
Abstract
The present study was designed to determine whether relaxations induced by hypercapnia depend upon nitric oxide (NO) derived from the endothelium, and whether NO-mediated relaxant response to electrical and chemical stimulation of vasodilator nerves is modulated by hypercapnia. In canine and monkey cerebral arterial strips contracted with K+, raising the level of CO2 of the aerating gas in the bathing media from 5 to 10% produced a moderate relaxation, together with an increased Pco2 (from 29.8 to 59.3 mm Hg) and a decreased pH (from 7.43 to 7.15). Relaxation was not influenced by endothelium denudation and treatment with NG-nitro-L-arginine. Contractions elicited by the NO synthase inhibitor were attenuated by the removal of the endothelium. Relaxations, caused by transmural electrical stimulation and nicotine, of canine cerebral arterial strips contracted with prostaglandin F2α were potentiated only slightly by hypercapnia, but the potentiation of the response to exogenous NO (acidified NaNO2) was clearly greater. It is concluded that as far as the arteries used are concerned, hypercapnia does not seem to liberate NO from the endothelium but does potentiate the effect of NO. The reason for lesser potentiation, by hypercapnia, of the response to nitroxidergic nerve stimulation than to NO action may be associated with an impairment by intracellular acidosis of NO synthase activation.
Hypercapnia has long been recognized as acting to vasodilate cerebral arteries and arterioles and to increase cerebral blood flow. Despite the effort of numerous investigators, the mechanism of these actions has not been clarified. Endothelium-derived relaxing factor (EDRF) and nitric oxide (NO) are widely recognized as physiologically important vasodilators in the vasculature, including cerebral arteries, of a variety of mammals (Kontos et al., 1988; Mayhan et al., 1990; Rosenblum and Nelson, 1988; Toda, 1990; Toda and Okamura, 1993; Whalley et al., 1987). NO, derived from perivascular nerves, also participates in the cerebroarterial dilatation in subprimate and primate mammals (Toda and Okamura, 1990a,b; Toda, 1993, 1995). Attention is currently directed to the role of NO in hypercapnia-induced vasodilation. Many in vivo studies have provided evidence that NO is involved in vasodilatation (Buchanan and Phillis, 1993; Dirnagl et al., 1993; Iadecola, 1992; Niwa et al., 1993; Pelligrinoet al., 1993; Wang et al., 1992); however, some investigations do not support this idea (Adachi et al., 1992; Goadsby and Hoskin, 1993). On the other hand, we did not find a dependence on the endothelium of the hypercapnia (15% CO2)-induced vasodilatation in isolated canine and monkey cerebral arteries (Toda et al., 1989, 1993a). The discrepancy between most in vivo studies and our in vitro study may be attributable to the degree of hypercapnia (50–60 mm Hg versus > 100 mm Hg Pco2, respectively), an idea postulated by Iadecola and Zhang (1994). Another explanation for the discrepancy might be the different degrees of functioning of the vasodilator nerves in which NO is involved as a neurotransmitter: There is no release of NO from the nerve, unless artificially stimulated, in isolated tissues, whereas a significant NO release is expected to be elicited in vivo on account of tonic efferent discharges from the brain (Toda et al., 1993d); vascular tone is controlled by the action of endogenous NO. Nerve-derived NO would be hypothesized to participate partially in hypercapnia-induced cerebral vasodilatation in vivo if an NO-mediated neurogenic response were proven to be potentiated by hypercapnic acidosis.
The present study was undertaken to determine whether the relaxation caused by moderate hypercapnia (∼50 mm Hg Pco2), induced by raising CO2 in the aerating gas from 5 to 10%, is mediated by NO from the endothelium in isolated monkey and canine cerebral arteries, and to examine the influence of hypercapnia on the cerebral arterial relaxation elicited by perivascular nerve stimulation, which liberates NO as a neurotransmitter in response to electrical pulses or nicotine (Toda and Okamura, 1990a,b).
METHODS
Japanese monkeys (Macaca fuscata) of both sexes, weighing 5–11 kg, were anesthetized with intramuscular injections of ketamine (40 mg/kg) and sodium thiopental (20 mg/kg) and killed by bleeding from the carotid arteries. The brain was then removed rapidly, and basilar and middle cerebral arteries (0.5–0.7 mm O.D.) were isolated. Mongrel dogs of both sexes, weighing 8–13 kg, anesthetized with intravenous injections of sodium thiopenthal (30 mg/kg), were killed by bleeding, and basilar and middle cerebral arteries were isolated from the brain. Arteries were cut into helical strips ∼20 mm long. Each arterial strip was fixed vertically between hooks in a muscle bath containing modified Ringer-Locke solution, which was maintained at 37 ± 0.3°C and aerated with a mixture of 5% CO2 and 95% O2. The hook anchoring the upper end of the strips was connected to the lever of a force-displacement transducer. Resting tensions were adjusted to 1.0 g for monkey arteries and 1.5 g for canine arteries; these tensions are optimal for inducing maximal contraction. Constituents of the solution were as follows (mM): NaCl, 120; KCl, 5.4; CaCl2, 2.2; MgCl2, 1.0; NaHCO3, 25.0; and dextrose, 5.6. Pco2 and Po2 in the bathing media were measured with a medical mass-spectrometer (Medspect II, Chemetron, Corp., St. Louis, MO, U.S.A.) and the pH was measured with a pH meter (H-7AB, Horiba Seisaku-sho, Kyoto, Japan). Before the start of experiments, strips were allowed to equilibrate for 90–120 min, during which time fluids were replaced every 10–15 min.
Isometric contractions and relaxations were recorded on an ink-writing oscillograph. The contractile response to 30 mM K+ was obtained first, and the strips rinsed repeatedly. Only one strip per dog per individual type of experiment was used. For experiments on hypercapnia-induced relaxation in monkey and canine cerebral arteries, arterial preparations were contracted with 20 mM K+. After the tension was stabilized, normal gas (5% CO2 and 95% O2) was replaced with a high CO2 gas (10% CO2 and 90% O2). For experiments on vasodilator nerve stimulation, canine cerebral arterial strips were placed between stimulating electrodes and transmurally stimulated by 0.2 ms electrical square pulses at a frequency of 5 Hz for 40 s. In order to stimulate the nerve chemically, we used nicotine in a submaximal concentration of 10−4 M (Toda and Okamura, 1991). This concentration of nicotine along with 10−7 M NO (acidified NaNO2) were applied successively to the bathing media. At the end, papaverine (10−4 M) was added to obtain maximal relaxation. Responses to transmural electrical stimulation, nicotine, and NO (acidified NaNO2) in prostaglandin (PG)F2α-contracted arterial strips exposed to control media were compared with those obtained at 30 min of exposure to hypercapnic media and at 30 min after returning to normocapnia. Relaxations induced by hypercapnia, and those by nerve stimulation and NO (acidified NaNO2) relative to those induced by 10−4 M papaverine were presented. From one of a pair of cerebral arterial strips obtained from the same monkey or dog, the endothelium was removed by gently rubbing the intimal surface with a cotton ball. Endothelial integrity was determined pharmacologically by the use of Ca2+ ionophore A23187, for the monkey artery (Toda et al., 1991), and substance P, for the canine artery treated with indomethacin (Onoue et al., 1988), in all of the strips used.
Results shown in the text, table, and figures are expressed as mean ± SD. Statistical analyses were made using Student's paired and unpaired t-tests for two groups and Tukey's method after one-way analysis of variance (ANOVA) for more than two groups. Drugs used were nicotine (base), hexamethonium bromide, L-arginine (Na-calai Tesque Ltd., Kyoto, Japan), NG-nitro-L-arginine (L-NA; Peptide Institute, Minoh, Japan), indomethacin (Sigma Chemical Co., St. Louis, MO, U.S.A.), beraprost (Toray Co., Tokyo, Japan), tetrodotoxin (Sankyo Co., Tokyo, Japan), PGF2α (Upjohn, Tokyo), Ca2+ ionophore A23187 (Boehringer Ingelheim Ltd., Elmsford, NY, U.S.A.), and papaverine hydrochloride (Dainippon Co., Osaka, Japan). Responses to NO were obtained by adding the NaNO2 solution adjusted to pH 2 (Furchgott, 1988). The same volume (20 μl) of the vehicle (pH 2) alone did not affect vascular tone.
RESULTS
Hypercapnia-induced relaxation in monkey and canine cerebral arteries
In helical strips of the monkey cerebral artery partially contracted with 20 mM K+, changes in the aerating gas from 5% CO2 and 95% O2 (normocapnia) to 10% CO2 and 90% O2 (hypercapnia) produced a slowly-developing relaxation that leveled off 10–20 min later, in association with an increase of Pco2 and a decrease of pH (Table 1). Reduction of CO2 to 5% restored the tension to a level approximately the same as that before application of high CO2 (Fig. 1). Responses to hypercapnia were reproducible. After the second response was determined to be identical to the first one, strips were treated with 10−5 M L-NA, which produced slight or moderate contractions (392 ± 138 mg, n = 7). Under treatment with the NO synthase inhibitor, the relaxation induced by hypercapnia was not attenuated, as compared with that in the control media (Fig. 1, upper tracing). L-arginine (3 × 10 −3 M) transiently increased the tension and then produced a relaxation that was stabilized to the level almost the same as that before addition of L-NA (393 ± 146 mg relaxation from L-NA-induced contraction). Response to hypercapnia was unaffected. Quantitative data are summarized in Fig. 2, upper panel. Increase in the concentration of L-NA to 10−4 M did not produce an additional contraction in the arterial strips nor did it inhibit the response to hypercapnia (n = 4).
Mean values of the pH, Pco2, Po2 and HCO3 in normocapnic and hypercapnic media
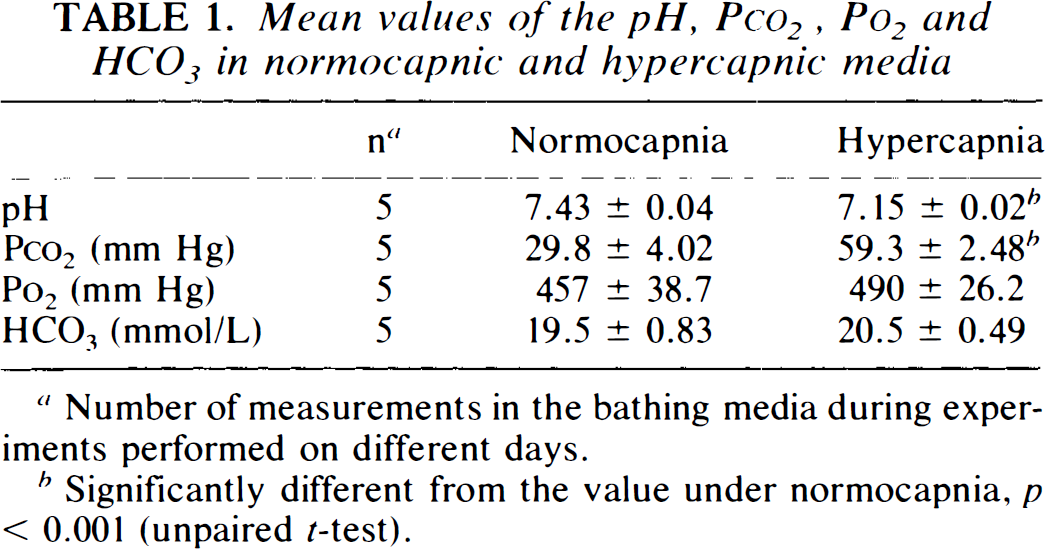
Number of measurements in the bathing media during experiments performed on different days.
Significantly different from the value under normocapnia, p < 0.001 (unpaired t-test).
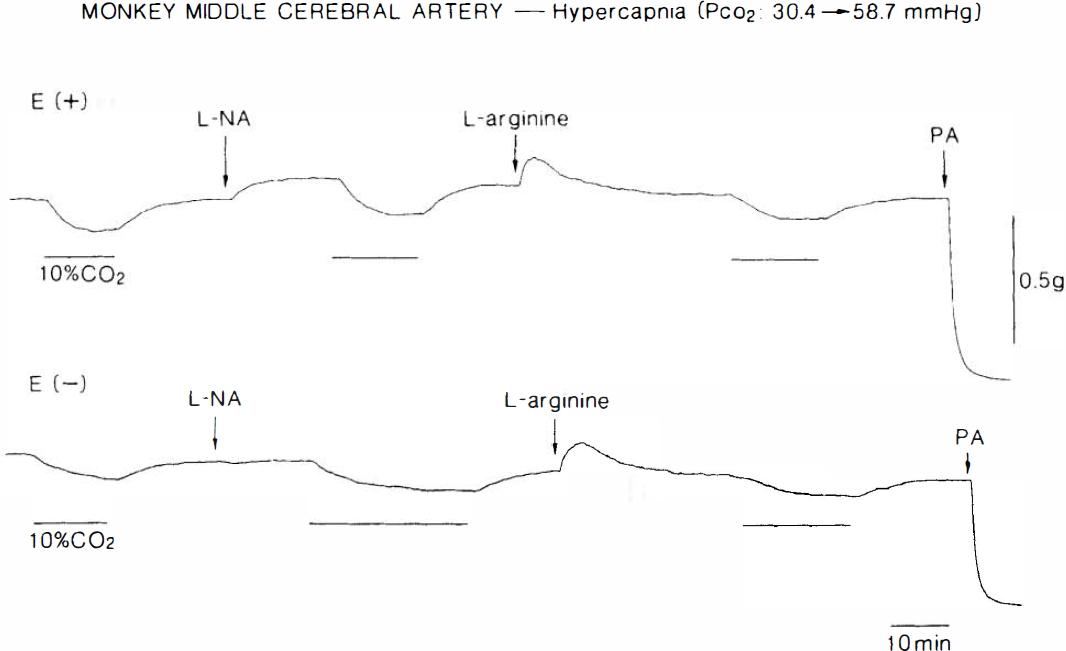
Typical recordings of the response to hypercapnia of monkey middle cerebral arterial strips with (E+) and without the endothelium (E-), partially contracted with 20 mM K+. Strips were obtained from the same monkey. Hypercapnia (10% CO2), applied at the horizontal lines shown under the tracings, produced relaxations. Concentrations of L-NA and L-arginine were 10−5 and 3 × 10−3 M, respectively. PA represents 10−4 M papaverine, which elicited the maximal relaxation.
Hypercapnia-induced relaxation was not influenced by removal of the endothelium (Figs. 1 and 2). Endothelial integrity was confirmed by the relaxation caused by 10−8 M Ca2+ ionophore A23187; mean values in endothelium-intact and -damaged strips were 78.0 ± 17.2 and 3.7 ± 6.1% (n = 7; p < 0.001, unpaired t-test), respectively, relative to papaverine (10−4 M)-induced relaxation. In endothelium-denuded arteries, the response to hypercapnia was not affected by L-NA and L-arginine (Fig. 1). L-NA-induced contractions were significantly attenuated by endothelium denudation; mean values in endothelium-intact and -denuded strips were 392 ± 138 mg (21.1 ± 11.6% relative to contraction caused by 30 mM K +, n = 7) and 244 ± 138 mg (8.0 ± 7.4%, n = 7; p < 0.05, unpaired t-test), respectively.
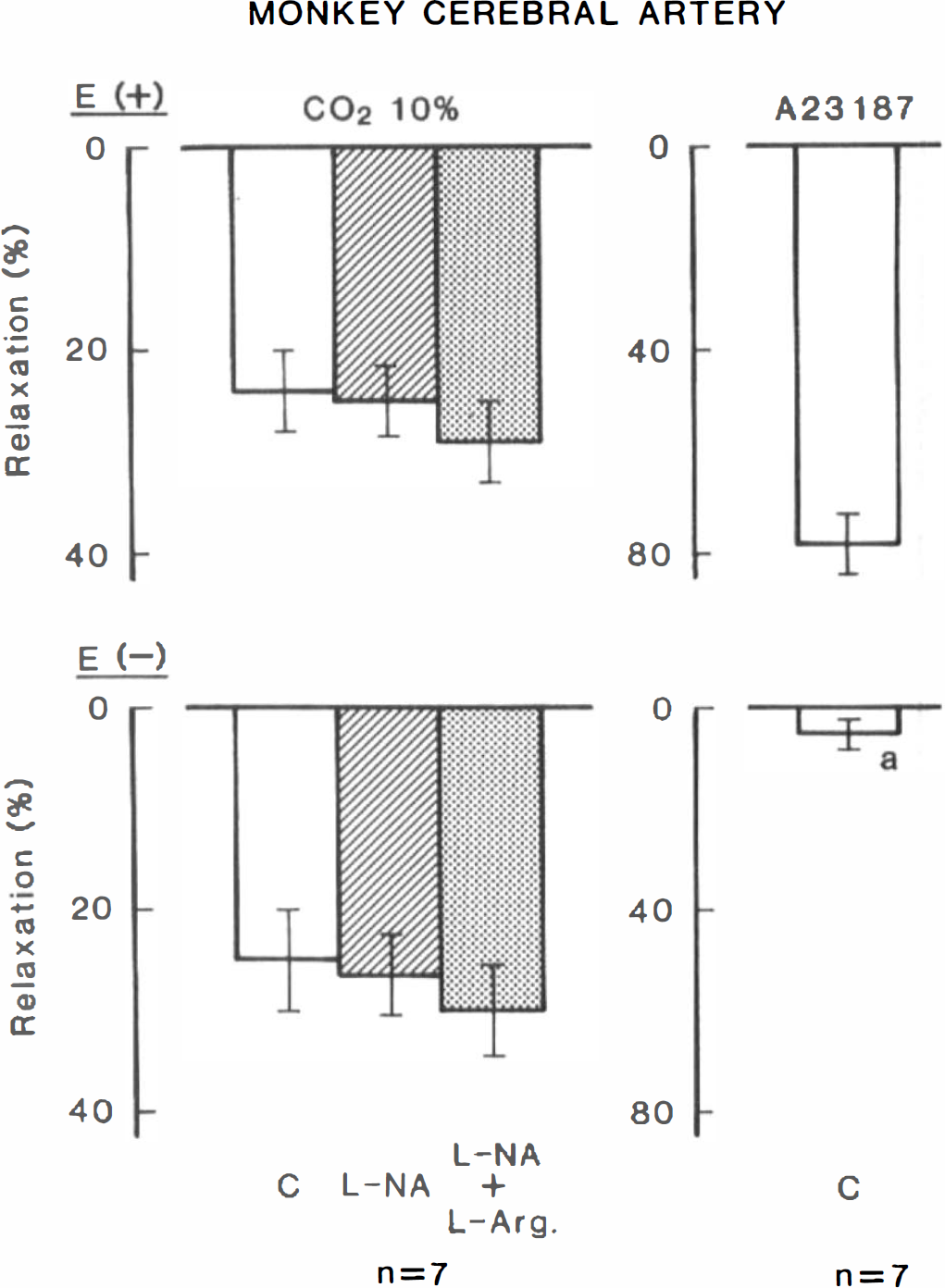
Modifications by L-NA (10−5 M), L-arginine (3 × 10−3 M) and endothelium denudation (E -) of the relaxant response to hypercapnia in monkey cerebral arterial strips contracted with 20 mM K+. Strips with and without the endothelium were obtained from the same monkeys (n = 7). Panels on the right illustrate the response to 10−8 M Ca2+ ionophore A23187, used to verify the presence of the endothelium. Relaxations induced by 10−4 M papaverine were taken as 100%. C, control; significantly different from the value in endothelium-intact strips, ap < 0.001 (unpaired t-test). Vertical bars represent SD.
In canine cerebral arterial strips contracted with K+, raising the concentration of CO2 in aerating gas from 5 to 10% produced a relaxation that was almost identical to that seen in monkey arteries. L-NA (10−5 M) contracted the arteries (552 ± 24.3 mg; 22.4 ± 12.9% relative to contraction by 30 mM K+), but did not inhibit the response to hypercapnia (Fig. 3). Indomethacin (10−6 M) also failed to reduce the response (n = 3). Endothelial denudation did not change the hypercapnia-induced relaxation (Fig. 3), although the L-NA-induced contraction was attenuated (376 ± 159 mg; 11.4 ± 6.3%, n = 9; p < 0.05, unpaired t-test). Endothelial functions were determined by the application of 10−8 M substance P in the presence of indomethacin; mean values in the control and de-endothelialized strips are shown in Fig. 3, left columns.
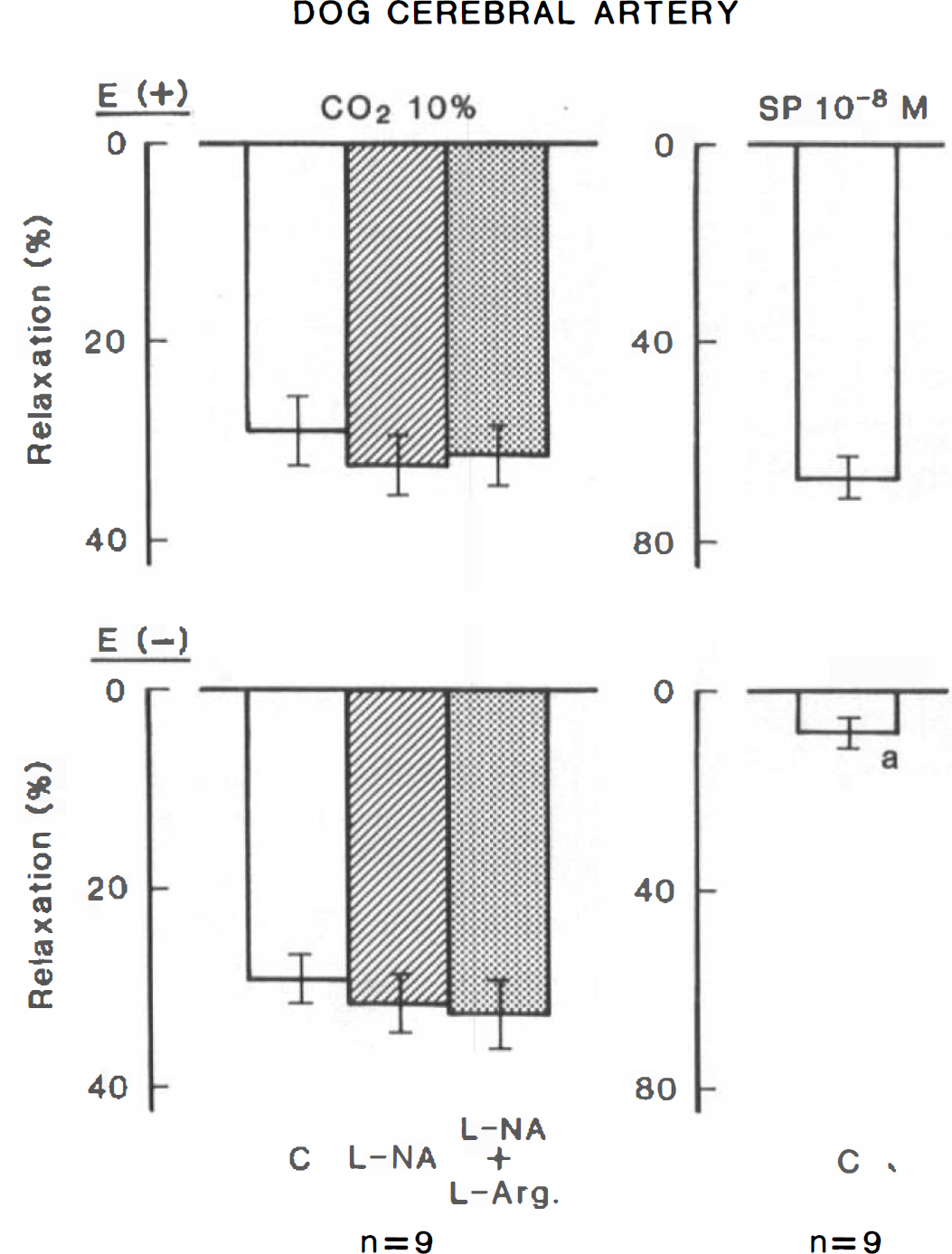
Modifications by L-NA (10 −5 M), L-arginine (3 × 10 −3 M) and endothelium denudation (E-) of the relaxant response to hypercapnia in canine cerebral arterial strips contracted with 20 mM K+. Strips with and without the endothelium were obtained from the same dogs (n = 9). Panels on the right illustrate the response to 10−8 M substance P (SP), used to verify the endothelial integrity. Relaxations induced by 10−4 M papaverine were taken as 100%. C, control; significantly different from the value in endothelium-intact strips, ap < 0.001 (unpaired ttest). Vertical bars represent SD.
Modification by hypercapnia of the response to vasodilator nerve stimulation
In canine cerebral arterial strips partially contracted with PGF2α, transmural electrical stimulation at 5 Hz for 40 s produced a transient relaxation that was abolished by tetrodotoxin (3 × 10−7 M) (Fig. 4) and L-NA (Toda and Okamura, 1991). Increase in the concentration of CO2 in aerating gas from 5 to 10% either did not alter or only slightly potentiated the response to nerve stimulation (Fig. 5). The differences in values for 5 and 10% CO2 are statistically significant when compared with the paired t-test (20.4 ± 21.7% increase, p < 0.05, n = 7).
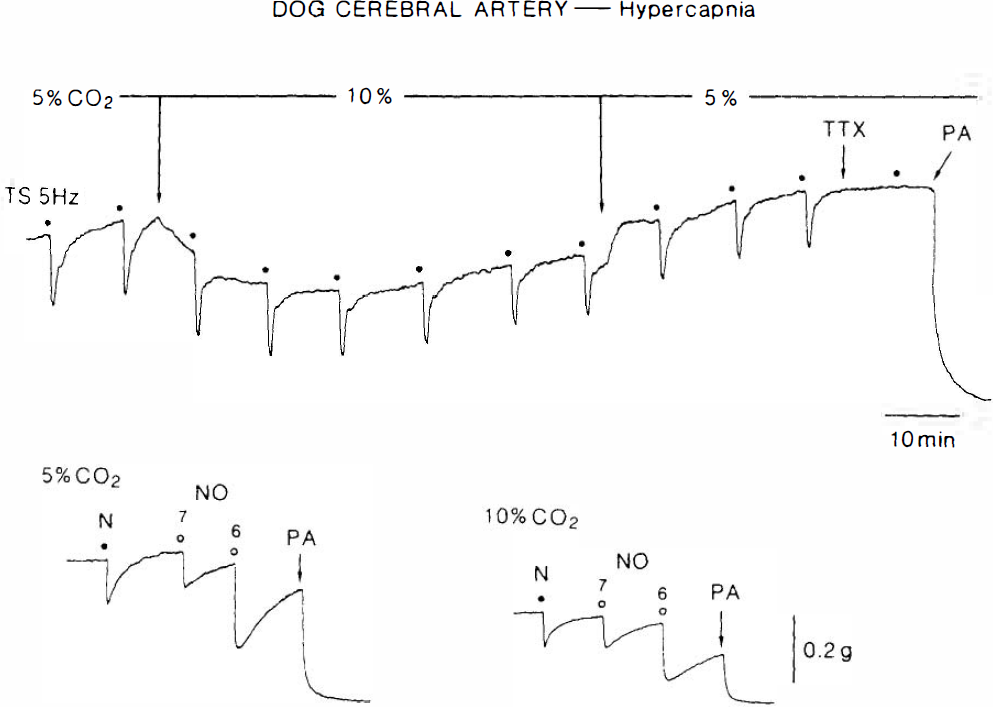
Actual recordings of the response to electrical nerve stimulation (5 Hz, upper tracing), nicotine (N) (10−4 M) and acidified NaNO2 (NO) (10−7 and 10−6 M) (lower tracing), as affected by hypercapnia (10% CO2) in canine middle cerebral arterial strips. Strips were contracted with PGF2α. TTX and PA represent 3 times; 10−7 M tetrodotoxin and 10−4 M papaverine, respectively.
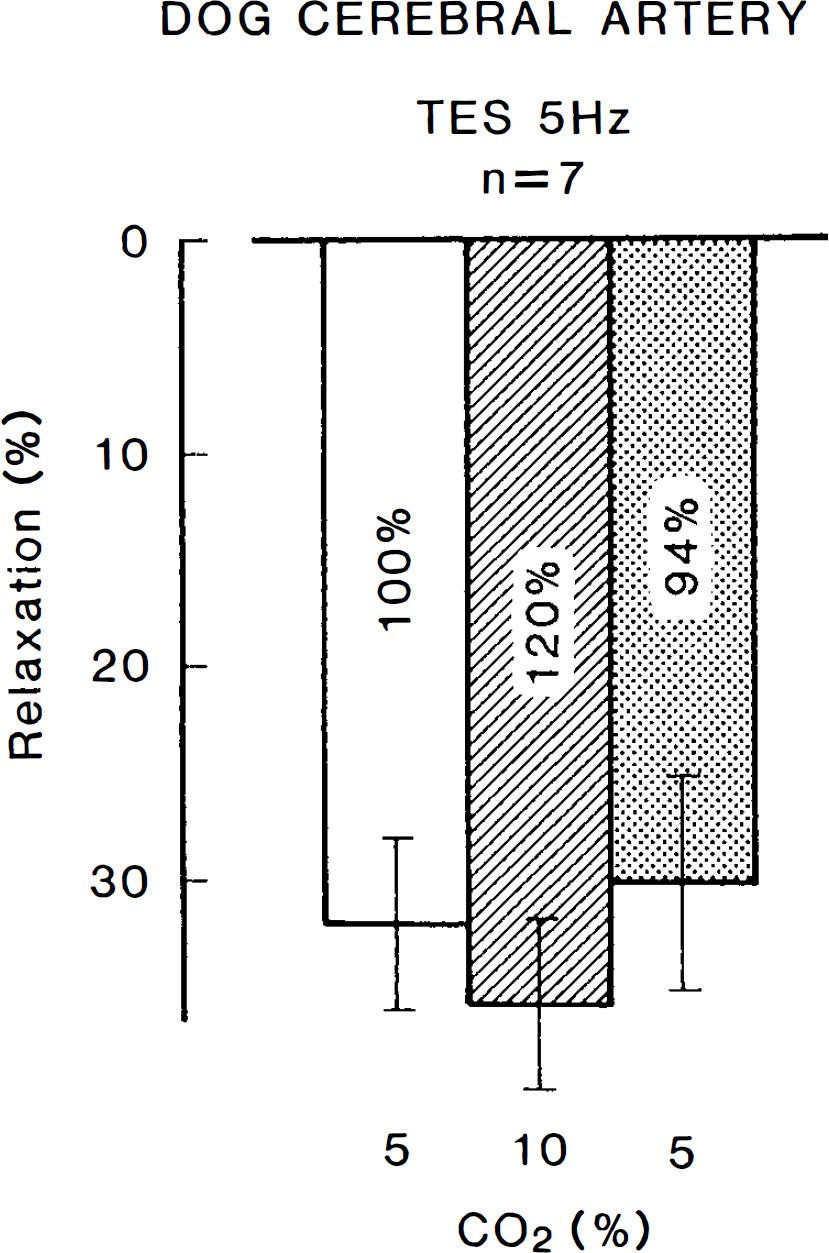
Modifications by hypercapnia (10% CO2) of the response to transmural electrical stimulation (TES) (5 Hz) in canine cerebral arterial strips contracted with PGF2α. Relaxations induced by 10−4 M papaverine were taken as 100%. The mean value during hypercapnia (120.4 ± 21.3%, n = 7) is significantly different (p < 0.05, paired t-test) from the value in control media (100%). Vertical bars represent SD.
In PGF2α-contracted strips, addition of nicotine (10−4 M) elicited a relaxation (Fig. 4) that was abolished by 10−5 M hexamethonium (n = 5) and by L-NA (Toda and Okamura, 1991). Hypercapnia increased the response to nicotine by 51.1 ± 58.3% (p < 0.05, n = 8, paired t-test), whereas the potentiation of the response to NO (acidified NaNO2, 10−7 M) was more evident (234.1 ± 224.9%, p < 0.02, n = 8); the difference in the potentiating effects was statistically significant (p < 0.05, unpaired t-test). Returning to normocapnia abolished the potentiation. Quantitative data are summarized in Fig. 6. The relaxant response to beraprost (10−8–10−6 M), a stable analog of PGI2, or to a low concentration of papaverine (10−6 M) was not significantly altered by hypercapnia; mean values of the relaxation at 10−7 M beraprost in normocapnia and hypercapnia were 34.0 ± 12.0 and 38.1 ± 7.3% (n = 6), respectively; those with papaverine were 47.8 ± 9.1 and 42.2 ± 10.0% (n = 6), respectively. These findings exclude the possibility of a nonspecific action of hypercapnia.
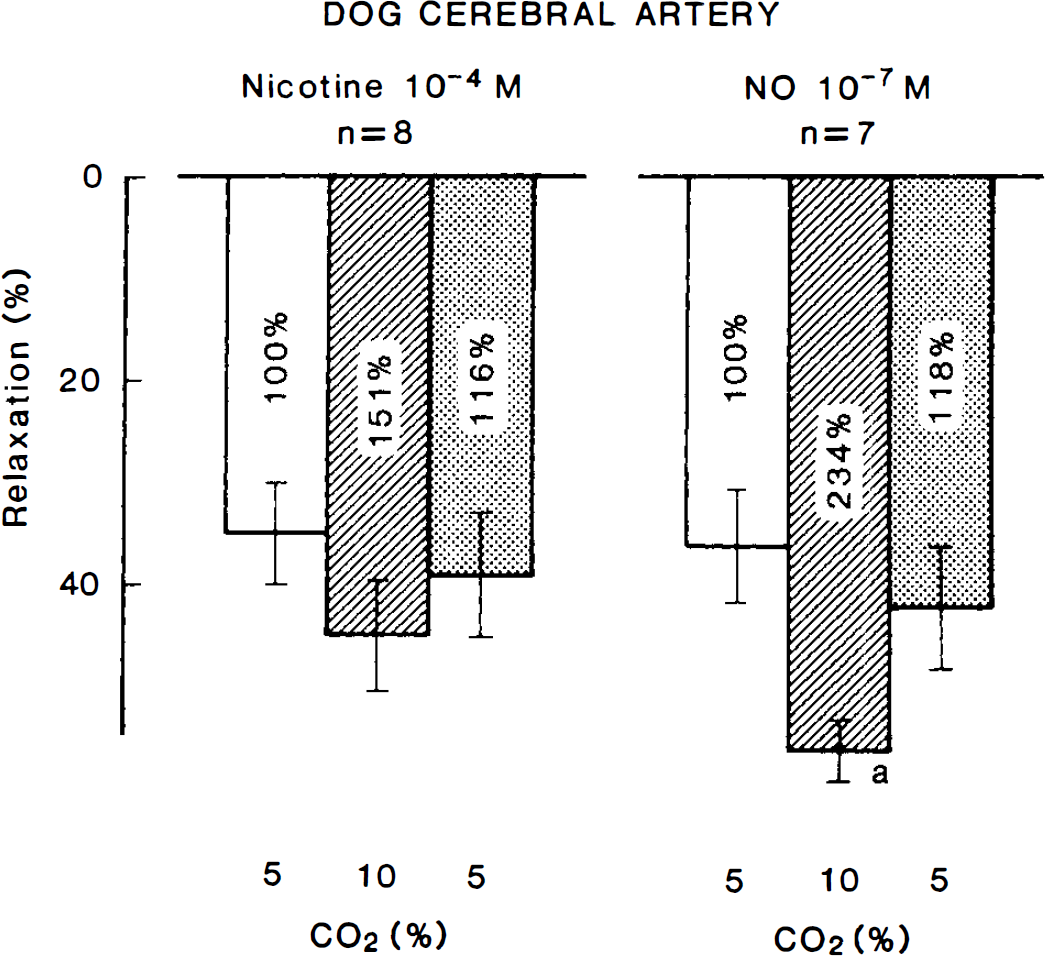
Modifications by hypercapnia (10% CO2) of the response to nicotine (10−4 M) and acidified NaNO2 (NO, 10−7 M) in canine cerebral arterial strips contracted with PGF2α. Relaxations induced by 10−4 M papaverine were taken as 100%. The mean value during hypercapnia (151.1 ± 57.7%, n = 8) is significantly different (p < 0.05, paired t-test) from the value in control media (100%). Significantly different from the value in control media, ap < 0.05 (n = 7, Tukey's method). Vertical bars represent SD.
DISCUSSION
Raising the concentration of CO2 in the aerating gas from 5 to 10% doubled the Pco2 value, significantly decreased the pH in the bathing media, and elicited a relaxation of monkey and canine cerebral arterial strips partially contracted with K +. Our earlier reports suggested that hypercapnia (15% CO2)-induced relaxation is associated with a reduction of extracellular pH and an activation of the electrogenic Na+ pump, but is not mediated by endothelium-derived vasodilators (Toda et al., 1989, 1993a). In contrast, cerebral vasodilatation and increase in cerebral blood flow by hypercapnia in anesthetized rats, rabbits, cats, dogs, and monkeys are reportedly inhibited by treatment with NO synthase inhibitors, suggesting the involvement of NO in the response (Iadecola, 1992; Iadecola and Xu, 1994; Faraci et al., 1994; Sandor et al., 1994; Saito et al., 1993; McPherson et al., 1995). The degree of hypercapnia was 50–70 mm Hg in these in vivo studies, whereas it was ∼110 mg Hg in our previous studies. Iadecola and Zhang (1994) postulated that the discrepancy between the in vitro and in vivo studies is derived from the different degrees of CO2 elevation, since they found that the attenuation by NO synthase inhibitors of hypercapnic vasodilatation is maximal at a Pco2 of 50–60 mm Hg and does not occur at a Pco2 of > 100 mm Hg. However, the present study revealed that the relaxation induced by hypercapnia of similar extent (from 29.8 to 59.3 mm Hg) to that applied in the in vivo study was not influenced by endothelium denudation and treatment with L-NA in a concentration sufficient to abolish the cerebroarterial response mediated by NO derived from the perivascular nerve and endothelium (Toda and Okamura, 1991; Toda et al., 1993b); this suggests that hypercapnia is unlikely to liberate NO from the endothelium. From the findings so far obtained, the discrepancy in the mechanism of hypercapnia-induced vasodilatation does not appear to depend on the degree of CO2 elevation, but, possibly, on the experimental conditions (particularly in vivo versus in vitro, in which basal releases of NO from the nerve and endothelium markedly differ).
Cerebral vasodilatation induced by nerve stimulation with electrical pulses or nicotine is mediated by NO or its analog, S-nitrosothiol, produced by constitutive NO synthase from L-arginine in nerve terminals (Toda and Okamura, 1990a,b, 1991); therefore, the nerve is called “nitroxidergic” (Toda and Okamura, 1992). Hypercapnia slightly increased the response to nerve stimulation, whereas the potentiation by high CO2 of the response to exogenous NO was evident. NO is stable in acidic solutions and is rapidly degraded in alkaline media. On the other hand, the optimal pH of cytosolic, constitutive NO synthase freshly isolated from pig endothelial cells is 7.2–7.3, and enzyme activity is reduced with decreasing pH (Hecker et al., 1994). These findings may indicate that the reason for less potentiation by hypercapnia of neurally induced relaxation than the response to NO is due to an impaired synthesis of NO in the nerve terminal in an intracellular acidic milieu.
In the present study, L-NA contracted monkey and canine cerebral arteries partially precontracted with K+, and the contraction was attenuated by endothelium denudation. As reported previously (Toda et al., 1993c), there is some basal release of NO from the endothelium in isolated monkey and canine cerebral arteries. Despite the fact that exogenous NO relaxed the arteries to a greater extent in hypercapnic acidosis (present study) and HCl acidosis (Toda et al., unpublished data) than in control media, hypercapnia-induced relaxations were not attenuated by removal of the endothelium and treatment with L-NA. This may be due to a depressed synthesis of NO by intracellular acidosis in the endothelium under resting conditions.
In summary, it seems likely that hypercapnia does not stimulate the release of NO from the endothelium of isolated monkey and canine cerebral arteries, but protects NO from degradation. The increased response to NO, because of being stabilized by acidosis, overcomes a decrease in the response by depressed NO synthesis in perivascular nerve terminals. Similar modulatory roles of NO in cerebral arterial function have been reported (Dreier et al., 1995; Bryan et al., 1995). Large cerebral arteries play an important role in the regulation of cerebral vascular resistance (Faraci and Heistad, 1990). In addition, neurogenic and endothelial NOs are expected to physiologically control intracerebral arteriolar caliber. Therefore, NO-dependent cerebral vasodilatation and the increase in the blood flow under hypercapnia seen in vivo may be associated with an increase in the net action of NO released from the nerve and endothelium. Whether increased Pco2may also stimulate nitroxidergic nerve pathways centrally, resulting in an increased discharge of efferent nitroxidergic nerves, remains to be answered.