. Category 2: gene transcripts expressed by peripheral EC and linked to BMEC by genomics experiments. Category 3: gene transcripts expressed by peripheral EC. Category 4: gene transcripts not previously shown to be expressed by EC. Category 5: unknown open reading frame.
ΔΔCt = ΔCt(in vivo)-ΔCt(urltreated in vitro) values were calculated as described in the Materials and methods section. A value of zero indicates that there is no difference between the gene expression levels of the two samples. Negative values imply lower expression in culture than in in vivo samples, and positive values imply higher expression in culture. The ΔΔCt values are the mean±s.d. of at least two independent experiments with independent mRNA samples. All values are statistically significant (P < 0.05) as determined by unpaired Student's t-test unless otherwise specified.
Quantiative ΔΔCt values were converted into approximate fold differences in gene expression by assuming 100% primer efficiency and using the equation 2ΔΔCt. A value of zero indicates that there is no difference between the gene expression levels of the two samples. Negative values imply lower expression in culture than in in vivo samples, and positive values imply higher expression in culture.
Gene detected by qPCR, but as a result of low expression levels in cultured BMEC samples and PCR artifacts at high cycle numbers, absolute quantitation was not possible. The numbers reported therefore represent the minimum amount by which expression was downregulated.
CPSF2 was the only SSH-derived gene that was not truly differentially expressed as assessed by qPCR. Expression levels were unchanged under all conditions examined.
Quantitative Real-Time Polymerase Chain Reaction
Quantitative real-time polymerase chain reaction (qPCR) was performed using a Bio-Rad iCycler thermal cycler and an iScript one-step qPCR kit with SYBR Green (Bio-Rad Laboratories, Hercules, CA, USA) according to the manufacturer's instructions. Each reaction was carried out with 1 ng of mRNA obtained from freshly isolated brain microvessels or from BMEC cultures. Messenger RNA samples were treated by the TURBO DNA-free™ kit to remove genomic DNA (Ambion). Gene-specific primers are shown in Table 1 and were designed using PerlPrimer software version 1.1.13 (http://perlprimer.sourceforge.net/). Three control genes included in the analysis encode GLUT-1, P-gp (MDR1A), and the transferrin receptor (TfR).
Gene expression was normalized solely to the reference gene β-actin, after an initial study where gene expression levels were normalized to β-actin or two other commonly used reference genes, glyceraldehyde-3-phosphate dehydrogenase and 60S acidic ribosomal protein P0 (RPLP0), yielded nearly identical values for all biological samples examined. Delta-delta cycle threshold values (ΔΔCt) were calculated according to the method developed by PE Applied Biosystems (Perkin Elmer, Waltham, MA, USA). The ΔΔCt value is a difference in gene expression levels between two samples, as measured by PCR cycles. The cycle threshold (Ct) was defined as the PCR cycle where fluorescence reached a defined threshold value. The normalized ΔCt was calculated by subtracting the gene cycle threshold value from the β-actin cycle threshold value (ΔCt = Ctβ-actin–Ctgene). Comparative gene expression between two independent samples, or ΔΔCt, was determined by subtracting the control delta cycle threshold from the sample delta cycle threshold (ΔΔCt = ΔCtsample–ΔCtcontrol). Throughout the paper, the ΔΔCt values are reported and serve as an accurate measure of differential gene expression. Where indicated, these ΔΔCt values were converted into approximate fold differences in gene expression by assuming 100% primer efficiency and using the equation 2ΔΔCt. Duplicate or triplicate data points were used to determine all ΔΔCt values and their associated error. In addition, all results were replicated in at least two separate experiments with independently derived in vivo, in vitro, puromycin-treated, or HC-treated mRNA samples.
Results
Preparation of In Vivo and In Vitro Samples for Genomic Comparison
The goal of this study was to identify differentially expressed gene transcripts resulting from the removal of BMEC from the brain microenvironment. As shown in Figure 1, this was accomplished by comparing in vivo and in vitro mRNA pools using SSH. The genomic technique yielded a gene panel representing the identity of differentially expressed genes between in vivo and in vitro BMEC, and qPCR was used to determine the corresponding comparative gene expression level profile.
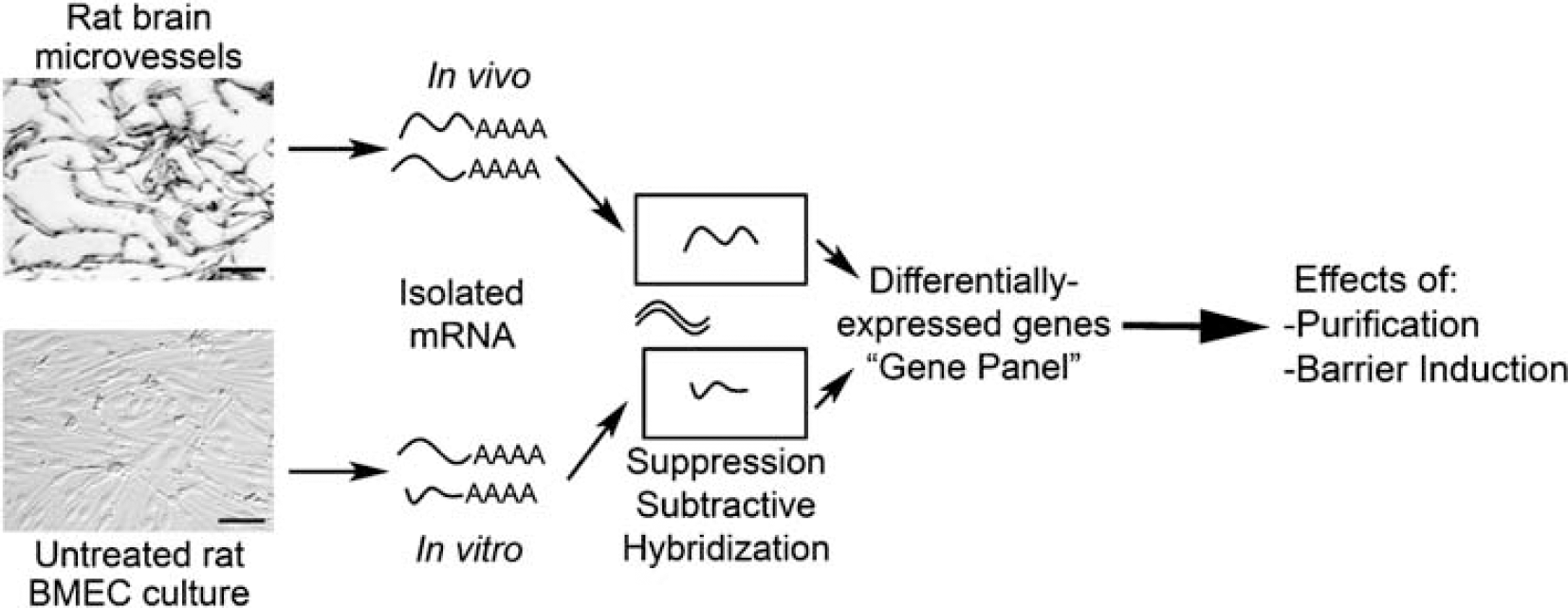
Flow chart detailing the steps performed to generate the SSH-derived gene panel. Messenger RNA was isolated from rat brain microvessels to generate a sample representing BMEC gene expression within the in vivo BBB. Messenger RNA was also isolated from cultured BMEC to generate a sample representing BMEC gene expression in the in vitro BBB. The genomic technique SSH was used to remove commonly expressed mRNA transcripts between these two samples, and to identify those transcripts that were differentially expressed. These differentially expressed genes comprised a gene panel representing the molecular level differences between in vivo and in vitro BMEC. The gene panel was further employed to assess the effects of two separate culture conditions. One condition involved the purification of BMEC cultures using puromycin. The other condition examined the effects of purified BMEC barrier induction with HC. Scale bars = 50 μm.
The first step in this procedure was to generate the starting materials for the genomic comparison (Figure 1). Rat brain microvessels were isolated using a mechanical homogenization procedure that enabled the sample to be maintained at 4°C at all times to prevent significant changes in the transcriptome during the isolation procedure. Care was taken to ensure that this procedure yielded a very pure microvessel preparation free of adjoining material and single cells (Figure 2A). Despite the high level of vessel purity, the brain microvessel preparation did contain pericytes in addition to BMEC, as a result of the practical consequence that both cell types are enclosed within a common basement membrane. A few smooth muscle cells were also inevitably present on precapillary arterioles. However, the majority of the contaminating fraction was pericytes, since their cellular abundance in vivo is approximately one pericyte for every 3 to 5 EC (Pardridge, 2001). The in vivo cellular population was thus approximately 75 to 83% BMEC. The in vivo BBB mRNA sample isolated from these microvessels therefore contained gene transcripts from multiple cellular origins, and was an important consideration in the design of our genomics experiment as described below.
For the comparative in vitro sample, primary BMEC were cultured for 4.0 DIV to achieve confluency. These cultures were probed for a variety of cellular markers to determine the identity and quantity of contaminating cell types, and the seeding density was tuned to achieve fractional pericyte abundance that approximated the in vivo distribution. This step was critical to ensure that the genomics analysis did not yield predominately pericyte transcripts, as would be the case if pure in vitro BMEC preparations were used. The results shown in Figure 2B and Table 2 indicate that BMEC comprised the target 69 to 79% of the cell population. The primary contaminating cell type in the cultures was α-actin-positive pericytes, with very few additional contaminating cell types including α-actin-positive smooth muscle cells (Table 2). Poly(A +) RNA was subsequently isolated from these cultures and represented the in vitro BBB sample. The quality of the in vivo and in vitro mRNA samples was analyzed by Northern blotting with an actin probe (Figure 2C), and integrity of the mRNA was confirmed.
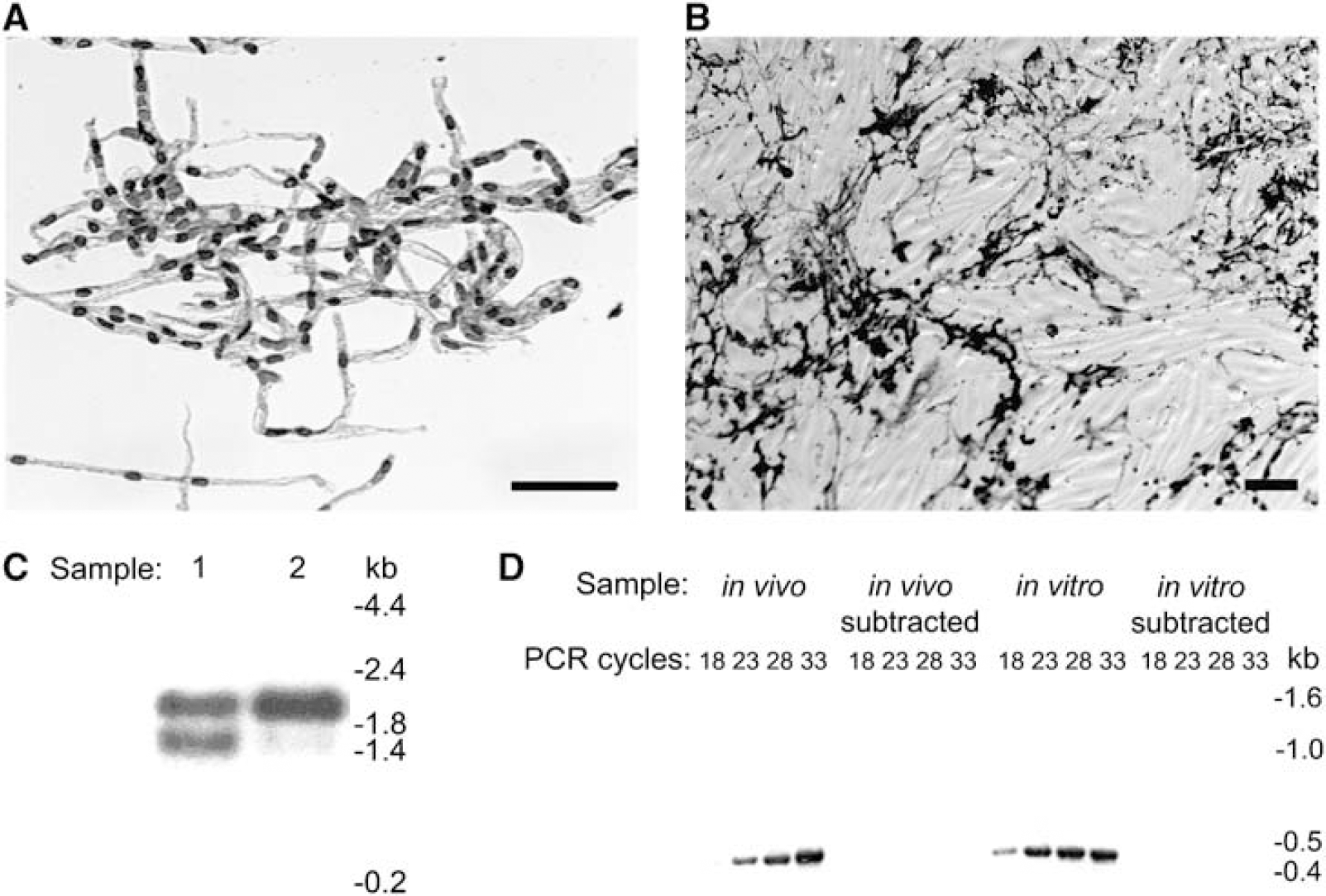
Sample preparation and validation of the suppression subtraction hybridization (SSH) procedure. (
Differential Transcript Profiling of In Vivo and In Vitro BMEC Using Suppression Subtractive Hybridization
In vivo and in vitro BBB mRNA samples were then compared using SSH. The subtraction was performed in both directions (in vivo minus in vitro and in vitro minus in vivo), and thus the procedure resulted in two transcript pools, one containing genes downregulated in vitro and the other containing genes upregulated in vitro. The success of the subtraction process was confirmed by verifying the absence of the housekeeping gene glyceraldehyde 3-phosphate dehydrogenase in the subtracted pools (Figure 2D). Subtracted gene products were then subjected to a Southern blot-based secondary screen to confirm their differentially expressed nature (see Materials and methods for details). In total, nonexhaustive secondary screens identified 128 clones as differentially expressed. Sequencing yielded 25 unique BMEC gene transcripts, with 21 being down regulated and four being upregulated upon in vitro culture (Table 3). Of the 21 genes downregulated in the BMEC cultures, 17 matched known gene sequences, two matched rat expressed sequence tags, and two matched rat genomic sequence not currently annotated to contain open reading frames. All four of the upregulated gene products matched known gene sequences. These differentially regulated genes mediate a variety of cellular functions, and many have not been previously identified as having altered expression levels as a result of BMEC culture.
Validating the Endothelial Origin of Differentially Expressed Transcripts
To validate the endothelial relevance of the SSH gene panel, we confirmed endothelial expression in two ways: by literature and database mining, and by qPCR with highly purified BMEC. First, known genes and rat expressed sequence tags identified by SSH could be organized into five expression-related categories after leveraging information available both from the literature and associated datasets archived on the Gene Expression Omnibus database (GEO; NCBI). Seven genes have been verified to be expressed by BMEC in specific detailed investigations (ATP1A2, FLT1, LKH14, OATP14, TSP1, SERPINE1, and SPOCK2) (Expression Category 1), seven have been shown to be expressed by peripheral EC and linked to the BBB by a genomic investigation(s) (CPE, CST3, IFNAR1, ITM2B, MYL9, NDRG2, and PLTP) (Category 2), and eight have been shown to be expressed by peripheral EC (APLN, CDGAP, CPSF2, MCP-3, NPR2, P28, RECK, and SEMA3G) (Category 3). One expressed sequence tag, a novel cDNA clone, has not previously been shown to be expressed by EC or the BBB (Category 4). The final two identified gene transcripts matched rat chromosomal sequences but did not correspond to known open reading frames (Category 5). As a further verification of BMEC transcript origin, highly purified (∼100% purity by puromycin treatment) BMEC cultures were shown to express all of the genes in expression categories 1 to 4 by qPCR (Figure 3, data not shown). Thus, the pericyte normalization procedure appeared successful in generating a gene panel that was of high endothelial relevance.
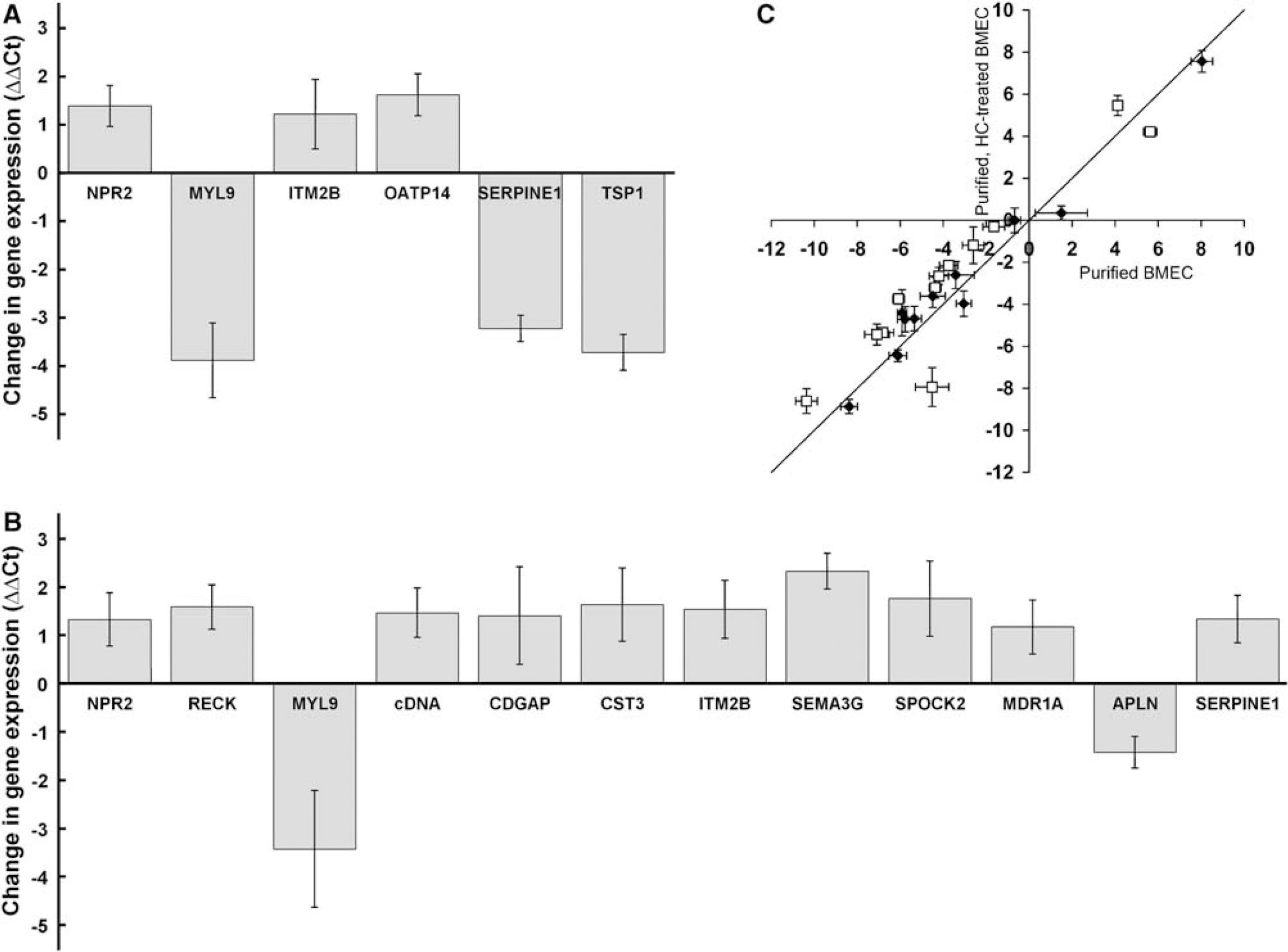
Analysis of BMEC culture conditions using the gene panel and qPCR. (
Quantitative Assessment of the Level of Differential Expression
The SSH procedure resulted in the identification of a panel of genes that were differentially regulated in BMEC when they were removed from the in vivo microenvironment and cultured. Since the SSH procedure is qualitative in nature, qPCR was employed to obtain quantitative data regarding the differences in gene regulation. Primer sets (Table 1) for 23 of the 25 gene transcripts identified by SSH (unknown open reading frame sequences were excluded) were utilized to examine relative gene expression levels. Besides the gene transcripts identified by SSH, three additional genes were added to the gene panel as controls: GLUT-1, MDR1A, and the transferrin receptor (TfR). These genes were chosen based on their frequent use during in vivo and in vitro BBB characterization and, because they have been previously reported to have downregulated expression in BMEC cultures (Boado et al, 1994; Gaillard et al, 2000). The ΔΔCt values from the in vivo and in vitro BMEC qPCR data are reported in Table 3, and the ΔΔCt values represent the quantitative differences in gene expression between the two samples in terms of a number of PCR cycles (see Materials and methods for details). The data indicated that 18 of the 23 gene products and the three control genes were significantly downregulated upon culture. Values of ΔΔCt for the in vivo versus in vitro comparison shown in Table 3 ranged from −11.4 to +8.6, and as a calibration, these ΔΔCt values correspond to approximate changes in fold expression levels ranging from 2,700-fold downregulated to 390-fold upregulated upon BMEC culture. Only one of the SSH-identified genes (CPSF2) proved not to be reproducibly differentially expressed between in vivo and in vitro BMEC (Table 3). The SSH gene panel therefore represents genes whose expression levels change dramatically as a result of removing BMEC from their microenvironment within the brain. In subsequent studies, the effects of different culture environments on BMEC gene expression were compared quantitatively and related back to the in vivo expression profile.
Effects of BMEC Purification on the In Vitro Transcriptome
High-purity BMEC cultures can be obtained using the protein translation inhibitor puromycin (Calabria et al, 2006; Perriere et al, 2005). Puromycin is a substrate of the efflux transporter p-glycoprotein (MDR1), which is more highly expressed in BMEC than in other contaminating cell types. The inclusion of puromycin in the culture medium therefore allows for the removal of contaminating cell types and the routine attainment of BMEC cultures with purities of 99.8% (Calabria et al, 2006). Thus, we wished to evaluate the effects of this purification method on the expression patterns within the gene panel by comparing the profiles generated by untreated versus puromycin-treated BMEC mRNA (Figure 3A). Brain microvascular endothelial cells were treated with puromycin, attained confluence after being maintained in culture for a total of 3.5 DIV, and were 99.9 ± 0.03% pure. In parallel, low purity, untreated BMEC cultures lacking puromycin (77 ± 4% BMEC), were maintained under otherwise identical conditions for 3.5 DIV. Poly(A ±) RNA isolated from these cultures was subjected to qPCR with primers for 23 of the SSH-derived genes and the three control genes (Table 1). The ΔΔCt values resulting from puromycin treatment of BMEC indicated that six of the 23 genes analyzed had statistically significant changes in gene expression, and none of the three control genes, including MDR1, was altered. Of the six genes with significant differences in expression, three were downregulated (MYL9, SERPINE1, and TSP1), as a direct result of the removal of pericytes and/or smooth muscle cells from the purified cultures. Three BMEC genes (ITM2B, NPR2, and OATP14) were upregulated upon purification, and it is important to note that these data represent true increases in BMEC gene expression and are not simply the result of the proportion of BMEC-derived mRNA being increased in the puromycin-treated case (from 77% BMEC in untreated case to ∼100% BMEC in puromycin-treated case; Figure 3A). Although subtle differences exist, the data indicate that the genomic fingerprints for untreated and puromycin-treated BMEC are quite similar.
Effects of Hydrocortisone on the In Vitro Genomic Fingerprint
Puromycin-purified BMEC cultures have previously been shown to respond optimally to HC barrier induction (Calabria et al, 2006). While changes in the localization of several tight junction proteins have been noted (Calabria et al, 2006), little is known about the molecular level events that accompany HC induction. To gain a better understanding of how HC induces barrier properties in BMEC and to see if this treatment resulted in a more in vivo-like genotype, the gene panel expression levels were analyzed after a 24 h induction of puromycin-purified BMEC with HC. As observed previously (Calabria et al, 2006), this treatment resulted in a significantly higher transendothelial electrical resistance (139 ± 1 Ω cm2) than control samples that were also puromycin-purified but lacking HC (38 ± 2 Ω cm2). As shown in Figure 3B, 12 of the 23 genes had statistically significant differences in gene expression upon HC induction. The differences in the ΔΔCt values ranged from 1.2 to 2.3 PCR cycles (∼2.3- to 5.1-fold) for the 10 upregulated genes (CDGAP, cDNA clone, CST3, ITM2B, MDR1A, NPR2, RECK, SERPINE1, SEMA3G, and SPOCK2) and 1.4 to 3.4 cycles (∼2.70- to 10.7-fold) for the downregulated APLN and MYL9 genes. Importantly, the changes in gene expression had the effect of moving 10 genes closer to in vivo gene expression levels in correlation with the improved barrier phenotype (Figure 3C). Only MYL9 and SERPINE1 were differentially regulated further from their in vivo values. Although the gene expression profile was significantly improved, the HC treatment did not restore the in vivo expression profile, and in most cases, the HC-treated BMEC expression levels were still well removed from the in vivo situation (Figure 3C, distance from the x axis).
Discussion
By using genomics approaches, this study offers a semiglobal view of changes in gene expression that occur upon removal of BMEC from the brain microenvironment and subsequent culture in vitro. Quantitative assessment of the changes in expression level indicated that the majority of the affected genes were substantially downregulated in vitro. Moreover, affected genes included those involved in functions of high BBB importance such as transport, angiogenesis, and neurogenesis. The resultant gene panel was then used to assess the effects of two culture treatments. The first treatment of BMEC using puromycin indicated that the effects of purification were minimal and could be mostly ascribed to removal of pericytes. Further treatment of purified BMEC with HC illustrated that along with a change in barrier phenotype, a fairly widespread change in genotype toward the in vivo situation was also observed. The roles of the specific genes in relation to BBB function will be discussed in more detail below.
Since microvessels represent only 0.1% of the total brain volume (Pardridge, 2001), the separation of brain microvessels (BMEC) away from total brain material was required to perform accurate comparisons of in vivo and in vitro BMEC gene expression (Figure 1). In addition, the in vivo BMEC had to be isolated in such a manner that their gene expression profile was maintained. Thus, a mechanical homogenization procedure was used to obtain rat brain microvessels in an ‘in vivo-like’ state. Although other BMEC isolation methods exist, this procedure was chosen because it maintained samples at 4°C to minimize endogenous RNase activity, required no specialized equipment, and provided relatively high yields of high-quality brain microvessel mRNA with no requirement for a potentially biasing mRNA amplification step (150 ng of mRNA per rat brain). The in vitro sample used for comparisons were purposefully manipulated to contain contaminating cells, in particular pericytes. We had previously showed that modulating the initial cell seeding density and the total time in culture can be used to control the percentage of contaminating cells in culture (Calabria et al, 2006). These cultures were therefore ‘tuned’ from 69 to 79% BMEC, which was similar to the value found in the in vivo BMEC sample (75 to 83% BMEC). Normalization of the contaminating cell percentage in the in vivo and in vitro BMEC samples minimized the isolation of genes from these other cell types during the SSH procedure. As a result, the BMEC relevance of the SSH gene panel was quite high and was confirmed in several ways. First, most of the isolated genes had been previously shown to be expressed in BMEC or peripheral endothelia. Second, BMEC in highly pure cultures were shown to express all of the isolated genes. Finally, many of the genes were responsive to BMEC treatment by HC, further confirming their endothelial origin. Thus, although some of the genes might be expressed by multiple cell types at the BBB, the gene panel proved useful for evaluating BMEC function under different culture conditions.
Twenty-five genes were identified by SSH as being differentially expressed in BMEC when these cells were removed from the brain microenvironment and cultured. Twenty-one of these clones were downregulated in vitro and four were upregulated. Quantitative PCR confirmed the differential expression of all the known genes and expressed sequence tags identified in this study except for CPSF2, whose gene expression values were not reproducibly different in culture as compared with in vivo (Table 3). The qPCR results indicated that the majority of the reported gene transcripts were not only differentially regulated, but experienced significant deviations (up to 2,700-fold) from the in vivo condition. In addition, many of the genes have not previously been identified as being differentially expressed in BMEC cultures. The largest functional set of genes identified was related to angiogenesis and vascular functions (Table 3). One example, TSP1, an inhibitor of angiogenesis, is expressed by a variety of cells including BMEC (Sheibani and Frazier, 1998), and was found to be upregulated in BMEC culture. Another is RECK, which is expressed by EC, and can inhibit their migration and angiogenic properties (Oh et al, 2001), and is downregulated in cultured BMEC. A third gene, FMS-like tyrosine kinase 1, also known as vascular endothelial growth factor receptor 1, plays an important role in promoting angiogenesis and was also downregulated in vitro. Notably, two other genes were substantially downregulated and encode the transporters Na+/K+ -ATPase, α-2 (ATP1A2) and BBB specific anion transporter (OATP14) (Betz et al, 1980; Li et al, 2001). In addition, the three control transporters, TfR, MDR1, and GLUT-1, were significantly downregulated as a result of culture. Phospholipid transfer protein (PLTP) was also downregulated and has been reported to be more highly expressed at the BBB compared with liver and kidney endothelium (Li et al, 2002), where it probably functions in transferring phospholipids and antioxidants from high- to low-density lipoproteins and endothelium (Desrumaux et al, 1999). Finally, the Golgi-associated snare (GOSR1) gene functions in the regulation of vesicle trafficking (Hay et al, 1997), and a downregulation of GOSR1 suggests there may be a general decrease in vesicular transport in cultured BMEC. Collectively, these findings are of particular importance since molecular transport is critical to BBB function.
Another subset of SSH-identified gene transcripts was of particular interest because they encode proteins that have been shown to have regulatory roles in the developing and adult central nervous system. Cystatin C is an ubiquitous, secreted cysteine protease inhibitor originally identified as an abundant protein of the cerebral spinal fluid with the choroid plexus as a major site of production. This gene was downregulated substantially upon BMEC culture, and in a previous study was identified to be one of the 25 most highly expressed transcripts at the BBB (Enerson and Drewes, 2007). Cystatin C has been reported to stimulate neural stem cell proliferation and neurogenesis in vivo (Taupin et al, 2000), in addition to promoting astrogenesis and suppressing oligodendrogenesis in vitro (Hasegawa et al, 2007), and has been linked to pathological processes such as cerebral amyloid angiopathy, cerebral hemorrhage, and Alzheimer's disease. The integral membrane protein 2b (ITM2B) was also downregulated in vitro. Integral membrane protein 2b is expressed in multiple tissue types including brain, and has also been shown to be expressed in infant brain (Vidal et al, 1999) indicating a possible role in nervous system development. Peptides derived from a genetic mutant of ITM2B are found in perivascular amyloid deposits associated with familial British dementia (Vidal et al, 1999). Another protein potentially involved in neural cell differentiation, the N-myc downstream-regulated gene 2 (Ndrg2) (Okuda and Kondoh, 1999), was downregulated to the point of the qPCR detection limit in vitro. The NDRG2 gene is part of a four-member gene family whose members are involved in cell differentiation, and NDRG2 has been shown to be expressed in the embryonic ventricular zone as well as in neurogenic regions in adult brain (Okuda and Kondoh, 1999). Also downregulated was sparc/osteonectin, cwcv and kazal-like domains proteoglycan 2 (SPOCK2), an extracellular protein also known as testican-2, which has functions that include regulation of protease activity (Nakada et al, 2003) and neurite outgrowth (Schnepp et al, 2005). Finally, semaphorin 3G (SEMA3G) was also identified as being downregulated in BMEC cultures and while little is known about this semaphorin isoform, it has been shown to have involvement in axonal guidance through neuropilin-2 (Taniguchi et al, 2005). All genes discussed above, except NDRG2, were shown to be expressed by a microarray study using human umbilical vein EC (Hiyama et al, 2005; GEO database profile number GDS1760(ACCN)). Except for SEMA3G and SPOCK2, all were also linked to the BBB by a comprehensive serial analysis of gene expression study of the rat BBB (Enerson and Drewes, 2006). Finally, combined with the confirmed expression of these genes by purified BMEC, these data suggest that BMEC may play an important role in the regulation of brain development and maturation, a theory that has been supported by various studies that indicate EC can regulate neurogenesis (Shen et al, 2004). Importantly, the expression level loss of these genes indicates that the brain phenotype of in vitro BMEC is significantly altered.
The puromycin BMEC purification methodology has been shown to yield in vitro BMEC cultures that are nearly pure, and importantly, these cultures respond optimally to inducing factors like glucocorticoids and cyclic adenosine monophosphate leading to increased electrical resistance and resistance to passive diffusion (Calabria et al, 2006; Perriere et al, 2005). However, to validate this approach on a more comprehensive scale, we employed the panel of differentially expressed genes to evaluate whether positive or deleterious changes in gene expression resulted upon puromycin treatment. Seventeen of the 23 SSH-derived genes and the three control genes examined did not experience a significant change in gene expression as a result of the puromycin treatment. As described previously, although the BMEC expression of the MDR1 (p-glycoprotein) gene product is instrumental in the puromycin purification method, transcript levels for MDR1 were not changed by puromycin treatment (Perriere et al, 2005). The downregulation of three genes by purification (MYL9, SERPINE1, and TSP1) can be explained, to a large extent, by the removal of contaminating cell types from the cultures. Of the six gene transcripts that experienced significant changes due to puromycin treatment, two have been shown to be expressed by pericytes and smooth muscle cells, in addition to BMEC. TSP1 has been shown to be expressed by retinal pericytes (Canfield et al, 1996), and myosin regulatory light chain 9 (MYL9 or MLC2) is a protein required for cell contractile activity in both smooth muscle and nonmuscle cells including endothelium (Kumar et al, 1989). Thus, assuming contaminating cell expression of TSP1 and MYL9 was higher than that found for BMEC, one explanation for the observed downregulation is simply the removal of these cells from the culture. MYL9 has also been shown to be upregulated in brain endothelium by pulsatile flow (Desai et al, 2002), which in part could also explain the dramatic downregulation of MYL9 upon static in vitro culture compared with the in vivo situation (Table 3). Another gene whose expression was significantly downregulated in puromycin-treated BMEC is serine (or cysteine) proteinase inhibitor (SERPINE1), also known as plasminogen activator inhibitor type 1 (PAI-1). The SERPINE1 protein and gene expression levels have been shown to be induced by pericytic factor(s) in both brain and non-brain EC (McIlroy et al, 2006), possibly accounting for the downregulation of SERPINE1 observed when pericytes were removed using puromycin. Finally, three gene transcripts, OATP14, ITM2B, and NPR2, had expression levels that increased in puromycin-treated cultures, and it appears that the contaminating cells were negative regulators of these BMEC gene products. Taken together, puromycin treatment of BMEC cultures looks to be a useful method to obtain highly pure BMEC cultures without significant change to the BMEC transcriptome.
The glucocorticoid, HC, has been shown to restore partially in vivo properties such as barrier tightness (Hoheisel et al, 1998), with optimal results seen with puromycin-purified BMEC (Calabria et al, 2006). These barrier improvements were accompanied by morphological changes and alterations in junctional structure (Calabria et al, 2006). In addition, previous studies have noted increased tight junction protein expression in immortalized BMEC as the result of glucocorticoid induction (Forster et al, 2005). To investigate more thoroughly the effects of HC induction and its ability to promote cultured BMEC to mimic their in vivo character, puromycin-purified BMEC were treated with HC and gene expression was compared with both untreated BMEC and in vivo BMEC. The treatment resulted in a 3.6-fold increase in transendothelial electrical resistance as compared with control samples, and Figure 3C demonstrates an overall shift of gene expression toward a more in vivo-like expression profile, as 10 of the 12 genes having altered expression levels moved toward the in vivo situation. Three functional categories of genes identified by SSH were regulated in what appeared to be a concerted fashion in HC-treated BMEC. First, HC treatment induced neurogenesis-related gene transcripts: CST3, ITM2B, SEMA3G, and SPOCK2. The capability of HC to induce expression of these genes further supports our finding that they are of BMEC origin and suggests an intriguing role for BMEC in regulation of the neurovascular unit. A second set of genes that were differentially regulated and have related functions are involved in angiogenesis. Corticosteriods are known antiangiogenic agents and SERPINE1 levels were increased by HC treatment. Physiological concentrations of SERPINE1 are proangiogenic while higher levels are antiangiogenic (Devy et al, 2002). RECK was also upregulated after HC treatment, and has been shown to function in an antiangiogenic capacity by inhibiting several membrane-type matrix metalloproteinases (Oh et al, 2001). Also, apelin (APLN), a protein that has vasodilatation/vasoconstriction roles and has been shown to promote gastric cell proliferation in vitro (Wang et al, 2004), was downregulated by HC. Taken together, SERPINE1, RECK, and APLN all responded in a manner that suggested HC treatment could regulate angiogenesis and/or proliferation of BMEC, possibly via regulation of the extracellular matrix, an effect that could be responsible for the BMEC tightening seen after HC treatment (Calabria et al, 2006; Hoheisel et al, 1998). Interestingly, we have shown that HC treatment does not affect the proliferation rate in these confluent cultures, as assessed by 5-bromo-2-deoxyuridine incorporation (Calabria et al, 2006). In contrast, other culture manipulations, such as the introduction of pulsatile flow, can cause a growth arrest of brain EC through downregulation of the cyclins (Desai et al, 2002). Thus, HC appears to be functioning in a fashion distinct from that promoted by shear stress, and these data indicate that the quantitative gene panel is composed of transcriptome elements capable of responding to various stimuli. A third category of genes, which was notable for its general lack of response to HC treatment, encodes proteins that function as molecular transporters or in vesicular trafficking processes. OATP14, GLUT-1, transferrin receptor, PLTP, and GOSR1 all remained unchanged by HC treatment, whereas MDR1A expression was increased. Finally, while HC produced a substantial shift toward the in vivo BMEC transcriptome, none of the differentially expressed genes examined in this study had expression levels that were completely restored to their in vivo levels by HC treatment. Thus, it would probably take re-introduction of other components of the in vivo microenvironment. As mentioned earlier, other in vitro models having specialized conditions, such as coculture with perivascular brain cells or the presence of flow are in use, and our assessment of in vitro BBB models was by no means exhaustive. However, much like we employed the in vivo versus in vitro quantitative gene panel to assess the effects of puromycin and HC on BMEC, this new tool could be used in an analogous fashion to evaluate other in vitro models by allowing direct comparison to the quantitative in vivo transcriptome. This process would also likely increase our understanding of the specific molecular level events that regulate BBB properties.