
Abstract
Large conductance, Ca2+-activated K+ (BK) channels control cerebrovascular tone; however, the regulatory processes influencing these channels remain poorly understood. Here, we investigate the cellular mechanisms underlying the enhancement of BK current in rat cerebral arteries by nitric oxide (NO) signaling. In isolated cerebral myocytes, BK current magnitude was reversibly increased by sodium nitroprusside (SNP, 100 μM) and sensitive to the BK channel inhibitor, penitrem-A (100 nM). Fostriecin (30 nM), a protein phosphatase type 2A (PP2A) inhibitor, significantly prolonged the SNP-induced augmentation of BK current and a similar effect was produced by sildenafil (30 nM), a phosphodiesterase 5 (PDE5) inhibitor. Using proximity ligation assay (PLA)-based co-immunostaining, BK channels were observed to co-localize with PP2A, PDE5, and cGMP-dependent protein kinase (cGKI) (spatial restriction < 40 nm); cGKI co-localization increased following SNP exposure. SNP (10 μM) reversibly inhibited myogenic tone in cannulated cerebral arteries, which was augmented by either fostriecin or sildenafil and inhibited by penitrem-A. Collectively, these data suggest that (1) cGKI, PDE5, and PP2A are compartmentalized with cerebrovascular BK channels and determine the extent of BK current augmentation by NO/cGMP signaling, and (2) the dynamic regulation of BK activity by co-localized signaling enzymes modulates NO-evoked dilation of cerebral resistance arteries.
Introduction
The cerebral circulation is responsible for supplying blood and nutrients to all regions of the brain in a manner that meets the metabolic needs of active neurons and glia. The intraluminal diameter of cerebral resistance arteries is a major determinant of the spatial and temporal pattern of blood flow within the brain and is regulated by signaling pathways that are intrinsic (e.g. myogenic mechanisms) and extrinsic (e.g. hormone/endothelium-mediated events) to the cerebrovascular smooth muscle. Large conductance, Ca2+-activated K+ (BK) channels are prominently expressed in cerebral arterial smooth muscle and play a prominent role in the regulation of intraluminal diameter in cerebral resistance arteries.1–4 Functionally, these channels can be activated by either membrane depolarization,5,6 driven by intraluminal pressure, or elevations in local free [Ca2+], due to intracellular Ca2+ sparks that originate from Ca2+ release channels/ryanodine receptors (RyRs) present in the SR membrane of vascular myocytes. 7 Pharmacologic inhibition of Ca2+ sparks decreases BK channel activity2,8 and leads to constriction of myogenically active cerebral arteries, indicating that BK channels and RyRs exist in activity-coupled, nanodomains that promote Ca2+-evoked BK channel opening. BK channels thus act as negative feedback regulators of pressure-induced arterial constriction in the cerebral vasculature.1,9
Nitric oxide (NO) is an important vasodilator in the cerebral circulation that is produced by the vascular endothelium and perivascular nerves innervating resistance arteries,10,11 and has been further implicated in the pathophysiology associated with cortical spreading depression. 12 BK channel activity is known to be regulated by NO signaling and, in the vascular wall, endothelium-derived NO stimulates smooth muscle guanylyl cyclase to elevate cGMP levels, leading to activation of the type I cGMP-dependent protein kinase (cGKI) and direct phosphorylation of the BK pore-forming α subunit (for reviews, see Kyle and Braun 13 and Shipston and Tian 14 ). Such modifications increase BK channel open probability, thereby enhancing BK channel-mediated membrane hyperpolarization and smooth muscle relaxation.
The observed co-localization of BK channels with RyRs in cerebral smooth muscle suggests that BK channels may also form local complexes with signaling molecules that regulate their activity. However, in cerebral resistance arteries, relatively little is known regarding the potential co-localization of BK channels with phosphorylation-related regulatory elements, such as components of the NO/cGMP cascade. To investigate this scenario, the aim of our study was to identify key proteins associated with the regulation of cerebral BK channels by the NO/cGMP/cGKI signaling pathway. Our novel findings reveal that BK channels in cerebral myocytes co-associate with enzymes that regulate cGMP levels (i.e. PDE5) and BK channel phosphorylation and dephosphorylation (i.e. cGKI and PP2A) and that PDE5 and PP2A activities modulate NO-mediated vasodilation of cerebral resistance arteries in a BK channel-dependent manner.
Methods and materials
Arterial pressure myography
The experimental procedures used in this study were approved by the University of Calgary Animal Care Committee, and conform to the guidelines for the care and use of laboratory animals established by the Canadian Council on Animal Care and the National Institutes of Health, USA. While our study cannot be considered in vivo research, we followed the ARRIVE guidelines as best as we could in reporting this work. Animals were housed at room temperature with free access to food and water under a controlled photoperiod (12 h light; 12 h dark) in the rat half-way house facility within the Cumming School of Medicine at the University of Calgary. Protocols for the isolation and cannulation of cerebral resistance arteries have been recently described in detail in Mishra et al. 3 Briefly, male Sprague Dawley rats (12–15 weeks of age) were injected intraperitoneally with sodium pentobarbital (50–60 mg/kg) to induce stage 3 anesthesia, followed by rapid decapitation. Following removal of the brain, middle cerebral arteries were isolated and cannulated on glass micropipettes and the vessel lumen was filled with Krebs' buffer (115 NaCl, 5 mM KCl, 25 mM NaHCO3, 1.2 mM MgCl2, 2.5 mM CaCl2, 1.2 mM KH2PO4 and 10 mM D-glucose) containing 1% bovine serum albumin, pH 7.4. One cannula end was closed and the other was connected to a hydrostatic pressure column. The cannulated vessel was superfused with Krebs' buffer at 7 ml/min using a peristaltic pump at 37 ℃ and the solution was gassed with 95% air/5% CO2. Intraluminal diameter was continuously tracked using a video camera-based system (IonOptix, Milton, MA). Drugs were added to the bath via the perfusion pump.
Drug-induced changes in intraluminal diameter are presented as a percentage of the developed myogenic tone at 70 mmHg and were calculated as follows
3
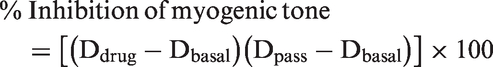
Dbasal = intraluminal diameter at basal myogenic tone, and
Dpass = maximal intraluminal diameter at 70 mmHg in the absence of external free Ca2+.
Passive arterial diameter was determined in the presence of a physiological saline solution containing zero added Ca2+ and 2 mM EGTA. To stimulate vessels with fixed or “clamped” concentrations of NO (see Supplementary Figures 4 and 5), we followed recently published protocols.15,16 In brief, vessels were first exposed to the NO scavenger carboxy-PTIO and the eNOS inhibitor L-N-nitro-arginine (L-NNA) to eliminate any existing NO and de novo NO synthesis, respectively. In the continued presence of these reagents, the spontaneous NO donor spermine NONOate was added to the bath solution at concentrations of 5 μM and 75 μM to generate free NO levels of 3 nM and 20 nM, as described by Villalba et al. 16
Enzymatic dissociation of rat cerebral arteries
Dissected artery segments (i.e. anterior, middle and posterior cerebral arteries) were transferred to a glass test tube containing a low [Ca2+] PSS (i.e. 140 mM NaCl, 5.5 mM KCl, 0.1 mM CaCl2, 1 mM MgCl2, 1.2 mM NaH2PO4, 2 mM Na pyruvate, 10 mM HEPES and 1 mg/ml BSA, pH 7.3), and incubated for 10 min at room temperature. The solution was decanted and replaced with the same low [Ca2+] PSS supplemented with papain (26 U/ml) and DTT (1 mg/ml) included, and incubated for 30 min at 37 ℃. This enzyme mixture was gently agitated every ∼7–8 min. Following this treatment, the solution was carefully decanted from the arterial tissue, and replaced with low [Ca2+] PSS containing collagenase (1.9 U/ml), soybean trypsin inhibitor (1 mg/ml) and elastase (10 U/ml), and incubated for 7–10 min at 37 ℃ until individual myocytes were liberated. This enzyme solution was carefully decanted and tissue was gently rinsed with cold low [Ca2+] PSS. Gentle trituration with a fire-polished glass pipette was applied to mechanically liberate cells from the tissue. Myocytes were seeded then onto glass cover slips treated with poly-L-lysine (0.1 mg/ml) for either voltage clamp experiments or immuno-staining.
Single cell voltage clamp
Whole-cell voltage clamp measurements were performed using conventional, ruptured-membrane patch clamp methodology, as described. 17 Whole cell currents were sampled at 5 kHz and filtered at 1–2 kHz. Borosilicate glass micropipettes (2–4 MΩ tip resistance) were filled with a solution containing 100 mM KOH, 30 mM KCl, 1 mM MgCl2, 2 mM NaATP, 1.5 mM NaGTP, 10 mM HEPES, pH 7.3 with methanesulfonic acid. Myocytes in the bath chamber were constantly superfused at ∼2 ml/min with a modified Ringer's saline solution containing 135 mM NaCl, 5 mM KCl, 1 mM MgCl2, 2.5 mM CaCl2, 5 mM glucose, and 10 mM HEPES, pH 7.3 with 1N NaOH. Solution changes were performed by gravity flow from elevated solution reservoirs using manually controlled solenoid valves. All recordings were performed at 35–37 ℃.
Immunocytochemistry and proximity ligation assay
For both immunostaining and proximity ligation assay (PLA) experiments, myocytes were dispersed and seeded onto glass cover slips as described above. Cells were fixed in PBS containing 4% (w/v) paraformaldehyde for 20 min at room temperature and washed five times with PBS. Cells were then permeabilized in PBS containing 0.3% Triton X-100 for 5 min and washed a further five times in PBS. Cells were then exposed to a blocking solution (3% v/v donkey serum in PBS) and incubated with individual primary antibodies overnight at 4 ℃. For immunocytochemical staining, the primary antibody solution was decanted; cells were rinsed with PBS and then incubated with PBS containing an appropriate, fluorescently tagged secondary antibody.
For PLA immunostaining,18,19 the PBS solution containing the first primary antibody was carefully decanted and replaced with PBS containing another primary antibody for 8 h. This solution was also then decanted and cells were rinsed with PBS. These cells were then treated with anti-mouse and anti-rabbit secondary antibodies coupled to complementary oligonucleotides from a Duolink PLA kit (Olink) and subsequent steps carried out according to the manufacturer's instructions.
Staining controls (see Supplementary Figure 2) were generated by omitting a particular primary antibody. All cover slips were mounted onto glass slides using 1 drop of mounting medium (ProLong Gold, Antifade; Life Technologies) containing DAPI nuclear stain and sealed with clear nail polish. Stained cells were viewed using an Olympus Fluoview FV10i confocal microscope equipped with a 60× oil immersion objective (NA = 1.35). For counting PLA (red) dots, a series of Z-stack images were recorded with approximately 500 nm slice intervals to obtain comprehensive cross-sections throughout each cell in x, y, and z dimensions.
Western Blot analysis
Rat aorta, cerebral arteries (i.e. middle, anterior and posterior), and lung tissue were dissected free and immediately frozen on dry ice, followed by storage at −80oC. Once thawed, a segment of aorta (50–100 mg wet weight) and piece of lung tissue (∼200 mg) was finely minced and then suspended (∼75 mg/ml) in a buffer containing (final): 25 mM Tris HCl, pH 6.8, 2.5% (w/v) sodium dodecyl sulphate, 1 mM benzamidine, 1 mM PMSF and 5 μg/ml each of leupeptin, pepstatin A and aprotinin. Aortic and lung tissues were homogenized for ∼10 s on ice using a small-probe Polytron. Middle cerebral arteries (6–7 vessels) were thawed, placed in a small Eppendorf tube and suspended in the same buffer using 15 μl per vessel. Tissue samples were then rotated overnight at 4 ℃, followed by centrifugation at 3000 r/min for 5 min to remove insoluble material. The supernatant fractions were collected and mixed with Laemmli sample buffer (4 × concentrate containing 100 mM DTT and ∼550 mM β-mercaptoethanol). Samples were then heated to 70 ℃ for 20 min prior to SDS-PAGE. A crude synaptosomal membrane fraction (P2) was isolated from whole mouse brain, as reported 20 and prepared for SDS-PAGE, as described above.
Proteins were separated by standard SDS-PAGE using either 8% or 10% polyacrylamide mini-gels, then electro-transferred to nitrocellulose membrane (0.2 μm pore size) using a Bio-Rad semi-dry device (Trans-Blot Turbo) and Towbin transfer buffer containing 20% (v/v) methanol + 0.01% (w/v) SDS. Membranes were blocked in 5% (w/v) skim milk powder dissolved in Tris-buffered saline + 0.05% Tween 20 (TBS-T) for ∼2 h at room temperature and then incubated overnight at 4 ℃ with primary antibody suspended in TBS-T + 1% skim milk powder. The membranes were washed (3–4 × 5 min) in TBS-T at room temperature and then incubated ∼2 h at room temperature with horseradish peroxidase-coupled secondary antibody suspended in TBS-T + 1% skim milk, followed by three to four washes. Bound antibodies on the membranes were detected by incubation with Supersignal West Dura chemiluminescence reagent, followed by detection using a FujiFilm LAS3000 instrument.
Reagents
Sildenafil citrate, fostriecin, penitrem-A, sodium nitroprusside (SNP), spermine NONOate, DMSO (dimethyl sulfoxide), pinacidil, ODQ (1H-[1,2,4] oxadiazolo [4,3-a]quinoxalin-1-one) and all required chemicals, as well as the PLA (Duolink®) Red Starter Kit (Mouse/Rabbit; cat #: DUO92101-1KT), were purchased from Sigma-Aldrich (Oakville, ON, Canada). Carboxy-PTIO (2-(4-Carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide, K+ salt) was obtained from Enzo Life Sciences. Euthanyl (sodium pentobarbital, 250 mg/mL) was purchased from Bimeda-MTC Animal Health Inc., Cambridge, ON, Canada. SKA-31 (naphtha[1, 2-d]thiazole-2-ylamine) was synthesized as previously described. 21 SKA-31, pinacidil, sildenafil citrate, ODQ, and penitrem-A were prepared as 10 mM stock solutions in DMSO and then diluted directly into the external bath solution. The final concentration of DMSO reaching the tissue was typically 0.05% (vol/vol) or less. In pilot experiments, bath exposure to DMSO at a final concentration of 0.2% (v/v) had no effect on the development of myogenic tone or drug-evoked vasodilation in cannulated cerebral arteries (data not shown).
The following primary antibodies were used: anti-BK α subunit (1:500 dilution, mouse monoclonal, BD Transduction Laboratories, San Jose, CA, USA; cat. # 611249), anti-BK α subunit (1:500 dilution, rabbit polyclonal, Millipore, Billerica, MA, USA; cat # AB5228), anti-cGKI (1:500 dilution, rabbit polyclonal, Enzo Life Sciences Inc., Farmingdale, NY, USA; cat. # ADI-KAP-PK005-D), anti-PP2A catalytic subunit (1:500 dilution, mouse monoclonal, BD Transduction Laboratories, San Jose, CA, USA; cat. # 610555), anti-PDE5A (1:250 dilution, rabbit polyclonal, FabGennix International Inc. Frisco; TX, USA; cat # PD5A-101AP) and anti-b-actin (1:5000 dilution, Sigma-Aldrich, St. Louis, MO, cat # A1978). Secondary antibody for standard immunostaining was Alexa488 (1:1000 dilution, donkey anti-rabbit IgG polyclonal, Jackson ImmunoResearch Laboratories, USA; cat # 711-545-152).
Statistical analysis
Data are presented as mean ± SEM. Statistically significant differences between different experimental conditions were evaluated using either a one-tailed Student's t-test or ANOVA, followed by Tukey's post hoc test. A calculated P value < 0.05 was taken to signify statistical significance.
Results
Inhibitors of PP2A and PDE5 augment the stimulation of BK current by NO signaling in cerebral arterial myocytes
The temporal regulation of macroscopic BK channel currents by the NO donor, SNP, was recorded from acutely isolated, adult cerebral myocytes and displayed using diary plots, as illustrated in Figure 1(a). Under baseline conditions, depolarizing voltage steps to +100 mV from the holding potential of −20 mV evoked robust outward currents (Figure 1(bi)), and brief exposure of cerebral myocytes to SNP (100 μM) significantly elevated the peak outward current magnitude. This stimulatory effect completely reversed ∼100 s following SNP wash-out (Figure 1(bi) and (ii)). Note that numerically labeled current tracings correspond to the time points denoted in the associated diary plot. Enhancement of BK current by SNP was reproducible following wash-out and subsequent re-exposure to SNP (Supplementary Figure 1(a)), indicating that cells did not readily desensitize to the stimulus. Importantly, the vast majority (∼90%) of outward current evoked by depolarizing steps was blocked by the selective BK channel inhibitor penitrem-A (100 nM; Supplementary Figure 1(b)).
22
Moreover, SNP was unable to enhance outward current in the presence of penitrem-A (Supplementary Figure 1(b)), demonstrating that the SNP-sensitive current component was mediated primarily by BK channel activity.
Augmentation of rat cerebral myocyte BK current is enhanced by fostriecin or sildenafil pre-treatment. (a) Schematic diagram illustrates the anticipated time course of SNP-evoked changes in BK current magnitude in absence and presence of fostriecin or sildenafil treatments. Following initial current measurements (i.e. basal), the voltage clamped cell is exposed to the NO donor SNP in either the absence or presence of a protein phosphatase (PP) or phosphodiesterase (PDE) inhibitor as indicated. The time course of the onset and reversal of enhanced BK current magnitude is plotted and the area under the curve (AUC) is calculated for each current magnitude vs. time plot. AUC thus reflects integrated BK channel activity under a given experimental condition. (bi) Representative tracings illustrate a typical response for SNP-evoked BK current enhancement measured ∼80 s following stimulus application. Current magnitude returned to basal levels ∼100 s after SNP was removed. (bii) Summary data expressed over time (i.e. diary plot) for SNP-evoked enhancement of BK current. Circled numbers on the plot indicate the experimental time points at which the representative traces shown in panel Bi were selected. (ci) and (di) Representative tracings illustrate the effect of either fostriecin or sildenafil pre-treatment, respectively, on SNP-evoked BK current enhancement. Note that BK current magnitude remained elevated ∼100 s following SNP washout. (cii) and (dii) Summary diary plots quantify the effect of fostriecin and sildenafil pre-treatment, respectively, on SNP-evoked enhancement of BK current magnitude. (e) Area under the curve analysis demonstrates that fostriecin or sildenafil significantly potentiated the enhancement of BK current magnitude by SNP. Average data are expressed as means ± SEM. * denotes a statistically significant difference vs. SNP alone (p < 0.05), as determined by one-way ANOVA and a Tukey's post hoc test. No significant difference was detected for SNP + fostriecin vs. SNP + sildenafil.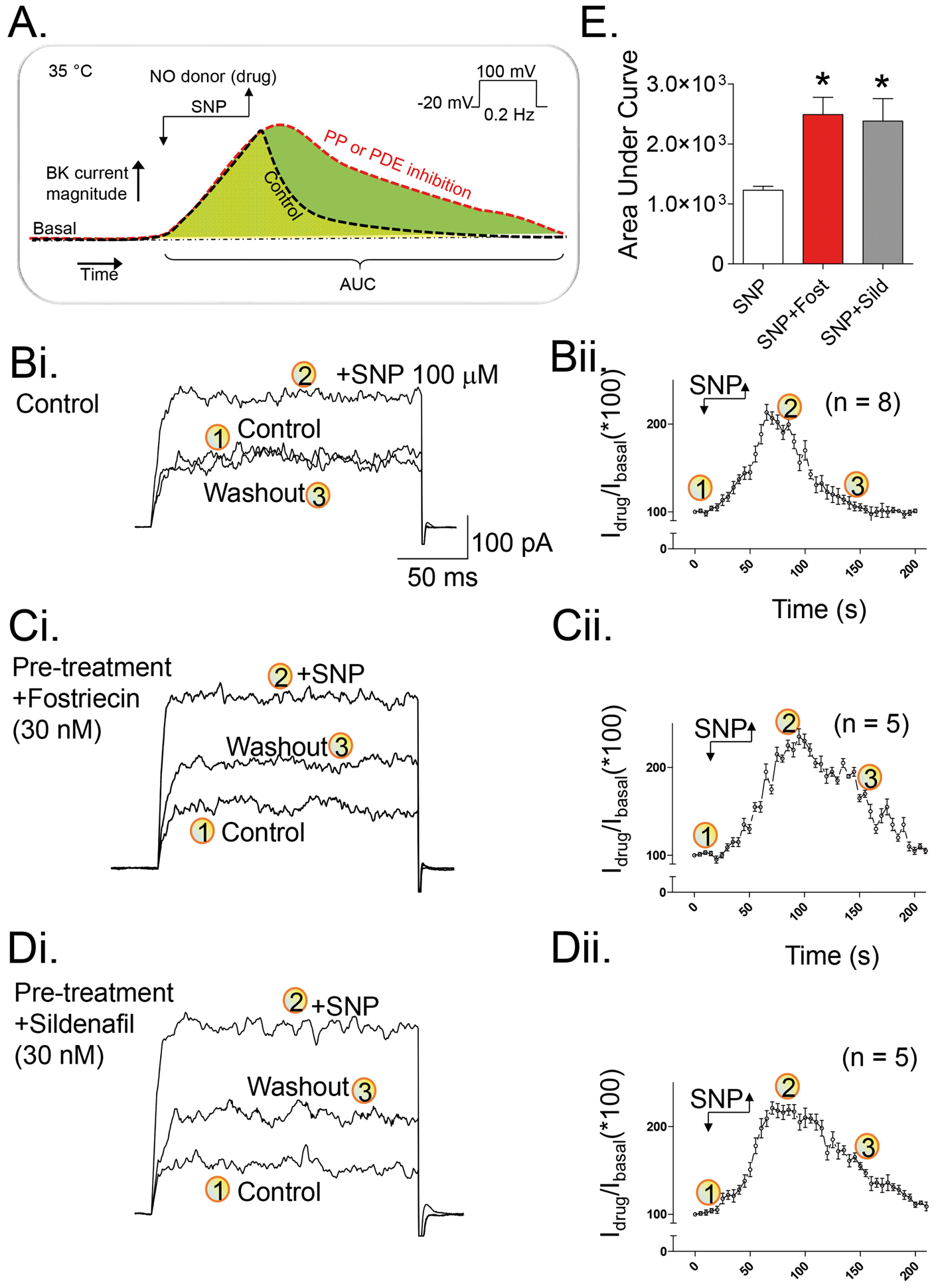
Compelling evidence has demonstrated a critical role for cGKI in the enhancement of BK channel activity by NO/cGMP signaling in vascular smooth muscle,23–26 which appears to be mediated by direct protein phosphorylation of the BK α subunit. Prolonging such modification would thus be expected to increase the duration of enhanced BK channel activity. In several tissues, protein phosphatase 2A (PP2A) is reported to modulate BK channel activity;14,27–29 however, its potential role in cerebral resistance arteries has not been described. We investigated this possibility by exposing isolated cerebral arterial myocytes to a low concentration of the selective PP2A inhibitor, fostriecin (30 nM, 10 min), and observed that this treatment significantly prolonged the enhancement of BK current following removal of SNP. Exposure of isolated myocytes to fostriecin alone did not elevate basal outward current (Supplementary Figure 1(cii)). These novel findings thus identify PP2A as an important regulator of BK channel activity by NO signaling in cerebral myocytes. Further contributing to the reversal of NO-mediated BK current enhancement is the deactivation of cGKI activity, driven by the degradation of intracellular cGMP levels via phosphodiesterase (PDE)-dependent catalysis. PDE5 represents a major isoform in vascular smooth muscle,30,31 and we treated myocytes with the highly selective PDE5 inhibitor sildenafil (30 nM, 10 min) to examine its effects on BK current augmentation by SNP (Figure 1(d)). Sildenafil treatment significantly prolonged the NO-mediated enhancement of macroscopic BK current, similar to that observed for fostriecin treatment. By integrating the BK current amplitude/time course data shown in panels Bii, Cii, and Dii of Figure 1 (area under the curve analysis, from time point 1 to 3), it is evident that fostriecin and sildenafil treatments significantly prolonged the duration of SNP-evoked enhancement of the BK current (Figure 1(e)).
The expression of BK channels, cGKI, PP2A and PDE5 is readily detected in cerebral arterial myocytes
To complement our functional studies demonstrating a role for PDE5 and PP2A activities in the regulation of NO-stimulated BK channel activity, we employed immunocytochemistry and immunoblotting to examine the expression of these key regulatory proteins in rat cerebral myocytes. As shown in Figure 2, isolated myocytes exhibited positive staining for the BK channel α subunit, PP2A, cGKI and PDE5 (Figure 2(a) to (d)). Western blot analysis using the same primary antibodies revealed protein bands corresponding to the predicted molecular masses of these target proteins in rat cerebral arteries, as well as aortic smooth muscle (Figure 2(f)). The two bands observed for cGKI staining in cerebral arteries and aortic smooth muscle are consistent with the expression of cGKIα and cGKIβ isoforms.
32
Immunocytochemical and western blot detection of key proteins in unstimulated rat cerebral myocytes and arteries, respectively. Primary antibodies detected BK channel α subunit (a), PP2A (b), cGKI (c), and PDE5 (D) protein in acutely isolated cerebral myocytes (green fluorescence). DAPI cell nuclear stain (blue fluorescence) is also shown in the merged images. The phase contrast image shown in the lower left corner of each panel demonstrates the myocyte shape and size. (e) No detectable fluorescent signal at 488 nm was detected following application of secondary antibodies in the absence of primary antibodies. (f) Western blot detection of BK channel α subunit, phosphodiesterase 5 (PDE5), type I cGMP-dependent protein kinase (cGKI) and protein phosphatase 2A (PP2A). Lanes 2 and 3 represent tissue homogenates from rat middle cerebral artery and aorta, respectively. For the upper row displaying anti-BKα subunit immunoreactivity, lane 1 represents mouse brain P2 fraction (∼10 μg protein loaded). For rows displaying immunoreactivity for PDE5, cGKI and PP2A, lane 1 represents rat lung homogenate (∼50 μg protein loaded). Following detection of BKα, PDE5, cGKI, and PP2A protein levels, blots were re-probed for β-actin immunoreactivity, which served as an internal loading control. The electrophoretic positions of molecular mass markers are provided on the right hand side of the image. Displayed data are representative of two experiments with similar results.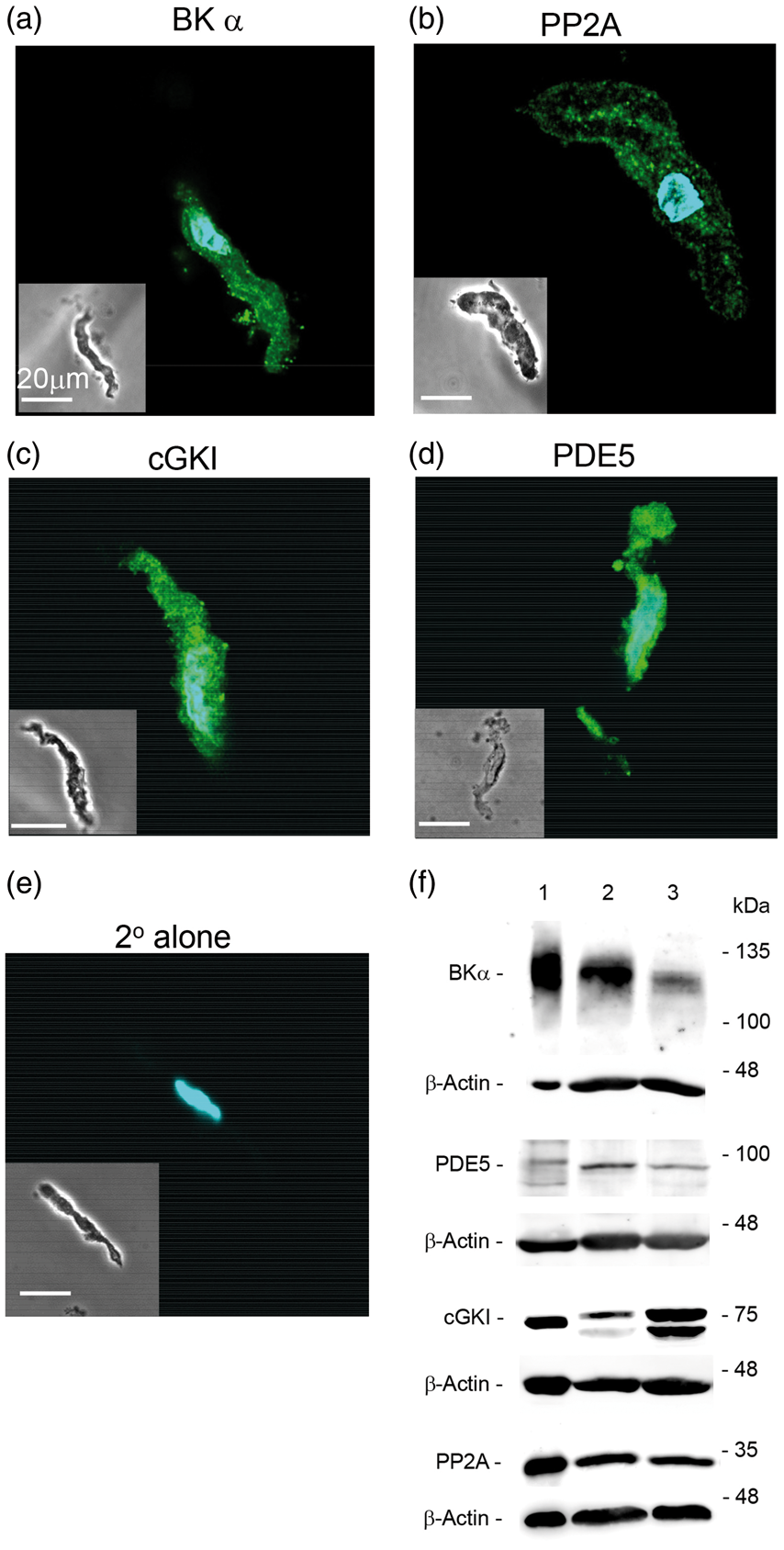
BK channels co-localize with PP2A, PDE5 and cGKI in cerebral arterial myocytes
We further determined whether these regulatory proteins were spatially co-localized with BK channels in cerebral arterial myocytes using the PLA immunostaining technique,18,19 which offered several advantages compared with conventional co-immunoprecipitation (co-IP) strategies. First, macromolecular complexes involving cytosolic proteins and integral membrane proteins may not be sufficiently stable to withstand detergent solubilisation, and would thus not be captured using standard co-IP. Second, PLA staining is readily quantifiable and allows spatial co-localization of two proteins within 40 nm under native cell conditions. Third, PLA staining could be applied to the same myocytes used for BK current recordings, thus providing insights relevant to functionally intact cells.
Positive PLA staining (red fluorescent dots) was clearly visible for BK-cGKI, BK-PP2A, and BK-PDE5 co-staining conditions in isolated cerebral myocytes (Figure 3), indicating that these pairs of protein targets are in close proximity to each other (i.e. within 40 nm). As a control, omission of one primary antibody from the labeling protocol led to a complete absence of red fluorescence, demonstrating the specificity of this immunostaining procedure (Supplementary Figure 2). Remarkably, we found that brief exposure of myocytes to SNP (90 s) followed by immediate fixation, resulted in significantly greater BK channel-cGKI co-localization, compared with basal conditions (Figure 3(a)). SNP treatment did not produce changes in the PLA fluorescence signals for either BK channel-PP2A or BK channel-PDE5 co-staining conditions (Figure 3(b) and (c)). In contrast to SNP, we did not observe any changes in PLA signals for any of these same protein target pairs following treatment of myocytes with either fostriecin or sildenafil (data not shown).
BK channels co-localize with key regulatory molecules in cerebral myocytes, as revealed by proximity ligation assay (PLA) detection. (ai) PLA signal (red fluorescent dots) demonstrates cellular locations where BK Channel α subunit and cGKI protein are co-distributed within a 40 nm distance. Analyses of Z-stack image series demonstrated ∼9 dots/cell under basal conditions. DAPI staining (blue fluorescence) for detection of nuclei has been merged with these data. The accompanying phase contrast image shown to the right illustrates the size and shape of the individual myocyte analyzed by PLA staining. Fluorescent signals associated with PLA and DAPI staining are superimposed (aii) Cells stimulated with SNP displayed more PLA dots (∼13 dots/cell) compared with the basal condition. (aiii) Histogram quantifying the number of PLA-associated red fluorescent dots detected in single cells under basal and SNP exposure conditions. (bi–iii) PLA fluorescent signals were detected (∼6 dots/cell) for myocytes co-immunostained for BK α subunit and PP2A protein. No quantitative difference was found between basal and SNP exposure conditions. (ci–iii) PLA fluorescent signals were detected (∼4 dots/cell) for myocytes co-immunostained for BK α subunit and PDE5 protein. No difference was found between basal and SNP exposure conditions. Average data are expressed as means ± SEM. * denotes a statistically significant difference vs. basal condition (p < 0.05), as determined by a Student's t-test.
Fostriecin treatment augments the NO-evoked vasodilation of cannulated, myogenically active cerebral arteries via a BK channel-dependent mechanism
Our electrophysiological and immunostaining data in isolated myocytes strongly suggest that cGKI, PP2A and PDE5 are intimately involved in the acute regulation of BK channel activity in cerebral arteries. To explore these interactions under more physiological conditions, we utilized arterial pressure myography to examine SNP-mediated vasodilation of myogenically-active, rat cerebral resistance arteries. In cannulated arteries, the average decrease in intraluminal diameter due to myogenic constriction at 70 mmHg was 74 ± 14 microns, which represents a ∼30% reduction relative to the maximal passive diameter (227 ± 36 µm, n = 14).
Under basal conditions, bath application of SNP (10 μM) evoked a robust inhibition of myogenic tone, as evidenced by the increase in intraluminal diameter that readily reversed upon SNP wash-out (Figure 4(a)). We also used an activator of endothelial KCa2.3 and KCa3.1 channels, SKA-31 (3 μM),
21
and the ATP-sensitive K+ (KATP) channel agonist, pinacidil (10 μM),
33
to dilate arteries independently of the NO/cGMP/cGKI signaling cascade; these responses served as internal reference responses for subsequent treatment conditions. Following baseline responses to all three vasodilatory agents, we re-applied these agents in the presence of the PP2A inhibitor fostriecin (100 nM), followed by a third application of the same agents in the presence of fostriecin with the BK channel blocker penitrem-A (2 μM). We have recently reported that SNP responses are readily reproducible in cerebral arteries.
3
Fostriecin treatment alone augmented the SNP-evoked vasodilation, but did not influence vasodilatory responses to either SKA-31 or pinacidil. Moreover, the SNP-evoked dilation was clearly blunted by the further addition of penitrem-A; however, vasodilatory responses to SKA-31 and pinacidil were unaffected. Quantification of these drug-evoked responses demonstrates that fostriecin treatment significantly enhanced the SNP-evoked vasodilation of rat cerebral arteries, without affecting responses to either SKA-31 or pinacidil (Figure 4(b)). Critically, further addition of penitrem-A selectively abolished the fostriecin-induced augmentation of SNP-evoked vasodilation, but did not influence responses to either SKA-31 or pinacidil. Notably, fostriecin exposure alone caused a slight inhibition of basal myogenic tone (i.e. increase in intraluminal diameter of 5.8 ± 1.9 μm, n = 5), which was reversed following the addition of the BK channel blocker penitrem-A. Importantly, in arteries treated with the soluble guanylyl cyclase (sGC) inhibitor ODQ (10 μM), SNP-evoked vasodilation was largely prevented, whereas responses to SKA-31 and pinacidil were unaffected (Supplementary Figure 3). These findings verify that SNP responses were mediated primarily via a cGMP/cGKI signaling pathway. In complementary experiments, we replaced SNP with a chemical strategy that allowed us to “clamp” the amount of NO in the bath solution at concentrations (i.e. ∼3 and 20 nM) approximating the presumed physiological range.
34
Using this approach,
16
we observed that fostriecin still augmented the NO-mediated vasodilation in a manner that was blocked by the BK channel inhibitor penitrem-A (Supplementary Figure 4). Collectively, these data demonstrate that PP2A activity augments the NO-evoked vasodilation in a BK channel-dependent manner and correlate strongly with our results obtained in isolated cerebral myocytes.
SNP-evoked dilation of pressurized cerebral arteries is significantly enhanced by fostriecin. Cannulated cerebral arteries subjected to stepwise increases in intraluminal pressure developed spontaneous myogenic tone and active vasoconstriction. Brief bath exposure to SKA-31 (3 μM), pinacidil (10 μM) or SNP (10 μM), as indicated by the horizontal bars above the tracing, caused reversible inhibition of myogenic tone (a). The vasodilatory actions of SKA-31, pinacidil and SNP were then re-examined following treatment of the artery with the PP2A inhibitor fostriecin (100 nM), as indicated by the horizontal line beneath the tracing. The artery was then treated with a combination of fostriecin + penitrem-A, as indicated beneath the tracing, and re-exposure to SKA-31, pinacidil and SNP was performed. Following washout of fostriecin and penitrem-A, the vessel was exposed to Krebs' solution containing 2 mM EGTA with CaCl2 omitted to determine the vessel's maximal passive diameter at 70 mmHg intraluminal pressure. Tracing is representative of the responses observed in 5 individual arteries. (b) Summary data showing the relative inhibition of myogenic tone by SKA-31, pinacidil or SNP under control conditions, in the presence of fostriecin alone and following treatment with both fostriecin and penitrem-A. Average data are expressed as means ± SEM. * denotes a statistically significant difference vs. the control vasodilatory response or response observed in the presence of fostriecin + penitrem-A (p < 0.05), as determined by one-way ANOVA and a Tukey's post hoc test.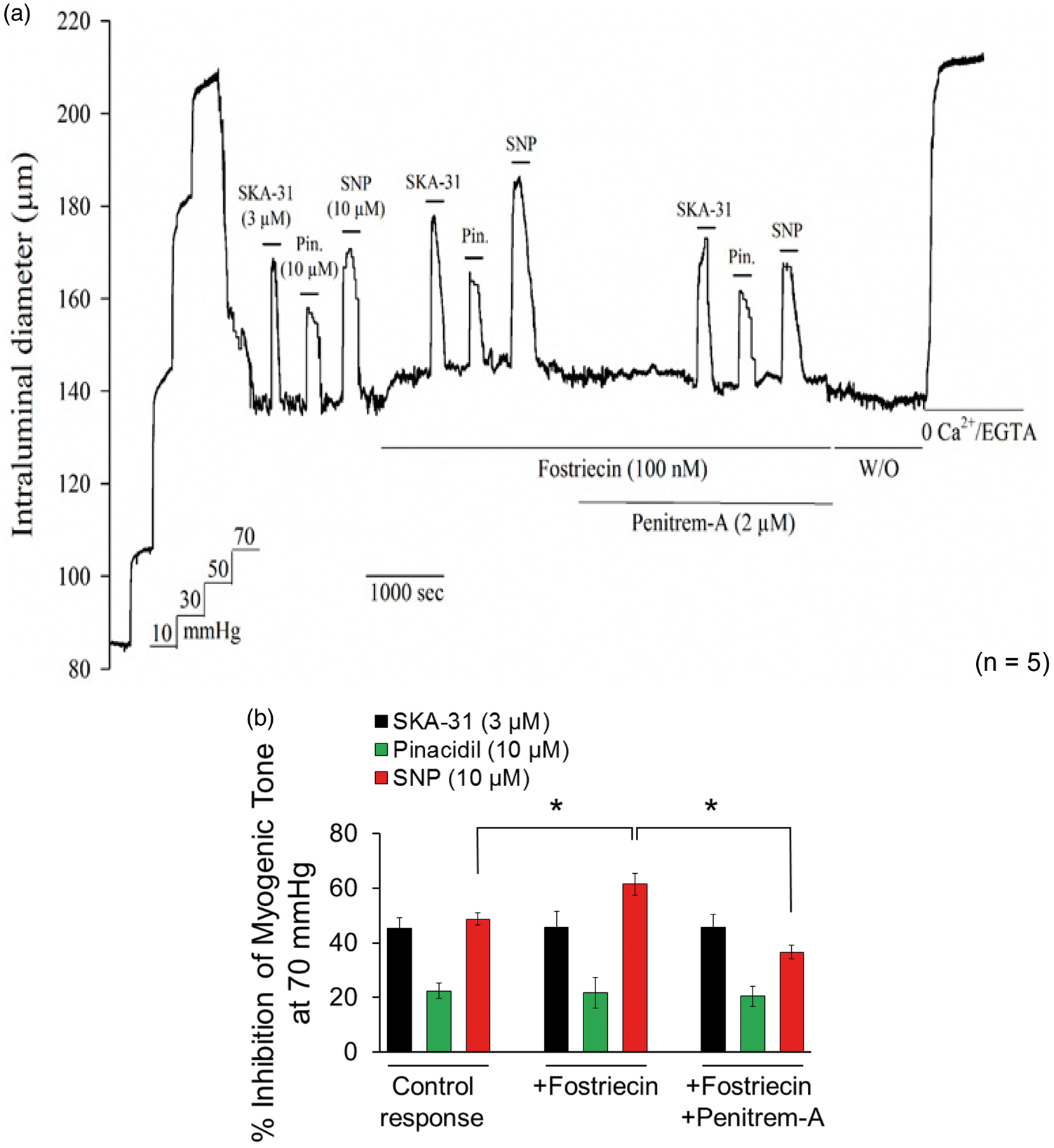
Sildenafil treatment augments the NO-evoked vasodilation of cannulated, myogenically active cerebral arteries via a BK channel-dependent mechanism
Treatment of myogenically active cerebral arteries with the PDE5 inhibitor sildenafil (50 nM) also significantly enhanced the SNP-evoked vasodilation, without affecting responses to either SKA-31 or pinacidil (Figure 5(a)). Further addition of penitrem-A selectively blunted the vasodilatory response in the presence of SNP + sildenafil, demonstrating an essential role for BK channel activity in mediating this enhancement. In related experiments, sildenafil treatment was able to potentiate NO-evoked vasodilation in arteries bath-exposed to ‘clamped’ amounts of NO (i.e. ∼3 and 20 nM),
16
which was prevented following bath addition of the BK channel blocker penitrem-A (Supplementary Figure 5). Similar to fostriecin, we also observed that sildenafil treatment alone was able to inhibit basal myogenic tone (i.e. increase in intraluminal diameter of 8.6 ± 2.6 μm, n = 6), which was reversed in the presence of penitrem-A.
SNP-evoked dilation of pressurized cerebral arteries is significantly enhanced by sildenafil. Stepwise increments in intraluminal pressure led to the development of spontaneous myogenic tone and active vasoconstriction in cannulated cerebral arteries. Bath application of SKA-31 (3 μM), pinacidil (10 μM) or SNP (10 μM) each reversibly inhibited myogenic tone (a). The vasodilatory actions of SKA-31, pinacidil, and SNP were then re-examined following treatment of the vessel with the PDE5 inhibitor sildenafil (50 nM), as indicated by the horizontal line beneath the tracing. Subsequently, the artery was treated with sildenafil + the BK channel blocker penitrem-A (2 μM) and the vasodilatory actions of SKA-31, pinacidil and SNP were re-examined. Following washout of sildenafil and penitrem-A, arteries were exposed to Krebs' solution containing 2 mM EGTA with CaCl2 omitted to determine the vessel's maximal passive diameter at 70 mmHg intraluminal pressure. The displayed tracing is representative of responses observed in 5 individual arteries. (b) Histogram quantifying the relative inhibition of myogenic tone by SKA-31, pinacidil, and SNP under control conditions, in the presence of sildenafil alone and the presence of both sildenafil and penitrem-A. Average data are expressed as means ± SEM. * denotes a statistically significant difference vs. the control vasodilatory response or response in the presence of sildenafil + penitrem-A (p < 0.05), as determined by one-way ANOVA and a Tukey's post hoc test.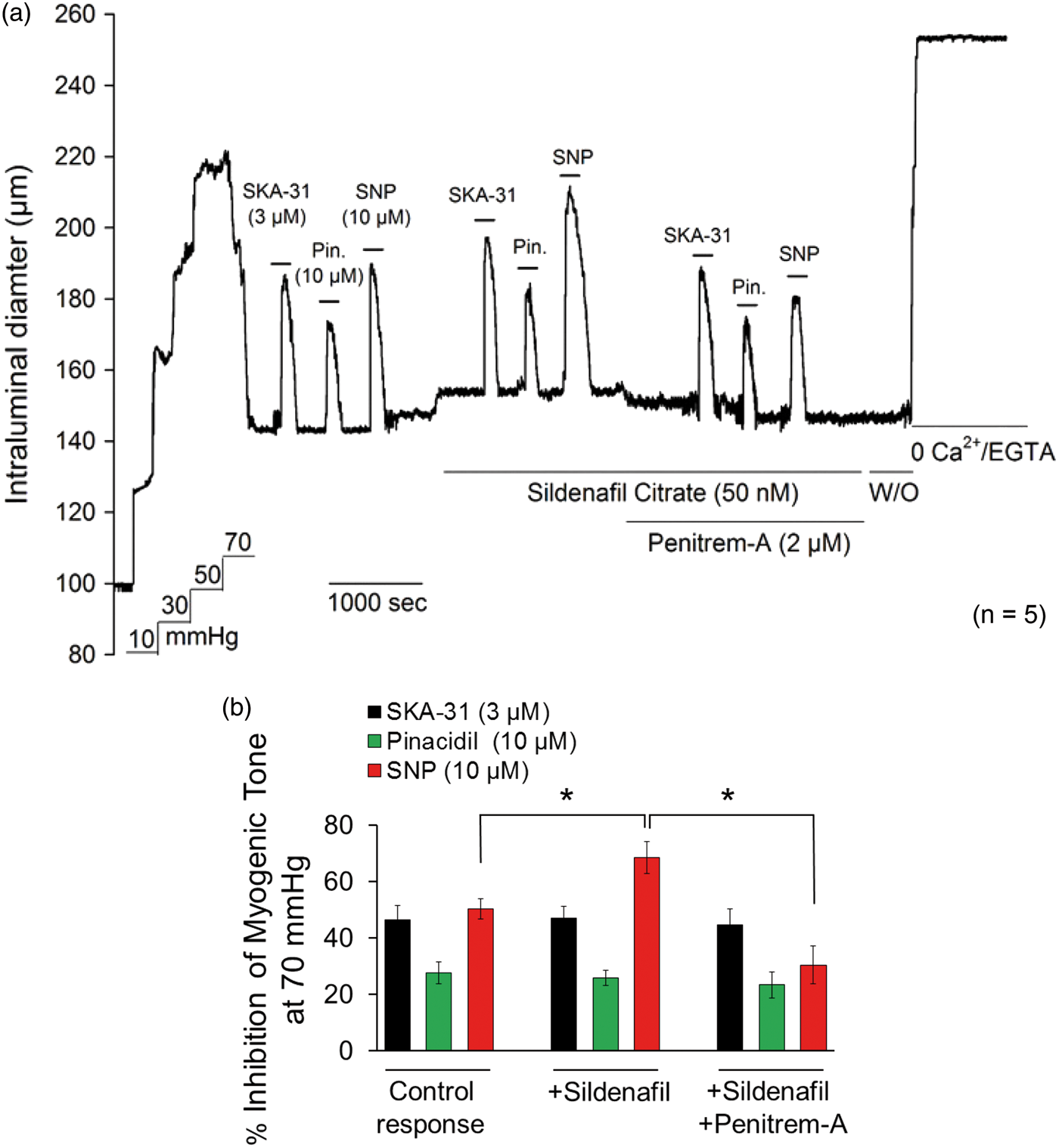
Discussion
BK channel activity directly impacts myogenic tone and intraluminal diameter in cerebral resistance arteries;1,2,9 however, the intracellular mechanisms regulating cerebrovascular BK current remain poorly defined. We and others have reported that NO-induced augmentation of BK current in smooth muscle can occur via cGKI signaling and direct channel phosphorylation,13,17,23,35,36 implicating an important role for BK channel phosphorylation and dephosphorylation in the regulation of cerebrovascular tone. In the present study, we have provided new information demonstrating that BK current in cerebral myocytes can be regulated by the co-localization of these channels with key components of the NO/cGMP signaling cascade, namely, cGKI, PP2A, and PDE5. At a more integrative level, it appears that these macromolecular complexes of BK channels and associated enzymes are functionally important for the control of intraluminal diameter by NO/cGMP signaling in myogenically active, cerebral resistance arteries.
Phosphorylation of BK channels by Ser/Thr protein kinases is known to increase macroscopic BK current (for review, see Kyle and Braun 13 and Shipston and Tian 14 ), and interventions that prolong this modification would be expected to alter the magnitude and/or duration of augmented current following a stimulus (see schema in Figure 1(a)). In response to the clinical nitrovasodilator SNP, cerebral myocyte BK current rapidly increased to a peak level, which is likely due to NO/cGMP-dependent stimulation of cGKI activity and BK channel phosphorylation (Figure 1(bi). This interpretation is supported by our recent data demonstrating that this SNP-mediated augmentation of BK current in rat vascular myocytes is prevented by a pharmacologic inhibitor of cGKI. 17 Withdrawal of the NO stimulus led to a complete reversal of augmented BK current to baseline within 2 min (Figure 1(bii)), which can be explained by a decline in cGKI activity and the enzyme-dependent dephosphorylation of modified BK channel complexes. In support of this interpretation, treatment of cerebral myocytes with the selective PP2A inhibitor fostriecin (30 nM) prolonged the augmentation of BK current in response to SNP (Figure 1(c) and (e)), most likely by slowing the dephosphorylation of cGKI-modified BK channel α subunits. A similar prolongation of SNP-induced BK current augmentation was observed following treatment with the PDE5 inhibitor sildenafil (30 nM) (Figure 1(d) and (e)), which would be expected to slow the degradation of intracellular cGMP, thereby prolonging cellular cGKI activity and maintaining BK channels in a phosphorylated state. This observed functional regulation of BK current is supported by immunocytochemical and Western blot data demonstrating the expression of BK channels, cGKI, PDE5, and PP2A in the cerebral myocytes utilized for patch clamp recordings (Figure 2).
As membrane ion channels are capable of co-localizing with key regulatory components, 37 we utilized PLA immunostaining as a novel strategy to quantify the spatial co-distribution of cerebrovascular BK channels with enzymes that directly influence their regulation by NO/cGMP signaling and phosphorylation. Critically, our data reveal that cGKI, PDE5, and PP2A exist in close spatial proximity to BK channels (i.e. inter-protein distance ≤ 40 nm) under resting conditions (Figure 3) and that the co-association of cGKI with BK channels can be increased by brief exposure to the NO donor SNP (Figure 3(a)ii). To our knowledge, this latter result represents the first demonstration that regulatory proteins in cerebral myocytes can be actively recruited to BK channels in a stimulus-dependent manner, and suggests a novel mechanism by which external signals can influence cerebrovascular excitability. Further investigation is needed to determine exactly how such recruitment occurs and identify the cellular machinery underlying this process. From a functional perspective, these data are supported by electrophysiological studies describing the regulation/co-association of BK channels by Ser/Thr protein kinases (e.g. cGKI and PKA) and phosphatases (e.g. PP2A) in various cell types,14,27,36,38–42 suggesting that such regulatory events are wide-spread. Cerebrovascular BK channels thus appear capable of assembling with key regulatory proteins under basal conditions to form macromolecular complexes or “signalosomes”. Our novel finding that the composition of these complexes is dynamic further suggests that magnitude of BK channel augmentation driven by these complexes may be scalable and vary with the magnitude of the vasoactive stimulus.
Quantitatively, PLA staining of BK channel complexes in cerebral myocytes revealed a modest number of fluorescent dots per cell (i.e. < 20), which would be consistent with the reported membrane clustering of these channels in smooth muscle43,44 and the likelihood that only a subset of these channels may co-associate with cGKI, PP2A and/or PDE5. Is it realistic that a limited number of complexes containing BK channels + cGKI, PP2A and/or PDE5 can account for the magnitude of macroscopic BK current observed in NO-stimulated vascular myocytes? We think the answer is yes, based on the following conditions. Under a physiologic K+ gradient, BK single channel conductance will be ∼100 pS, 45 yielding a single channel amplitude of 10-12 pA at the step voltage of +100 mV. Given that channel open probability at this voltage approximates 0.5 46 and NO/cGMP signaling should augment BK channel open probability by ∼2-fold, 47 we estimate that the amplitudes of basal and NO-stimulated whole cell BK current reported in our study could arise from the activity of 20–25 channels. Moreover, as smooth muscle BK channels can exist in clusters, 44 it is conceivable that a modest number of clusters each containing 4–5 BK channels together with cGKI, PP2A, and PDE5 may be sufficient to account for our observed current amplitudes. However, the literature suggests that additional cellular mechanisms may contribute to the observed augmentation of whole cell BK current by NO/cGMP signaling, including the stimulated membrane trafficking of BK β1 subunits to smooth muscle BK channels 48 and the elevation of cytosolic cAMP by cGMP-dependent inhibition of PDE3 and ensuing activation of cAMP-dependent protein kinase. 30 Co-localization of BK channels with enzymatic machinery may thus represent one of several mechanisms by which NO/cGMP signaling can augment macroscopic BK current in vascular myocytes to promote relaxation.
In myogenically active middle cerebral arteries, brief treatment with either fostriecin (Figure 4) or sildenafil (Figure 5) augmented the NO-evoked vasodilation, and this augmentation was completely blocked in the presence of the highly selective BK channel inhibitor penitrem-A. 22 Fostriecin and sildenafil produced a similar pattern of augmentation on NO-evoked dilation under conditions in which arteries were exposed to low nanomolar concentrations of NO (i.e. ∼3 and 20 nM) using a chemical “clamping” protocol15,16 (see Supplementary Figures 4 and 5). Collectively, these findings strongly suggest that the observed enhancement of BK current by fostriecin and sildenafil in isolated myocytes directly contributes to the vasodilatory responses evoked by NO in intact cerebral arteries. It is noteworthy that a significant fraction of NO-mediated vasodilation still remained following BK channel blockade with penitrem-A (Figures 4(b) and 5(b), Suppl Figures 4(b) and 5(b)). Such observations emphasize that BK current-dependent hyperpolarization is one of several cellular mechanisms (e.g. promotion of myosin phosphatase activity, enhanced re-uptake of cytosolic Ca2+ by the SR) that contribute to NO/cGMP-evoked smooth muscle relaxation.49,50 As anticipated, fostriecin and sildenafil did not impact the vasodilatory events elicited by either pinacidil or SKA-31, indicating that PP2A and PDE5 activities do not acutely modulate the vasodilatory effects associated with the activation of K-ATP channels or endothelial Ca2+-activated K+ channels, respectively. Interestingly, exposure of cerebral arteries to either fostriecin or sildenafil alone caused a modest inhibition of basal myogenic tone, which was reduced by penitrem-A treatment. This finding is thus in line with earlier data demonstrating that BK channels actively contribute to the regulation of resting tone in pressurized, cerebral resistance arteries.1,3
Figure 6 presents a schematic cartoon highlighting the key findings of our study in the physiological context of the cerebrovascular wall. Mechanistically, endothelial-derived NO activates sGC and increases intracellular cGMP in cerebral myocytes, leading to activation and mobilization of cGKI to BK channels. Activated cGKI then directly phosphorylates the BK α subunit,13,14,17,36 increasing BK channel-mediated K+ efflux and smooth muscle hyperpolarization. Active recruitment of cGKI to BK channels may further provide a means to upregulate BK channel activity in proportion to stimulus intensity. As smooth muscle BK channels are also known to co-localize with type 2 RyRs and be activated by SR-mediated Ca2+ sparks,7,44 one could envision how the actions of NO/cGMP signaling machinery would enhance the functional coupling of BK channels with RyR2s. Confirmation of such macromolecular assemblies awaits the development of more advanced imaging techniques to detect the spatial co-localization of ≥ 3 target proteins at a given locus. Reversal of NO-evoked enhancement of BK current via protein dephosphorylation and cGMP degradation is mediated, in part, by co-localized PP2A and PDE5 enzymes, respectively; these latter reactions would be expected to reduce BK channel activity and restore membrane potential to the basal level. In myogenically constricted cerebral arteries, pharmacologic manipulations designed to prolong the lifetime of phosphorylated BK channels resulted in an augmented vasodilatory response to the NO donor SNP (Figures 4 and 5), implicating these cellular mechanisms in the functional control of intraluminal diameter in cerebral resistance arteries. Ultimately, these effects will impact cerebral blood flow and neuronal activity in the brain.
Schematic cartoon highlighting key proteins regulating the phosphorylation status of BK channels in cerebral arteries. Nitric oxide generated by endothelial cells activates soluble guanylyl cyclase in vascular smooth muscle and increases the level of intracellular cGMP. Increased cGMP is expected to activate cGKI and leads to a greater amount of cGKI protein in close proximity (i.e. within 40 nm) to BK channels. Co-associated cGKI proteins promote the phosphorylation of BK channels, increasing their open probability. PDE5 enzymatically cleaves cGMP, reducing its local concentration within the cytosol and promoting de-activation of cGKI. PP2A enzymatically dephosphorylates BK channels modified by cGKI, thereby reducing its open probability. Note that additional pathways known to contribute to agonist-evoked vasodilation, such as endothelium-dependent hyperpolarization, the generation of prostaglandins, H2O2, etc. have been omitted from this simplified cartoon.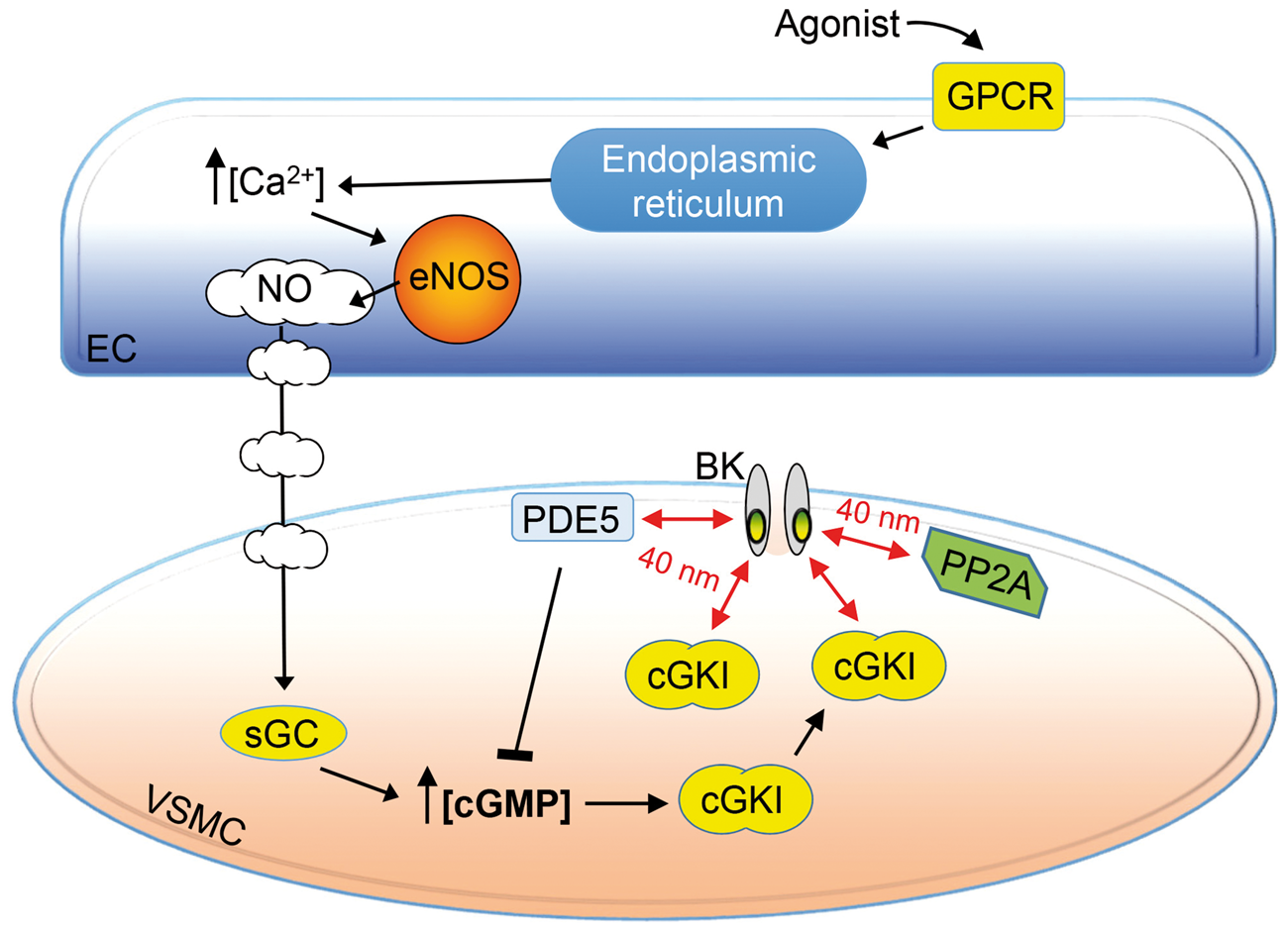
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by research funding to AP Braun from the Canadian Institutes of Health Research (MOP 97901) and the Natural Sciences and Engineering Research Council (RGPIN/312240).
Acknowledgements
The authors thank Drs. Mike Walsh, Bill Cole and Mike Hill for their critical reading of the manuscript and Dr. Heike Wulff (Dept. of Pharmacology, UC Davis) for generously providing SKA-31.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors' contributions
BDK, RCM and APB designed and performed the experiments, carried out analyses and prepared figures. BDK and APB wrote and revised the manuscript.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.