
Abstract
Cigarette smoking is a significant risk factor in the incidence of cerebrovascular disorders. Among the many compounds in cigarette smoke, nicotine is considered to most significantly affect cerebral arterial tone. The purpose of this study is to investigate precise pharmacological effects of nicotine on the regulation of cerebral arterial tone. To mimic smoking, a low concentration of nicotine (10−6 mol/L), which is equivalent to the serum level of habitual smokers, was treated for 1 hour in an isometric tension study and for 24 hours in a study using cultured vascular endothelial cells (VECs). Using the canine basilar artery, the effect of nicotine on uridine 5′-triphosphate (UTP)-induced vasoconstriction was examined in the isometric tension study. Protein kinase C (PKC) activity in the canine basilar artery was measured by enzyme immunoassay. Endothelial function was assessed by endothelium-dependent vasodilatation and endogenous nitric oxide (NO) synthesis in VECs using a fluorescent indicator, diaminofluorescein-FM diacetate (DAF-FM/DA). Nicotine significantly enhanced UTP-induced contraction and PKC activity in the artery, and attenuated endothelium-dependent vasodilatation and NO synthesis in VECs. Because PKC activity was increased by de-endothelialization itself, endothelial dysfunction by nicotine enhances PKC activity. Because PKC was further activated by nicotine even in the de-endothelialized artery, nicotine directly affects PKC activities in smooth muscle. These results indicate that nicotine potentiates contractile response through direct and indirect PKC activation in the canine basilar artery.
Introduction
It is well known that cigarette smoking is one of the major risk factors in the incidence of cerebrovascular disorders (Kurth et al, 2003; Ohkuma et al, 2003; Moncayo et al, 2000; Kool et al, 1993). Cigarette smoke contains a number of chemical compounds affecting cerebral vascular tone, such as nicotine, carbon monoxide, nitric oxide (NO), and cyanogen. Among these compounds, nicotine is known to have the most significant pharmacological effect on various tissues, including the cerebral arteries (Charles, 1995).
A high concentration of nicotine causes dose-dependent constriction in some arteries (Kurahashi et al, 2001; Shirahase et al, 1988). However, a high concentration of nicotine also induces transitory vasodilatation in precontracted arteries via the perivascular nitroxidergic nerve (Toda, 1975). It has also been reported that nicotine activates the sympathetic nervous system (SNS), and induces the vasoconstrictive effect in many organs via cathecholamine, an SNS neurotransmitter (Suzuki et al, 1973). The influence of cathecholamine and the SNS activation on the cerebral arteries are, however, considered to be comparatively small, because cerebral arteries are less sensitive to norepinephrine.
Smoking and the application of nicotine patches have been reported to induce significant cerebral arterial narrowing, and decreased cerebral blood flow (CBF) in vivo (Jackson, 1993; Cruickshank et al, 1989). Nevertheless, opposite results have been reported in that nicotine infusion induced cerebral pial arterial dilatation (Iida et al, 1998) and increased CBF (Kodaira et al, 1993). According to these previous reported data in vivo, the pharmacological effects of nicotine on the regulation of cerebral arterial tone remain controversial.
To clarify the direct pharmacological effects of nicotine on cerebral arterial tone, we examined how nicotine affects arterial contractility, protein kinase C (PKC) activity, and endothelial function, including NO synthesis, using isolated canine basilar arteries and primary human microvascular cultured endothelial cells (VECs). In previous literature, the pharmacological effects of nicotine were investigated using high concentrations such as 10−5 and 10−4 mol/L, which were concentrations inducing acute nicotine intoxication (Wesley et al, 1992). Furthermore, acute effects of nicotine after its application were examined (Ayajiki et al, 1994; Shirahase et al, 1988). To mimic cigarette smoking, we used nicotine at lower concentrations based on the arterial concentration level of nicotine in habitual cigarette smokers (Armitage et al, 1975), and incubated the artery for 1 hour and VECs for 24 hours.
Materials and methods
Animals
All experiments were performed according to the Rules of Animal Experimentation and the Guide for the Care and Use of Laboratory Animals of the Hamamatsu University School of Medicine. Thirty-six adult mongrel dogs of either sex, weighing 10 to 16 kg, were used. A total of 24 dogs were used for the isometric tension study and 12 dogs for the PKC assay. The dogs were killed with an intravenous injection of an overdose of pentobarbital sodium (50 mg/kg), and the basilar arteries were immediately excised.
Uridine 5′-Triphosphate-Induced Vasoconstriction
An isometric tension study was performed using an originally designed apparatus, and the details have been described in our previous paper (Nishizawa et al, 1992b). The basilar arteries were cut into pieces 4 mm long, and the arterial rings were suspended in the chamber and incubated without any stretching for 20 mins. Thereafter, the resting tension was set at 1.0 g. High K+ solution (containing 76.3 mmol/L of K+) was next applied, and the magnitudes of the developed tension in the following studies were expressed as a percentage of high K+-induced tension (=100%).
The arterial rings were randomly divided into four groups (control, nicotine-treated, de-endothelialized, and de-endothelialized/nicotine-treated groups), and each arterial ring taken from one dog was used in different groups (number of arterial rings; n=5, each group). Arterial rings in the control group were endothelium-intact and incubated without nicotine, and those in the nicotine-treated group were endothelium-intact and preincubated with nicotine (10−6 mol/L) for 1 hour before uridine 5′-triphosphate (UTP) application. In the de-endothelialized group, the endothelium was gently rubbed off (Nakayama, 1988) and incubated without nicotine. Arterial rings in the de-endothelialized/nicotine-treated group were de-endothelialized and preincubated with nicotine (10−6 mol/L) for 1 hour. For each arterial ring, UTP was cumulatively added into the chamber from 1 × 10−8 mol/L up to 1 × 10−2 mol/L. The concentration of nicotine, 10−6 mol/L, was determined by the arterial blood level of nicotine in habitual smokers (Armitage et al, 1975).
Measurement of Protein Kinase C activity
Protein kinase C activities were measured in the following three studies of experiments: UTP stimulation, nicotine-treatment, and de-endothelialization. One isolated canine basilar artery was cut into four or five pieces of arterial rings, and arterial rings were randomly divided into each group. It was set that the arterial rings in one group were derived from different dogs. In the UTP stimulation study, 24 arterial rings were randomly divided into four groups (n=6, each group): nonstimulated (Non), UTP stimulation (UTP), nicotine treatment and UTP stimulation (nicotine/UTP), and nicotine treatment and UTP stimulation in the presence of PKC inhibitor, calphostin C (nicotine/UTP/calph. C). In the UTP group, arterial rings were incubated with UTP (10−5 mol/L) for 10 mins. In the nicotine/UTP group, arterial rings were incubated with nicotine (10−6 mol/L) for 1 hour before UTP stimulation, and then UTP (10−5 mol/L) was added and incubated for 10 mins in the presence of nicotine (10−6 mol/L). For the nicotine/UTP/calph. C group, calphostin C was added for 10 mins before nicotine treatment, and also added during nicotine treatment and UTP stimulation. In the Non group, arterial rings were incubated for 1 hour without any reagents.
In the nicotine-treatment study, 16 arterial rings were divided into 4 groups (n=4, each group): incubated without nicotine for 1 hour, and incubated with nicotine at 10−8, 10−7, and 10−6 mol/L for 1 hour.
In the de-endothelialization study, 12 arterial rings were randomly divided into 3 groups (n=4, each group): intact arterial rings incubated without nicotine for 1 hour, and de-endothelialized arterial rings (Nakayama, 1988) incubated without or with nicotine (10−6 mol/L) for 1 hour.
Because activated PKC translocates from the cytosol to the membrane fractions (Nishizuka, 1984), PKC activities in the membrane fraction were measured using a nonradioisotopic protein kinase assay kit (MESACUP Protein Kinase Assay Kit, Medical and Biological Laboratories, Nagoya, Japan). The methods of sample collection and making a standard curve using canine cerebellar cytosol fractions in detail were as described previously (Nishizawa et al, 1995). The multidiluted cytosol fractions of the canine cerebellum were set and PKC activities were measured for making a standard curve. The PKC activities in the membrane fraction of the artery were computed using a standard curve and expressed as a percentage of those in the cerebellum fraction. Because PKC activity depends on the protein concentration in samples, the protein concentration was measured by the modified Lowry Protein Assay Reagent Kit (PIERCE, Rockford, IL, USA), and PKC activity of each sample was standardized according to the protein concentrations.
Endothelium-Dependent Vasodilatation by Substance P
Endothelium-dependent vasodilatation was measured in the isometric tension study. The arterial rings were randomly divided into two groups (n=5, each group). It was set that the arterial rings in each group were derived from different dogs. One was preincubated with nicotine (10−6 mol/L; nicotine-treated group) and the other was incubated without nicotine for 1 hour (control group). The precontractions by UTP at 3 × 10−3 mol/L in the control group and at 1 × 10−4 mol/L in the nicotine-treated group were obtained to make the magnitude of contraction in each group equivalent. Following the precontraction, substance P was cumulatively applied from 1 × 10−8 mol/L up to 10−6 mol/L. At the end of the study, papaverine (10−4 mol/L) was added to obtain the maximal relaxation, and each magnitude of substance P-induced vasodilatation was expressed as a percentage of the maximal relaxation (=100%).
Sensitivity of the Smooth Muscle to Nitric Oxide after Nicotine Administration
To investigate whether the sensitivity of the smooth muscle to NO after nicotine administration was altered, arterial dilatations induced by NO donors NOC 7 (1-hydroxy-2-oxo-3-(N-methyl-3-aminopropyl)-3-methyl-1-triazene) (Zhang et al, 1996) and NOR 1 ((±)-(E)-4-methyl-2-[(E)-hydroxyimino]-5-nitro-6-methoxy-3-hexenamide) (Kita et al, 1994) (both are noted NO donors) were examined using de-endothelialized arteries in the isometric tension study. The arterial rings were gently rubbed off by a steel wire (Nakayama, 1988), and were randomly divided into four groups (n=4, each group): control/NOC 7, nicotine-treated/NOC 7, control/NOR 1, and nicotine-treated/NOR 1. It was arranged that arterial rings in each group were derived from different dogs. Arterial rings were incubated without (control group) or with (nicotine-treated group) nicotine (10−6 mol/L) for 1 hour. Arterial rings were precontracted by UTP at 3 × 10−4 mol/L in the control group and at 1 × 10−4 mol/L in the nicotine-treated group, to obtain the equal magnitude of contraction in each group. After precontraction by UTP, NO donor was cumulatively applied from 1 × 10−11 mol/L up to 3 × 10−6 mol/L. The magnitudes of vasodilatation were expressed as a percentage of precontraction by UTP (=100%). In the study using NOC 7, the pH of PSS in the chamber was checked by pH test paper after the application of the maximal concentrated NOC 7, because NOC 7 was dissolved in 1 × 10−1 mol/L NaOH.
Biochemical Nitric Oxide Imaging by Diaminofluorescein-FM Diacetate
The effect of nicotine on NO synthesis was assessed in primary human microvascular endothelial cells (VECs; cell-system BME cell; Dainippon Pharmacy, Osaka, Japan). The VECs were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum, 0.3 mg/mL glutamic acid, 100 U/mL penicillin, 100 μg/mL streptomycin, and 10 ng/mL basic-fibroblast growth factor (bFGF). To avoid the effect of the fluorescent substance in DMEM on NO imaging, an artificial cerebrospinal fluid (aCSF; mmol/L: Na+ 140.00, K+ 5.0, Cl− 151.4, Ca2+ 2.0, Mg2+ 1.20, HEPES 10.0, and glucose 10.0, pH 7.2) was used during the measurement.
For direct measurement of intracellular NO synthesis, diaminofluorescein-FM diacetate (DAF-FM/DA) (5 × 10−6 mol/L), a fluorescent NO indicator, was loaded into the VECs at 37°C for 15 mins in the dark. Diaminofluorescein-FM diacetate fluorescence was excited at 500 nm. Emitted cellular fluorescence was recorded at 515 nm with an inverted fluorescence microscope (Axiovert 10, Carl Zeiss, Oberkochen, Germany) and a three-chip CCD camera (WV-BD800, Panasonic, Osaka, Japan). Diaminofluorescein-FM diacetate-loaded VECs were stimulated with substance P (10−6 mol/L), and fluorescent images were recorded every 5 mins after the stimulation. Zero minute was defined as just before the stimulation. While obtaining images, DAF-FM/DA fluorescence was excited for only a few seconds to avoid dye fading and cell damage. Fluorescence images were analyzed with computer analyzing software (MetaMorph Imaging System, Universal Imaging System, Downingtown, PA, USA), and the cellular fluorescence intensity was calculated. The fluorescence intensity was expressed as a percentage of that before the stimulation in the same cell (=100%).
To assess the pharmacological effect of nicotine on endogenous NO synthesis, some culture dishes of VECs were treated with nicotine for 24 hours, and were treated with N-nitro-L-arginine methyl ester (L-NAME, 10−4 mol/L) for 30 mins before NO imaging. During NO imaging, nicotine or L-NAME was present in the dish.
Drugs
Pentobarbital sodium (Somnopentyl, Kyoritsu Shoji, Tokyo, Japan), nicotine, papaverine, L-NAME (Sigma, St Louis, MO, USA), substance P (Peptide Institute, Osaka, Japan), NOC 7, NOR 1 (Dojindo Laboratories, Kumamoto, Japan), DAF-FM/DA (Daiichi Chemicals Co., Tokyo, Japan), DMEM (Nissui, Tokyo, Japan), fetal calf serum (Trace Scientific Ltd., Melbourne, Australia), glutamic acid (Katayama, Osaka, Japan), penicillin, streptomycin (Meiji Seika, Tokyo, Japan), and bFGF (PeptoTech, London, UK) were commercially purchased. The other reagents and salts in this study, all of the highest commercial quality, were purchased from Wako Pure Chemical Industries (Osaka, Japan).
Nicotine, papaverine, L-NAME, UTP, and substance P were dissolved with physiological saline solution (mmol/L: Na+ 144.44, K+ 4.10, Cl−, 127.2, Ca2+ 2.49, Mg2+ 1.19, PO43− 1.54, SO42− 1.12, HCO3− 24.9, and glucose 5.0, pH 7.4). NOC 7 was dissolved in 1 × 10−1 mol/L sodium hydroxide (NaOH), and NOR 1 in dimethylsulfoxide, to give the additional volume of less than 1% in the total volume.
Statistical Analysis
All data were expressed as the mean±s.d. Paired or unpaired Student's t-tests were used for the analysis of statistical difference between the two groups. The statistical significance of differences among groups was established according to Dunnett's multiple comparison test after an analysis of variance (ANOVA). P-values less than 0.05 were considered to be statistically significant.
Results
Effect of Nicotine Exposure on Uridine 5′-Triphosphate Induced Vasoconstriction in Endothelium-Intact Canine Basilar Arteries
The arterial rings of canine basilar arteries with endothelium were exposed to nicotine (10−6 mol/L) for 1 hour; however, no contractile responses were observed (data not shown). After the incubation without or with nicotine, Uridine 5′-triphosphate-induced vasoconstrictions were measured (n=5 each). UTP-induced arterial contraction in the nicotine-treated group was significantly greater than that in the control group above 1 × 10−5 mol/L of UTP (P<0.05 at 1 × 10−5 mol/L, P<0.01 above 3 × 10−5 mol/L). Summarized data are shown in Figure 1.
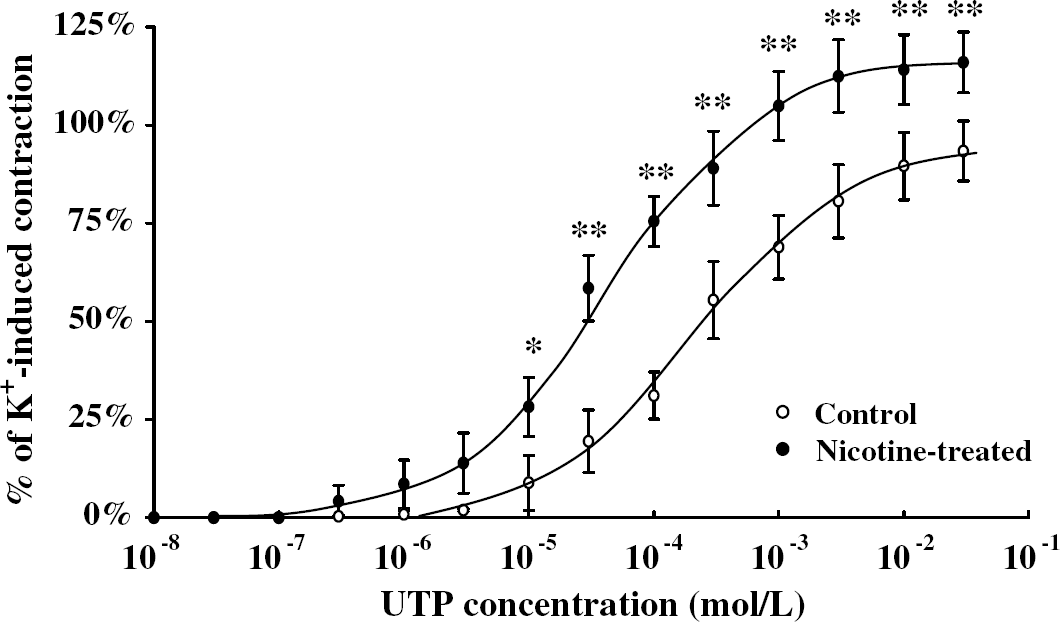
Dose–response curves showing the effects of nicotine exposure on uridine- 5′-triphosphate (UTP)-induced contraction in the canine basilar artery. After the arteries were incubated without (open circle) or with (closed circle) nicotine (10−6 mol/L) for 1 hour, UTP was cumulatively applied. The developed tensions were expressed as a percentage of the high K+-induced tension (=100%), and the data were expressed as the mean±s.d. (n=5 each). *P<0.05, **P<0.01 versus the contractile value of the arteries in the control group.
Effect of Nicotine Exposure on Uridine 5′-Triphosphate-Induced Vasoconstriction in De-Endothelialized Canine Basilar Arteries
The effect of nicotine exposure on UTP-induced vasoconstriction in the de-endothelialized canine basilar artery was examined. The arterial rings of de-endothelialized canine basilar arteries were incubated with nicotine (10−6 mol/L) for 1 hour; however, significant contractile responses were not observed (data not shown). After the incubation without or with nicotine, Uridine 5′-triphosphate-induced vasoconstrictions were measured. UTP-induced arterial contraction in the de-endothelialized group was significantly enhanced against that in the control group (P<0.05 at 3 × 10−6 mol/L and at 3 × 10−5 mol/L, P<0.01 above 1 × 10−4 mol/L, n=5 each). In de-endothelialized arterial rings, nicotine significantly augmented UTP-induced contraction (P<0.05 at 1 × 10−6 mol/L and at 3 × 10−4 mol/L, P<0.01 from 3 × 10−6 mol/L to 1 × 10−4 mol/L, n=5 each). Summarized data are shown in Figure 2.
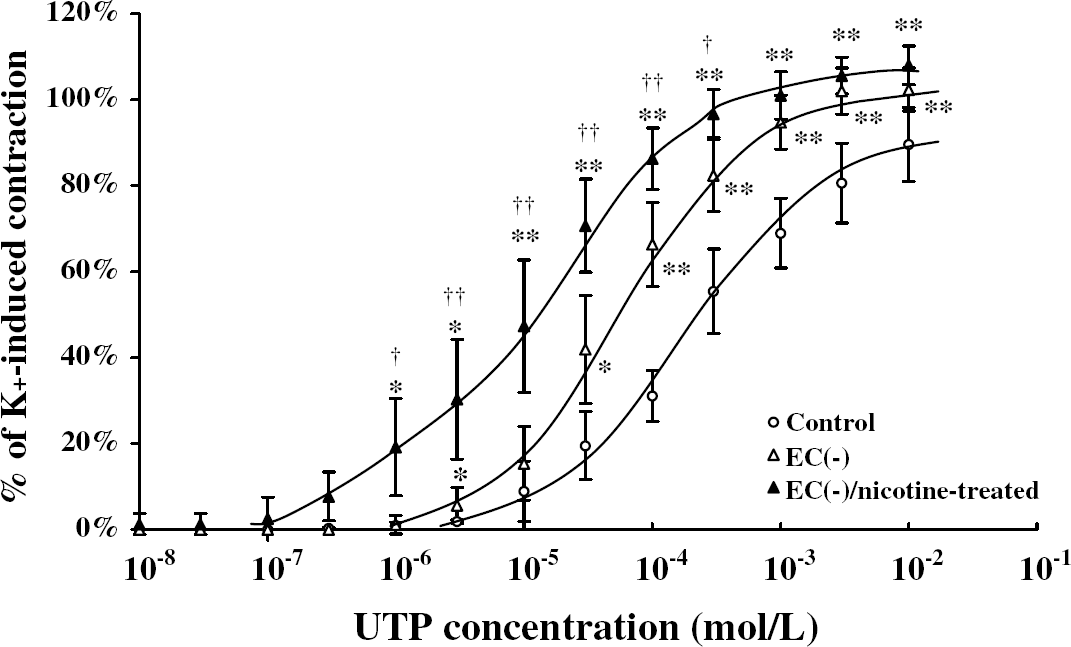
Dose–response curves showing the effects of nicotine exposure and/or de-endothelialization on uridine- 5′-triphosphate (UTP)-induced contraction in the canine basilar artery. In the de-endothelialized group described as EC (−), the endothelium was gently rubbed off. After the arteries were incubated without or with nicotine (10−6 mol/L) for 1 hour, UTP was cumulatively applied. The developed tensions were expressed as a percentage of the high K+-induced tension (=100%), and the data were expressed as the mean±s.d. (n=5 each). *P<0.05, **P<0.01 versus control, †P<0.05, ††P<0.01 versus EC (−).
Effect of Nicotine on Protein Kinase C Activity in Canine Basilar Arteries
Protein kinase C activities in canine basilar arteries were measured in the following conditions. In the UTP-stimulation study, arterial rings were divided into four groups (Non, UTP, nicotine/UTP, and nicotine/UTP/calph. C) as described in Materials and methods, and Protein Kinase C activities in each group were 11.5%±4.0% in the Non group, 38.7%±9.0% in the UTP group, 66.7%±12.0% in the nicotine/UTP group, and 23.9%±6.4% in the nicotine/UTP/calph. C group (n=6 each, Figure 3). Protein Kinase C activity in the UTP group was significantly higher than that in the Non group (P<0.01). Furthermore, PKC activity in the nicotine/UTP group was significantly greater than those in the Non and UTP groups, respectively (P<0.01 each). Protein kinase C activity in the nicotine/UTP/calph. C group was significantly inhibited compared with that in the nicotine/UTP group.
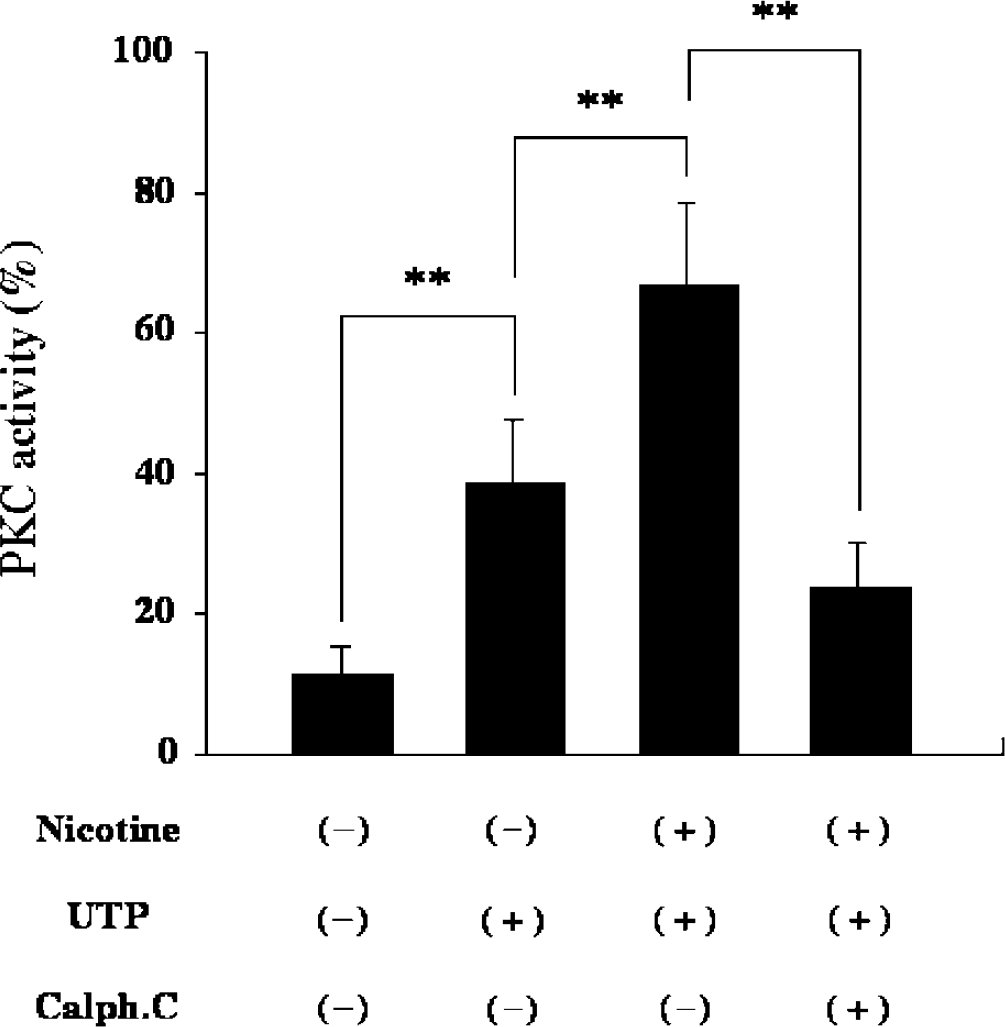
Protein kinase C (PKC) activity in canine basilar arteries. Canine basilar arteries with endothelium were incubated without or with nicotine and/or uridine 5′-triphosphate (UTP) and/or calphostin C in the manner described in Materials and methods. The PKC activities in the membrane fraction were measured by an enzyme immunoassay, and expressed as a percentage of the activity in the cytosol fraction of the cerebellum (=100%). The data were expressed as the mean±s.d. (n=6 each). **P<0.01 between the groups.
In the following experiment (nicotine-treatment study), it is examined whether nicotine itself enhances PKC activity. Protein kinase C activity in the artery incubated in the absence of nicotine was 8.7%±2.8% (n=4). Protein kinase C activities in the artery incubated with each concentration of nicotine were as follows: 9.8%±1.9% at 10−8 mol/L, 17.1%±2.1% at 10−7 mol/L, and 21.6%±2.8% at 10−6 mol/L, respectively (n=4 each). Protein kinase C activity in the artery incubated at 10−8 mol/L of nicotine was not significantly different from that incubated without nicotine. However, PKC activities in the artery incubated with 10−7 and 10−6 mol/L of nicotine were significantly enhanced compared with the activity in the artery incubated without nicotine, respectively (P<0.05 at 10−7 mol/L, P<0.01 at 10−6 mol/L, Figure 4).
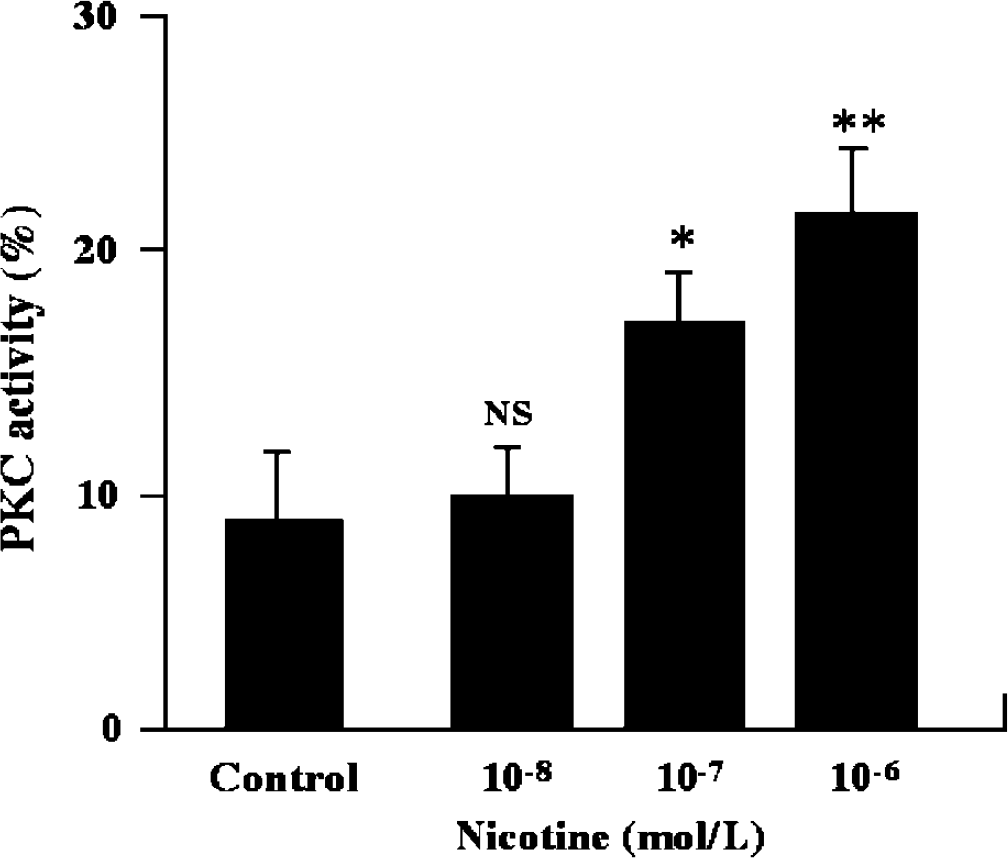
Effect of nicotine exposure on protein kinase C (PKC) activity in canine basilar arteries. Canine basilar arteries with endothelium were incubated without or with nicotine for 1 hour. The PKC activities in the membrane fraction were measured by an enzyme immunoassay, and expressed as a percentage of the activity in the cytosol fraction of the cerebellum (=100%). The data were expressed as the mean±s.d. (n=4 each). Control: incubated without nicotine. NS: not statistically significant. *P<0.05, **P<0.01 versus control.
In the de-endothelialization study, PKC activities in the de-endothelialized artery were measured. The effect of nicotine on Protein kinase C activity in the de-endothelialized arteries was also examined. Protein kinase C activity in the artery with endothelium was 9.0%±2.5% (n=4). In the de-endothelialized artery, PKC activity was 16.6%±1.3% (n=4). Protein kinase C activity was significantly enhanced by de-endothelialization itself (P<0.05). Nicotine exposure to the de-endothelialized artery further increased PKC activity to 24.3%±1.4% (n=4, P<0.01). Summarized data are shown in Figure 5.
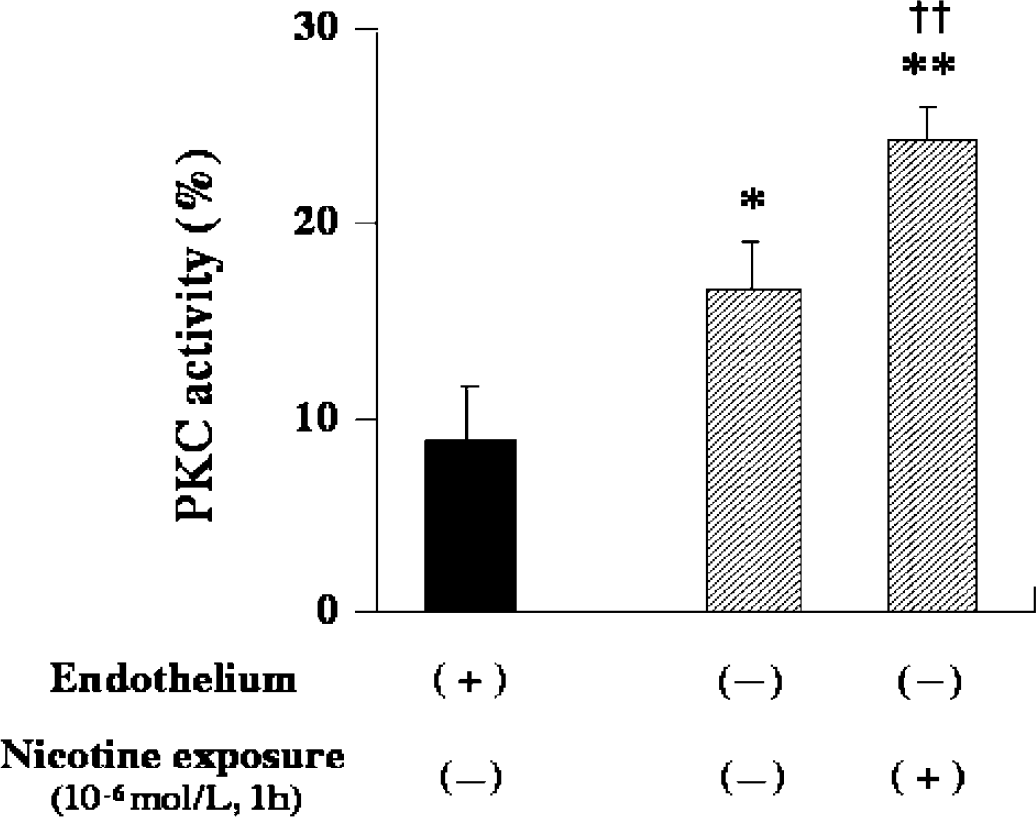
Effect of nicotine exposure and/or endothelium removal on protein kinase C (PKC) activity in canine basilar arteries. Canine basilar arteries with (closed columns) or without endothelium (shaded columns) were exposed to nicotine (10−6 mol/L) for 1 hour. The PKC activities in the membrane fraction were measured by an enzyme immunoassay, and expressed as a percentage of the activity in the cytosol fraction of the cerebellum (=100%). The data were expressed as the mean±s.d. (n=4 each). *P<0.05, **P<0.01 versus PKC activity in the artery with endothelium, without nicotine exposure. ††P<0.01 versus PKC activity in the artery without endothelium, without nicotine exposure.
Effect of Nicotine on Substance P-induced Vasodilatation in Canine Basilar Arteries
To obtain the equivalent precontraction, arterial rings were contracted by 3 × 10−3 mol/L of UTP in the control group, and 1 × 10−4 mol/L of UTP in the nicotine-treated group. The obtained pre-contractions were 77.9%±10.3% in the control group and 71.9%±13.6% in the nicotine-treated group (n=5 each, high K+-induced tension=100%). There was no significant difference between the two groups. After the arterial ring was precontracted by UTP, substance P was cumulatively applied to the arterial ring. The artery showed dose-dependent dilatation by substance P, and the magnitudes of the dilatation were as follows: 7.8%±5.5% at 10−8 mol/L, 31.3%±4.7% at 10−7 mol/L, and 55.2%±5.9% at 10−6 mol/L in the control group (n=5). For the nicotine-pretreated group, the magnitudes of substance P-induced vasodilatation on UTP-induced precontraction were as follows: 5.2%±1.6% at 10−8 mol/L, 17.4%±2.1% at 10−7 mol/L, and 29.5%±3.2% at 10−6 mol/L (n=5). Substance P-induced vasodilatation in the nicotine-treated group was significantly attenuated (P<0.05 at 10−7 mol/L, P<0.01 at 10−6 mol/L). The dose–response curves of substance P-induced vasodilatation are shown in Figure 6.
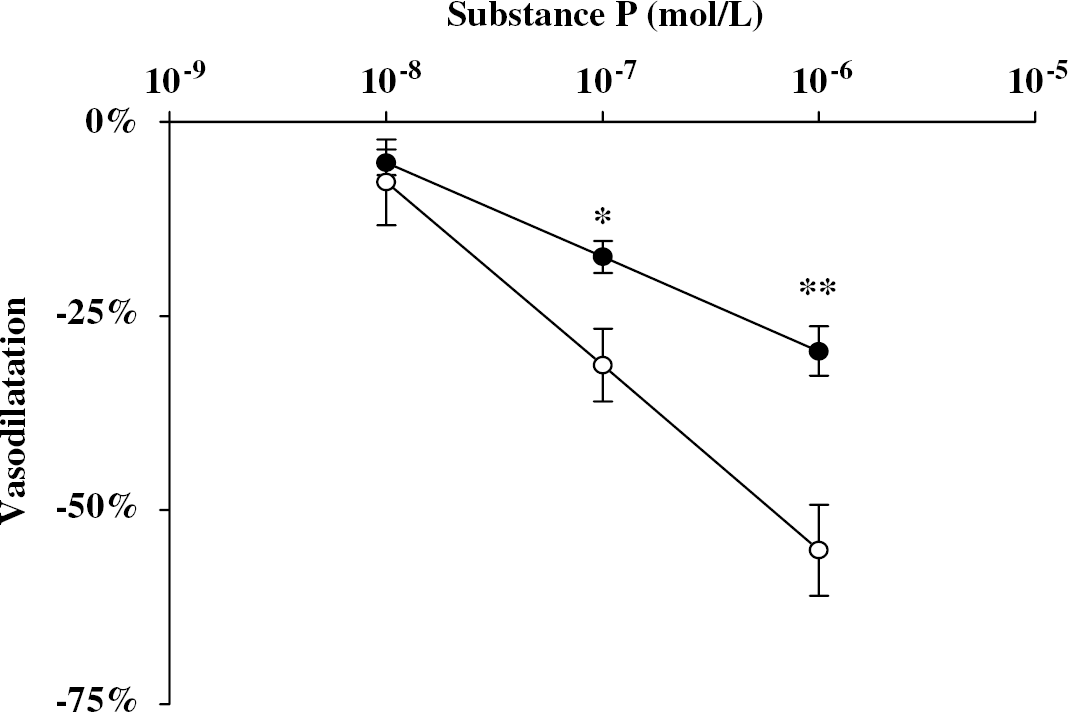
Effect of nicotine exposure on substance P-induced vasodilatation in canine basilar artery. The artery was incubated without (open circle) or with (closed circle) nicotine (10−6 mol/L) for 1 hour before the measurement of SP-induced vasodilatation. After the arterial ring was precontracted with uridine 5′-triphosphate (UTP), substance P was applied cumulatively. The value of substance P-induced vasodilatation was expressed as a percentage of the papaverine-induced maximal relaxation (=100%). The data were expressed as the mean±s.d. (n=5). *P<0.05, **P<0.01 versus the value of the group without nicotine treatment.
Sensitivity of the Smooth Muscle to Nitric Oxide After Nicotine Administration
To assess the sensitivity of the smooth muscle to NO after nicotine administration, NO donor (NOC 7 or NOR 1)-induced vasodilatations were measured in de-endothelialized artery. De-endothelialization was confirmed by the application of substance P on UTP-induced precontraction. Using other de-endothelialized arterial rings, precontractions by UTP at 3 × 10−4 mol/L in the control group and at 1 × 10−4 mol/L in the nicotine-treated group were obtained. The magnitudes of precontraction in each group were made equivalent. The obtained contractions were 76.7%±15.5% in the control group and 80.6%±11.5% in the nicotine-treated group (n=8 each, high K+-induced tension =100%), and there was no significant difference between the two groups. NOC 7 or NOR 1 was cumulatively applied on the precontraction by UTP in the absence (control group) or presence (nicotine-treated group) of nicotine, and arterial rings dilated dose-dependently. There were no statistically significant differences between control/NOC 7 and nicotine-treated/NOC 7, and between control/NOR 1 and nicotine-treated/NOR 1. After the application of the maximal concentrated NOC 7, the pH of PSS in the chamber was 7.2 to 7.5. The dose–response curves with NOC 7-induced and NOR 1-induced vasodilatation are shown in Figure 7.
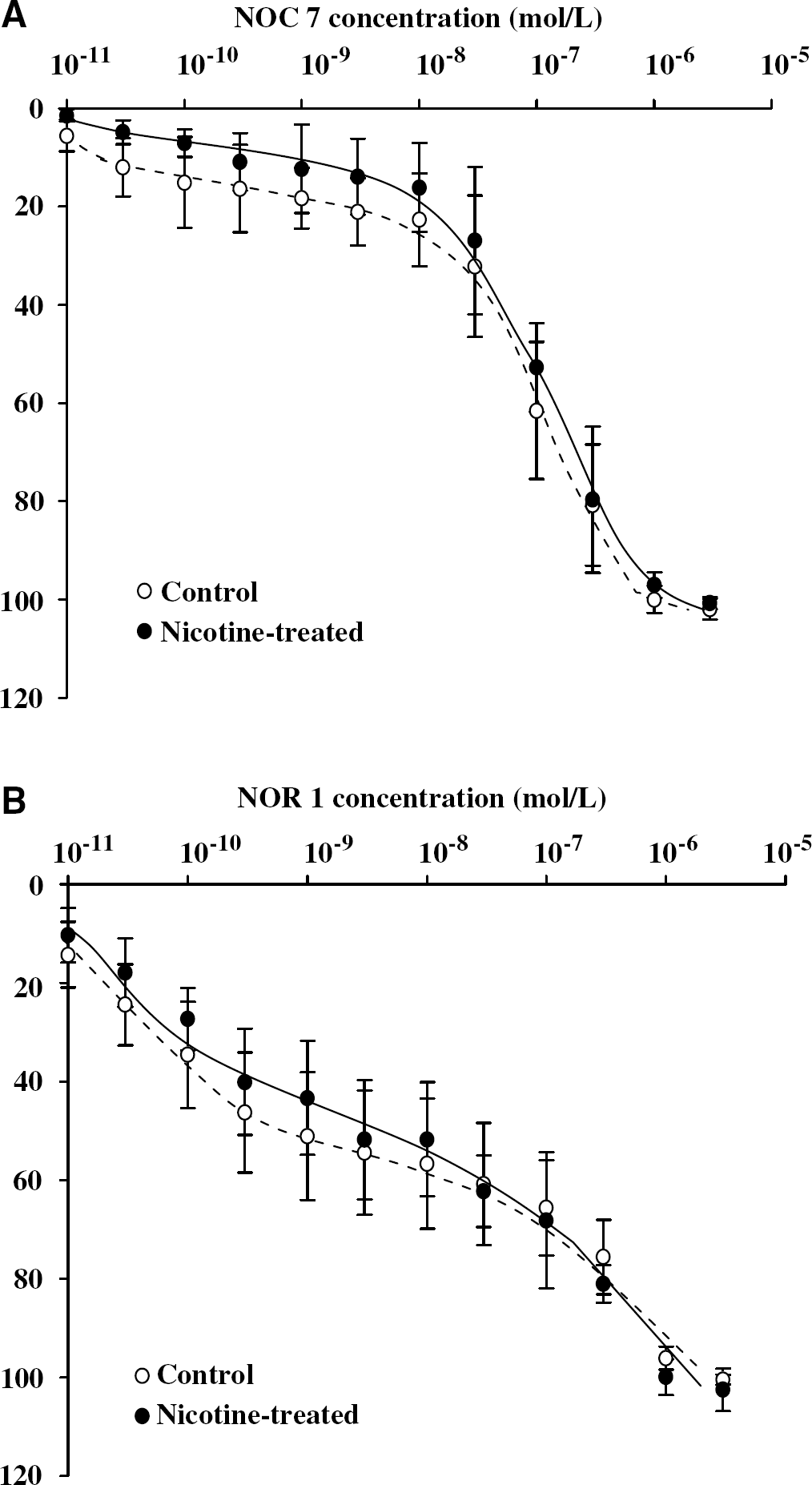
Effect of nicotine treatment on the sensitivity of the smooth muscle to nitric oxide (NO). The de-endothelialized arteries were incubated without (control, open circle) or with (closed circle) nicotine (10−6 mol/L) for 1 hour, and then NO donors, NOC 7 (
Effect of Nicotine on Nitric Oxide Synthesis in Vascular Endothelial Cells
The cell morphology of VECs in aCSF was observed with differential interference contrast microscopy (Axiovert 10, Carl Zeiss, Oberkochen, Germany), and no change was detected for 30 mins (data not shown).
At first, the fluorescent intensity of VECs without any simulation was examined, and no changes were observed at 5 and 10 mins after the start of recording. The fluorescence intensity in VECs without stimulation was 113.0%±4.9% at 5 mins, and 112.6%±4.8% at 10 mins (n=5), and there was no statistically significant difference between the intensities at 5 mins and 10 mins.
Figure 8A shows the change of fluorescence intensity in VECs after stimulation with substance P. In the VECs without nicotine pretreatment, the fluorescence intensities after substance P stimulation showed a significant increase and their values were as follows: 164.0%±3.4% at 5 mins and 207.3%±5.0% at 10 mins after (n=14, Figure 8B). Between the intensities at 5 and 10 mins, there was a statistically significant difference (P<0.01). For the nicotine-treated VECs, the fluorescence intensities were also increased by substance P stimulation. The values of fluorescence intensity at 5 and 10 mins were 123.1%±3.0% and 147.3%±4.7%, respectively (n=11, Figure 8B). There was a statistically significant difference between the intensities at 5 mins and 10 mins (P<0.01). Comparing the values of fluorescence intensity between the groups without or with nicotine treatment, differences at both 5 and 10 mins were statistically significant (P<0.01 each).
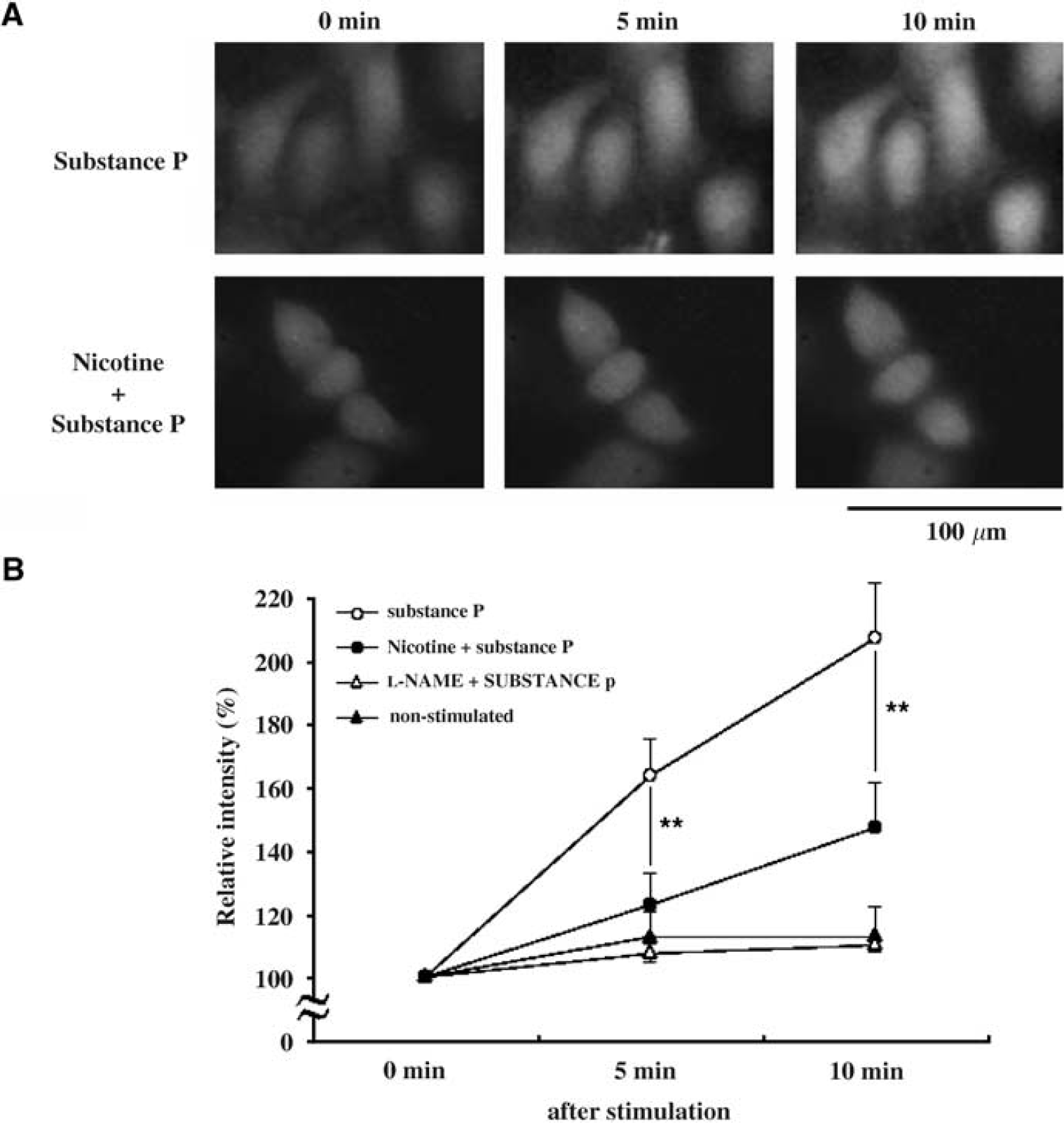
Effect of nicotine exposure on substance P-induced nitric oxide (NO) synthesis in primary human microvascular endothelial cells (VECs). (
In the presence of L-NAME, substance P stimulation did not increase the fluorescence intensity in VECs, and the values were 107.6%±0.5% at 5 mins and 110.1%±0.6% at 10 mins (n=7).
Discussion
Effects of Nicotine on Uridine 5′-Triphosphate-Induced Arterial Contraction and Protein Kinase C Activity
According to previous reports in vivo, the effects of nicotine on regulation of cerebral arterial tone have been controversial (Jackson, 1993; Cruickshank et al, 1989; Iida et al, 1998). The aim of this study was to clarify the direct pharmacological effects of nicotine, mimicked smoking, on cerebral arterial tone and endothelial function using an isolated canine basilar artery and cultured VECs. To mimic smoking, nicotine was treated at low concentration based on the serum concentration level of nicotine in habitual cigarette smokers (Armitage et al, 1975), and for 1 hour in the isometric tension study using canine arterial rings and for 24 hours in the study using VECs. Nicotine exposure itself did not affect resting tension in the isometric tension study; however, it significantly enhanced UTP-induced vasoconstriction in the endothelium-intact artery (Figure 1). These results indicate that nicotine exposure itself has no arterial contraction effect in the canine basilar artery with endothelium, but potentiates the arterial contractility.
We have previously shown that PKC plays a pivotal role in the regulation of cerebral arterial tone (Nishizawa et al, 1992a, 1992b, 1996) in the canine basilar artery. It was reported that UTP-induced contraction involves the activation of PKC in rat aorta (Lopez et al, 2000). We hypothesized that PKC activation is involved in this contractile potentiation by nicotine, and PKC activities in the artery were actually measured. Uridine 5′-triphosphate significantly increased PKC activity in the canine basilar artery (Figure 3). Uridine 5′-triphosphate further incteased PKC activity in the nicotine-treated artery, and this enhancement was inhibited by calphostin C (Figure 3). These results suggest that nicotine enhances UTP-induced contraction by PKC activation. Besides, nicotine dose-dependently increased PKC activities, although it did not induce arterial contraction in the canine basilar artery (Figure 4). That is, a low concentration of nicotine, such as in smoking, would make the artery prone to contract via PKC activation.
Effects of Nicotine on Endothelial Function and Protein Kinase C activity
It was reported that nicotine infusion attenuated endothelium-dependent vasodilatation in vivo (Mayhan and Patel, 1997), suggesting that endothelial dysfunction is caused by nicotine. We examined the effect of nicotine on endothelium-dependent vasodilatation using an isolated canine basilar artery. The isolated artery showed dose-dependent dilatation by substance P; however, the magnitudes of the dilatation were significantly attenuated in the nicotine-treated artery (Figure 6). Because NO plays a role in endothelium-dependent vasodilatation, we visualized and quantified NO synthesis using DAF-FM/DA in cultured VECs derived from the cerebral artery. Substance P significantly increased the fluorescence intensity of DAF-FM/DA in VECs (Figure 8). Such substance P-induced enhancement of fluorescence intensity was abolished by the pretreatment and the presence of L-NAME, that is the substance P-induced increment of fluorescence intensity showed endogenous NO synthesis through activation of NO synthase. In nicotine-treated VECs, substance P also increased fluorescence intensity; however, the magnitude of this increase was significantly attenuated (Figure 8). A low concentration of nicotine, mimicked smoking, diminished NO synthesis in VECs. Besides, we assessed whether the sensitivity of the smooth muscle to NO was altered following nicotine administration. Using NO donors, NO-induced relaxations of smooth muscle were measured in de-endothelialized artery without or with nicotine treatment. There was no significant difference of NO-induced relaxation between the control group and the nicotine-treated group, with the result that the sensitivity of the smooth muscle to NO was not changed by nicotine treatment (Figure 7). These results show that nicotine causes endothelial dysfunction, resulting in the attenuation of endothelium-dependent vasodilatation. This is consistent with our pre-noted data about endothelium-dependent vasodilatation and previous papers (Tsuchiya et al, 2002; Miller et al, 2000).
We previously reported the close relationship between endothelial function and PKC activation in the regulation of cerebral arterial tone (Nishizawa et al, 1996). Endothelial dysfunction enhanced PKC activity in the canine basilar artery (Nishizawa et al, 1997, 1998). We examined the relationship between nicotine-caused endothelial dysfunction and nicotine-induced PKC activation. In this study, PKC activity was significantly increased by de-endothelialization itself, and was further enhanced by nicotine even in the de-endothelialized artery (Figure 5). These results show that nicotine enhances PKC activity not only by endothelial dysfunction but also directly. In addition, we also showed that UTP-induced contraction in the de-endothelialized artery was significantly greater than that in the endothelium-intact artery, and nicotine caused the further potentiation of UTP-induced vasoconstriction in the de-endothelialized artery (Figure 2). These results also support our investigation that nicotine enhanced UTP-induced vasoconstriction via PKC activation not only by endothelial dysfunction but also directly.
Effects of Nicotine on the Regulation of Arterial Tone
Figure 9 schematically denotes a possible mechanism inducing contractile potentiation by nicotine exposure, mimicked smoking. The serum concentration of nicotine increases with by smoking. The nicotine causes endothelium dysfunction, with a decrease in NO synthesis and an increase in endothelium-dependent vasodilatation. Besides, nicotine enhances PKC activity by endothelial dysfunction and directly.
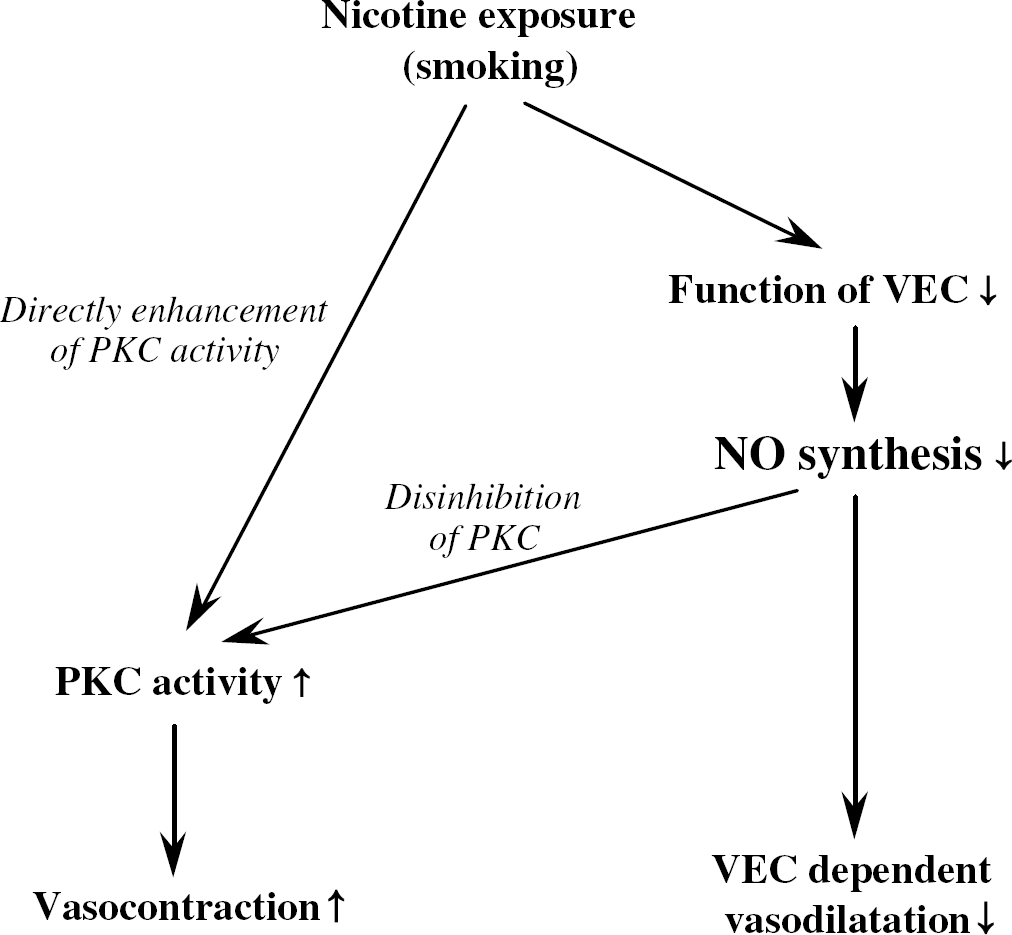
Schematic representation showing a possible mechanism inducing contractile potentiation by nicotine exposure, mimicked smoking. The serum concentration of nicotine increases with smoking. Nicotine causes dysfunction of vascular endothelial cells (VECs), with a decrease in nitric oxide (NO) synthesis in VEC and an increase in endothelium-dependent vasodilatation. Besides, nicotine enhances protein kinase C (PKC) activity via endothelial dysfunction and directly. These enhancements of PKC activity potentiate contractile response in the artery.
In conclusion, the present experimental data indicate that nicotine itself at the same concentration as reported in smokers does not cause cerebral arterial contraction per se, but significantly impairs endothelial function and enhances PKC activity, resulting in the artery being prone to constriction.