
Abstract
The role of inflammation in neurological disorders is increasingly recognised. Inflammatory processes are associated with the aetiology and clinical progression of migraine, psychiatric conditions, epilepsy, cerebrovascular diseases, dementia and neurodegeneration, such as seen in Alzheimer’s or Parkinson’s disease. Both central and systemic inflammatory actions have been linked with the development of brain diseases, suggesting that complex neuro-immune interactions could contribute to pathological changes in the brain across multiple temporal and spatial scales. However, the mechanisms through which inflammation impacts on neurological disease are improperly defined. To develop effective therapeutic approaches, it is imperative to understand how detrimental inflammatory processes could be blocked selectively, or controlled for prolonged periods, without compromising essential immune defence mechanisms. Increasing evidence indicates that common risk factors for brain disorders, such as atherosclerosis, diabetes, hypertension, obesity or infection involve the activation of NLRP3, NLRP1, NLRC4 or AIM2 inflammasomes, which are also associated with various neurological diseases. This review focuses on the mechanisms whereby inflammasomes, which integrate diverse inflammatory signals in response to pathogen-driven stimuli, tissue injury or metabolic alterations in multiple cell types and different organs of the body, could functionally link vascular- and neurological diseases and hence represent a promising therapeutic target.
Introduction
The role of innate immune and vascular inflammatory mechanisms in common neurological disorders
Induction of inflammation in any organ or tissue includes rapid vascular changes in parallel with the activation of innate immune cells that sense pathogens or tissue injury via pattern recognition receptors (PRRs). Recognition of pathogen-derived molecules, and those released by the compromised cells and tissues of the host (which induce sterile inflammation), triggers an inflammatory response that initially includes the activation of largely overlapping immune cell populations in different organs, such as monocytes, macrophages, dendritic cells or granulocytes. These cells express multiple PRRs, and some PRRs recognise both host- and pathogen-derived molecules. As an example, Toll-like receptor 4 (TLR4) is activated by both bacterial lipopolysaccharide (LPS) and high mobility group box protein 1 (HMGB1), which is released from injured cells and acts as an endogenous danger molecule.1,2 In this way, monocytes, neutrophils or tissue macrophages can react rapidly to harmful stimuli of different origin and mount an inflammatory response that has several common features, which are not stimulus-specific. Injured tissues contribute to sterile inflammation via releasing danger signals and alarmins such as ATP, interleukin-α (IL-1α), HMGB1 and many other cytoplasmic proteins as well as nuclear or mitochondrial DNA. Following the development of the appropriate immune response, and when the harmful stimulus is no longer present, resolution of inflammation takes place and tissue regeneration is initiated.3,4 The normal inflammatory response to injury or infection commonly involves systemic changes including increases in circulating inflammatory cytokine and acute phase protein levels, activation of the HPA axis and the autonomic nervous system, which resolve soon after tissue regeneration or clearance of infection. In contrast, non-resolving infection, impaired wound healing or lasting metabolic alterations such as high blood glucose, uric acid or triglyceride levels are associated with chronic inflammation. Excessive or uncontrolled inflammation can result in tissue injury and is often associated with systemic inflammatory changes. These include prolonged upregulation of circulating cytokines, acute phase proteins and inflammation in different vascular beds of the body. Systemic inflammation is also associated with vascular changes and activation of glial cells in the nervous system and perhaps not surprisingly, diabetes, hyperlipidaemia, atherosclerosis, hypertension, obesity and infection are risk factors for cerebrovascular and neurodegenerative diseases. Preceding systemic inflammation also leads to worse clinical outcome in acute cerebrovascular diseases.5–9 Evidence indicates that systemic inflammation is associated with inflammatory changes in the brain before neurological symptoms develop. For example, patients with multiple risk factors for stroke and chronically elevated C-reactive protein (CRP), but without history of a previous cerebrovascular event and in the absence of any obvious brain pathology as assessed by neuroradiologists on MR scans, show microglial activation in the brain. This also occurs in mice or rats with chronic vascular disease. 10 Vascular and microglial activation and lipid deposition in the brain are also seen in mice fed an atherogenic diet.10,11 Acute or chronic inflammation in the CNS could also be induced by local signals. ATP, IL-1α, HMGB1 and DNA are released from injured neurons or glia, whereas amyloid-beta (Aβ) oligomers, tau and aggregated α-synuclein are potent inducers of glial activation and inflammatory cytokine production.12–17 Emerging data indicate that blockade of inflammation is protective in common neurological diseases. Interestingly, non-steroidal anti-inflammatory drugs (NSAIDs) and statins, which have diverse anti-inflammatory actions, appear to be protective in Alzheimer’s (AD) and Parkinson’s disease (PD).18–25
IL-1 and IL-18: Mediators of inflammation, vascular disease and brain injury
One of the main inflammatory mediators that contributes to a wide range of vascular, metabolic and neurological diseases is IL-1. IL-1 family cytokines are key mediators of inflammation, and the proinflammatory forms IL-1α and IL-1β are involved in both central and systemic inflammatory mechanisms.3,8,9,16,17,21,22,26 Blockade of IL-1 actions is markedly protective against brain injury in animal models.27,28 IL-18 is another inflammatory cytokine whose involvement in acute and chronic inflammatory conditions and common brain diseases has been documented.
29
In particular, IL-18 is elevated in the brain and/or the circulation of patients with AD
Structure and activation of inflammasomes
Inflammasomes are large multi-molecular protein complexes that form in response to inflammatory stimuli and that are responsible for the processing of the inactive precursors of IL-1β and IL-18. 33 Inflammasomes are composed of a sensor molecule, adaptor proteins and pro-inflammatory caspases. The sensor molecule is a PRR that senses pathogen (PAMPs), or damage associated molecular patterns (DAMPs), which are markers of infection or cell stress/injury, respectively. Inflammasomes are defined by the PRR, and although several have been described, 33 those that have been associated with brain injury are NLRP1,34–36 NLRP3,37–39 NLRC4 40 and AIM2.40,41 The endogenous ligand for NLRP1 (NLR family pyrin domain containing 1) is unknown. NLRP3 (NLR family pyrin domain containing 3) senses a diverse array of both PAMP and DAMP stimuli. 33 NLRC4 (NLR family CARD domain containing 4) senses bacterial ligands through co-receptors called NAIP proteins (NLR family apoptosis inhibitory protein),42,43 and AIM2 (absent in melanoma 2) directly binds to double stranded DNA.44,45 Upon PAMP or DAMP sensing NLRP3 nucleates the ASC (apoptosis-associated speck-like protein containing a caspase recruitment domain) adaptor molecule to oligomerise and form a large inflammasome speck within the cell based on a prion like aggregation.46,47 Pro-caspase-1 is also recruited to this complex where it becomes activated. Active caspase-1 then processes pro-IL-1β and pro-IL-18 to mature forms that are rapidly secreted from the cell. 48 Once formed these inflammasome specks can also become extracellular and propagate further inflammatory responses.49,50 The mechanism of IL-1β release after inflammasome activation has been poorly understood but is now known to require cleavage of the cytoplasmic protein gasdermin D (Gsdmd).51,52 Gsdmd is cleaved by caspase-1 and results in a destabilisation of the plasma membrane facilitating release of IL-1β.51–53 Similar to the effects of knocking out Gsdmd, we have recently reported that membrane stabilising agents such as the complex polyphenol punicalagin are able to inhibit the release of IL-1β specifically without affecting inflammasome activation. 54 Interestingly, punicalagin has also been shown to be neuroprotective in rodent models of stroke.55,56
Emerging role for inflammasomes in vascular disease, neuroinflammation and brain injury
Recent studies suggest that inflammasomes are involved in a wide range of pathophysiological processes in the brain and in chronic diseases that are risk factors for neurodegenerative or cerebrovascular disease. IL-1β and/or NLRP3 polymorphisms and changes in gene expression are associated with depression, migraine, MS, AD, PD and ischaemic stroke in patients.28,57–63 Similarly, NLRP3 polymorphisms are associated with risk of hypertension and age-related increase of blood pressure, type 2 diabetes, coronary heart disease and acute vascular events.64–67 Increased caspase-1 activation is seen in the brain of AD patients and the NLRP3 inflammasome was found to contribute to disease progression and changes in cognitive function in rodents.21,68 The NLRP3 inflammasome is also involved in the pathogenesis of CNS demyelination and prion disease in animal models69–72 and contributes to amyotrophic lateral sclerosis (ALS). 73 Acute brain injury following cerebral ischaemia and traumatic brain injury (TBI) as well as spinal cord injury involve ASC-dependent mechanisms mediated by the NLRP1, AIM2 or NLRC4 inflammasomes.34–36,40 Since IL-1- and inflammasome-mediated actions contribute to inflammation and injury in both the periphery and the brain, it is important to understand the cellular mechanisms involved in order to develop effective therapeutic strategies for CNS disease.
Cell type-specific activation of inflammasomes links systemic inflammation and brain disease
Inflammasome research has largely focused on monocytes and macrophages, since these cells not only respond rapidly to a diverse array of DAMPs and PAMPs, but are also considered to be key inducers of inflammation via IL-1 and IL-18 in different tissues. However, recent research has revealed that endothelial cells, granulocytes, neurons and astrocytes (and other cells) also express functional inflammasomes that contribute to both systemic inflammation and pathophysiological processes in the CNS (Figure 1). Because of the extensive literature available, this section focuses on inflammasome-mediated actions in different cell types in the context of neuroinflammation and risk factors for brain disease. Inflammasome signalling in monocytes and macrophages are not discussed in detail here but are reviewed extensively elsewhere.3,33,74,75
Inflammasome signalling in different cell types involved in brain inflammation and injury. Because of the extensive literature data available, inflammasome signalling in monocytes and macrophages is not discussed in detail only pathways confirmed in the case of microglia. See detailed explanation in the text. Please note that triggers, pathways and downstream events of inflammasome activation were depicted in the figure only if experimental evidence is available for the given cell type.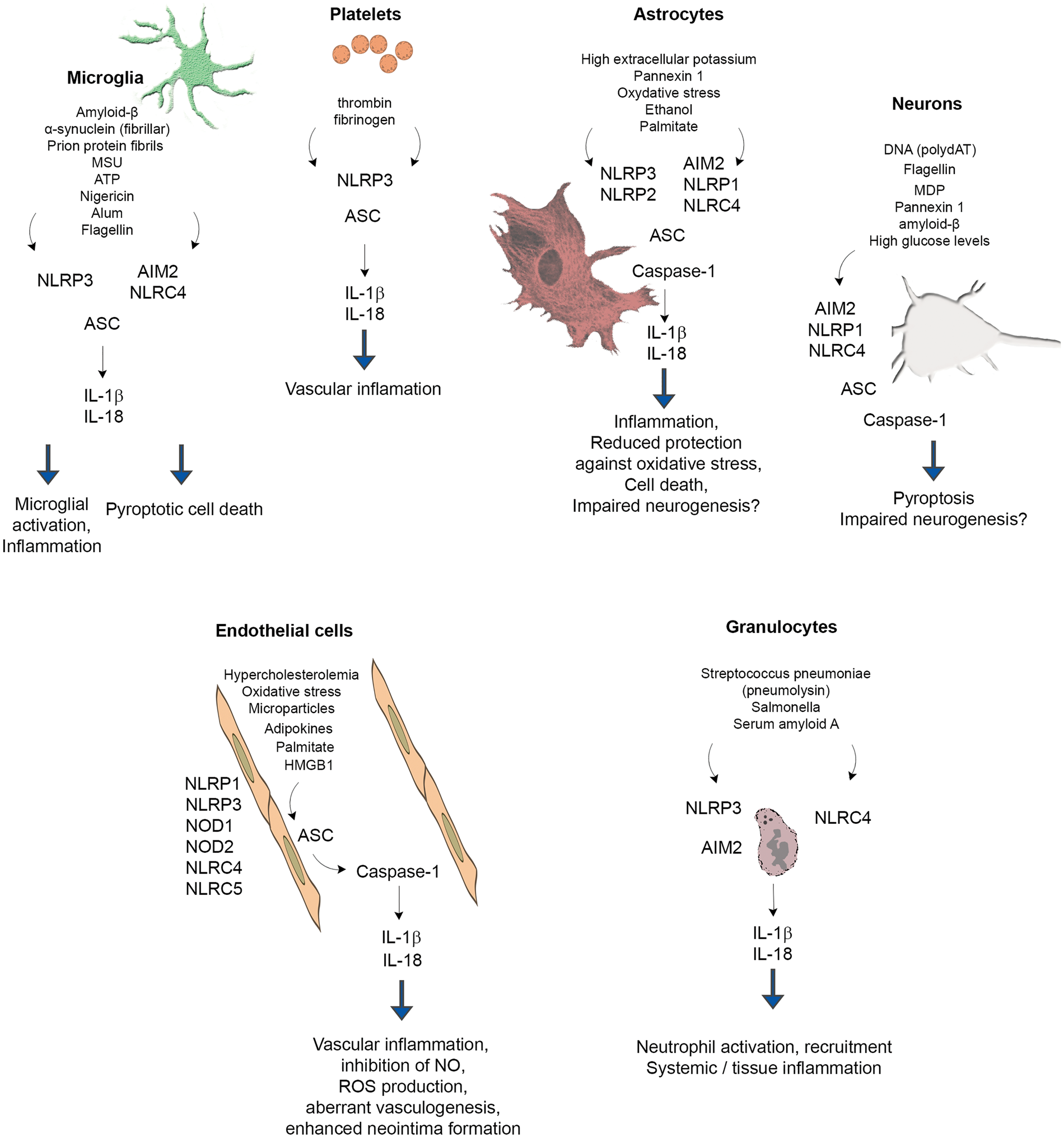
Microglia
Similarly to other organs, injury in the CNS causes the release of DAMPs leading to microglial activation and IL-1 production. Although a comprehensive comparison of microglial inflammasome signalling with that of monocytes and macrophages has not been performed, the available evidence indicates that common PAMPs and DAMPs activate microglial inflammasomes. For example, LPS, HMGB1 and the acute phase protein serum amyloid A act as a priming stimulus for microglia, whereas classical DAMPs such ATP and monosodium ureate (MSU) crystals induce the activation of the NLRP3 inflammasome, leading to the release of mature IL-1β.76–79
Aggregated amyloid-β can activate the NLRP3 inflammasome in microglia, which release IL-1β upon stimulation following LPS priming. This is associated with lysosomal damage and release of cathepsin B.78,80 In turn, caspase-1 expression is observed in the human brain in patients with mild cognitive impairment and AD, whereas NLRP3- or caspase-1-deficient mice carrying mutations associated with familial AD are largely protected from loss of spatial memory. 68 Deposition of α-synuclein is one of the main pathological changes seen in the brain in PD. Both monomeric and fibrillar forms of α-synuclein were found to induce the synthesis of IL-1β through TLR2, whereas fibrillar α-synuclein induced IL-1β secretion via NLRP3 in microglia. 12 In another study, stimulation of microglia with α-synuclein was sufficient to induce a modest priming effect, but did not result in the release of active IL-1β. 78 α-synuclein added to THP1 cells (a human monocyte cell line) induces activation of TLR2 and the NLRP3 inflammasome leading to IL-1β secretion.81,82 Prion protein fibrils induce the activation of NLRP3 inflammasome and IL-1β production from microglia via lysosome destabilisation, ultimately leading to the release neurotoxic mediators and neuronal death. 83 In brain tissues from patients with Rasmussen’s encephalitis (an inflammatory encephalopathy of unknown cause defined by seizures with progressive neurological disabilities), increased expression inflammasome-associated genes (IL-1β, IL-18, NLRP1, NLRP3 and CASP1) was found. IL-1β and caspase-1 expression was associated with white matter myeloid cells. 84
Neurons and astrocytes
Neurons express inflammasome components both in vivo and in vitro and neuronal inflammasome activation occurs in response to acute injury, brain trauma, stress and in animal models of neuroinflammation and neurodegeneration. For example, NLRP1, NLRC4, AIM2, caspase-1 and ASC mRNAs are present in cultured neurons, and treatment with the NLRP1 agonist muramyl dipeptide (MDP), the AIM2 agonist poly-(dA:dT) or the NLRC4 agonist recombinant flagellin, induces activation of caspase-1 and caspase-6. 85 NLRP1 immunopositive neurons are increased 25- to 30-fold in AD brains compared with non-AD brains. 85 Neuronal NLRP1 levels are also upregulated in APPswe/PS1dE9 transgenic mice, and NLRP1-mediated pyroptosis (a caspase-1 dependent, inflammatory form of cell death 86 ) is induced in cultured cortical neurons in response to amyloid-β. 87 The NLRP1 inflammasome is also activated in patients with medial temporal lobe epilepsy, and NLRP1 inflammasome activation contributes to neuronal pyroptosis in the amygdala kindling-induced rat model. 88
Traumatic brain injury induces expression of caspase-1, caspase-11, ASC and NLRP1 in cortical neurons, which is associated with production of mature IL-1β. Activation of the AIM2 inflammasome is also seen in the human brain after traumatic brain injury and neurons stimulated with synthetic double-stranded DNA (poly-(dA:dT)) undergo AIM2 inflammasome-dependent pyroptosis, which is prevented by inhibition of pannexin-1. 41 Systemic metabolic and/or inflammatory changes could also exert direct impact on neuronal inflammasomes. For example, high glucose levels induce the release of mature IL-1β and IL-18 from cultured neurons, whereas increased expression of NLRP1, ASC and caspase-1 is found in neurons both in vitro and in vivo in the cerebral cortex of streptozocin (STZ)-induced diabetic rats. 89
Astrocytes are capable of producing IL-1β both in vitro and in vivo and express NLRP3, AIM2, NLRP1 and NLRC4 inflammasomes.90–92 Expression of the NLRP2 inflammasome by astrocytes has also been suggested. 93 A growing body of literature suggests that inflammasome activation in astrocytes could contribute to brain inflammation after acute injury and in neurodegenerative diseases. High extracellular potassium levels open pannexin 1 channels leading to caspase-1 activation and IL-1β and IL-18 release from primary neurons and astrocytes. 94 Astrocytes play an important role in protection against oxidative stress in the brain and this is linked with inflammasome activation. Lack of uncoupling protein 2 (UCP2), which regulates ROS production, leads to the aggravation of inflammation via activating the NLRP3 inflammasome and at the same time increased endoplasmic reticulum stress in astrocytes. UCP2 knockout mice show exacerbated dopaminergic neuron loss in the 1,2,3,6-methyl-phenyl-tetrahydropyridine (MPTP) murine model of PD, which is accompanied by increased astrocyte activation. 95 Ethanol also induces NLRP3 activation and cell death in cultured astrocytes, which is blocked by a mitochondrial ROS scavenger. 96 Increased expression of both NLRP1 and NLRP3 is seen in hippocampal neurons and astrocytes in post mortem alcoholic human brain, and inhibition of neurogenesis by ethanol is reversed by a neutralising antibody to IL-1β, or blockade of the IL-1β receptor with IL-1R antagonist in organotypic slice cultures. 97 Expression levels of NLRC4 and ASC are significantly elevated in a subgroup of sporadic AD patients, and palmitate (saturated fatty acid, a major component of high fat diet) induces the activation of NLRC4 and production of mature IL-1β in cultured astrocytes. 98 In post mortem tissues from patients with amyotrophic lateral sclerosis, increased NLRP3, ASC, caspase-1 and IL-18 levels are found compared with control tissues, and NLRP3 is expressed in spinal cord astrocytes of male SOD1(G93A) mice carrying a mutant human superoxide dismutase 1 (SOD1) variant. In these mice, NLRP3 expression and production of IL-1β were already detectable at a pre-symptomatic stage of the disease. 90 Interestingly, another study failed to find substantial IL-1β or IL-18 release from astrocytes after inflammasome activation. 78 Thus, it is important to note that some of the neuroprotective effects of inflammasome inhibition could be due to direct actions on neurons or astrocytes and not necessarily to blockade of microglia- or macrophage-dependent mechanisms in different models of brain injury.
Endothelial cells
Human cerebral endothelial cells express various NOD-like receptors (e.g. NOD1, NOD2, NLRC4, NLRC5, NLRP1 and NLRP3) and release IL-1β in a caspase-1-dependent manner upon LPS priming and stimulation with muramyl dipeptide. 99 Importantly, inflammasomes in endothelial cells contribute to chronic inflammation and altered vascular responses in different vascular beds. Hypercholesterolemia and oxidative stress result in NLRP3 inflammasome activation in endothelial cells, leading to inhibition of endothelial nitric oxide synthase (eNOS) and coronary endothelial dysfunction.100,101 Adipokines such as visfatin activate the NLRP3 inflammasome in endothelial cells, leading to enhanced neointima formation. 102 Similarly, uraemic sera from patients with chronic kidney disease induces NLRP3 activation, caspase-1 activation and ROS production in cultured endothelial cells. 103 Palmitate in vitro and high-fat diet in vivo result in NLRP3 inflammasome activation in endothelial cells via thioredoxin-interacting protein (TXNIP)-mediated actions. 104 It has also been postulated that the NLRP1 inflammasome is involved endothelial activation as human aortic endothelial cells exposed to sera from patients with peripheral arterial disease show significantly higher expression of NLRP1 than those exposed to sera from healthy individuals. 105 Inflammasome activation may lead to aberrant vasculogenesis. In autoimmune diseases like systemic lupus erythematosus (SLE), IL-18 inhibits the differentiation of endothelial progenitor cells and circulating angiogenic cells into mature endothelial cells, which is reversed by antibody neutralisation of IL-18. 106 Hemorrhagic shock leads to the activation of the NLRP3 inflammasome in lung endothelial cells in response to HMGB1 stimulation of TLR4, leading to secretion of IL-1β. 107 Although the contribution of endothelial inflammasomes to neuroinflammation is presently unclear, circulating DAMPs and PAMPs are known to induce inflammatory changes in multiple organs, including the brain, via direct endothelial signalling mediated by TLRs.108–111
Granulocytes
Granulocytes are key drivers of inflammation and tissue injury in both the periphery and the injured brain. IL-1 is a potent inducer of granulocyte migration to inflamed tissues and granulocytes undergo profound functional changes after transendothelial migration. In the brain, transmigrated neutrophils release proteases and decondensed DNA contributing to neurotoxicity, which can be transferred by neutrophil-conditioned medium in vitro.20,112,113 Interestingly, IL-1 is a key mediator driving the recruitment of neutrophils in the mouse brain but is dispensable in extracerebral tissues including the lung and peritoneum. 114 Human and mouse neutrophils also express key inflammasome components and release IL-1β and IL-18. Purified human neutrophils were found to have functional NLRP3 and AIM2 inflammasomes. 115 However, unlike macrophages, neutrophils appear to be resistant to pyroptotosis and sustained IL-1β production is not associated with reduced neutrophil viability. 116 Pneumolysin (a pore-forming toxin of Streptococcus pneumoniae) induces IL-1β processing in neutrophils through NLRP3 and ASC, a process that takes place independently of pyroptotic cell death or lysosomal destabilisation. 117 Similarly, Salmonella infection triggers the activation of the neutrophil NLRC4 inflammasome, which selectively promotes IL-1β maturation without pyroptosis. 116 Inflammasomes in neutrophils are also involved in the pathophysiology of non-infectious diseases, such as asthma. 118 Interestingly, acute phase proteins can directly induce inflammasome activation in neutrophils. In neutrophils isolated from healthy donors, the acute phase protein serum amyloid A (which also induces microglial priming, see above) promotes the expression of pro-IL-1β, followed by the activation of the NLRP3 inflammasome and caspase-1, leading to the secretion of mature IL-1β. 119 Recruitment of granulocytes into the cerebral ventricles as well as microglial and vascular activation in response to diet induced atherosclerosis in mice is also dependent on IL-1β. 11 Thus, inflammasome activation in endothelial cells, microglia and granulocytes could link systemic inflammatory actions with diverse neuropathological processes in the brain.
Platelets
Platelets are a major source for IL-1α, IL-1β and IL-18 and contribute to circulating levels of these cytokines. The role of platelet-derived IL-1 in systemic and cerebrovascular inflammation has also been demonstrated.120–123 Blockade of platelet-mediated actions and IL-1 signalling has been found to protect against brain injury after cerebral ischaemia in both naive mice and under systemic inflammatory conditions, when stroke is preceded by Streptococcus pneumoniae infection.124,125 Platelets constitutively express the inflammasome components NLRP3 and ASC and have functional inflammasomes, enabling caspase-1 activation and IL-1β processing. 126 Similarly to nucleated cells, platelets can be primed by TLR2- and TLR4-mediated signals, but caspase-1-dependent IL-1β processing does not require a second stimulus. Platelet IL-1β mRNA is induced by thrombin or fibrinogen and also released through platelet microparticles. 127 Although the functional role of platelet inflammasomes in disease remains to be investigated in detail, interactions between platelets, circulating immune cells and the vasculature in response to various inflammasome activating stimuli is expected to occur in a wide range of diseases.
Inflammasomes in comorbidities and risk factors for brain disease
Diabetes
Inflammasomes in vascular diseases and risk factors for brain disease.
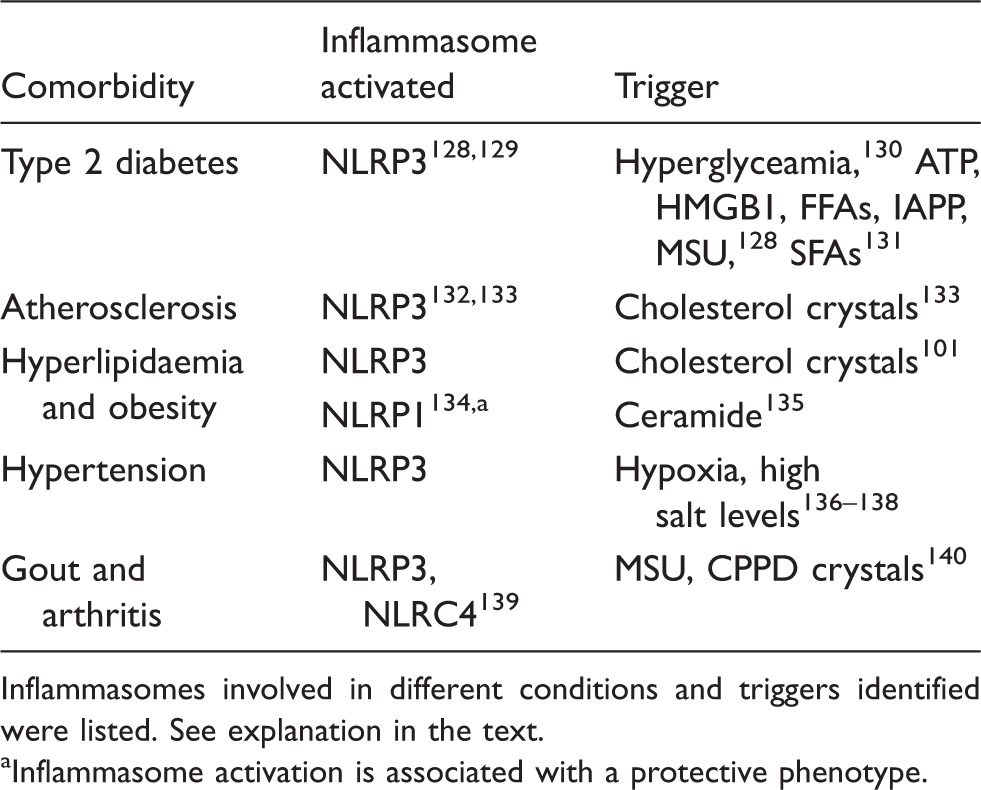
Inflammasomes involved in different conditions and triggers identified were listed. See explanation in the text.
Inflammasome activation is associated with a protective phenotype.
Atherosclerosis
Atherosclerosis is a progressive artery disease which involves inflammation of the vessel walls and the deposition of lipid-rich plaques. One major component of atherosclerotic lesions is cholesterol. Cholesterol crystals can induce inflammation via activating the NLRP3 inflammasome in vitro and in vivo, a process associated with phagolysosomal damage. It has been demonstrated that NLRP3 is overexpressed in the aorta of patients with atherosclerosis or hypertension. 132 LDLR−/− mice fed a high cholesterol diet show markedly decreased early atherosclerosis in parallel with reduction in IL-1β and IL-18 levels after transplantation with NLRP3−/−, ASC−/− or IL-1α/β−/− deficient bone-marrow, whereas mice lacking NLRP3 or IL-1 are resistant to diet-induced atherosclerosis. 133 It has been shown that cholesterol crystals could function as both a priming stimulus, and a stimulus to induce IL-1β release. Cholesterol crystals also trigger formation of neutrophil extracellular traps (NETs), which prime macrophages to release cytokines, further amplifying the immune cell recruitment in atherosclerotic lesions. 142 Selective blockade of the NLRP3 inflammasome in ApoE2.Ki mice results in the reduction of IL-1β production in peritoneal macrophages and reduced serum lipid (total cholesterol and triglyceride) levels are observed with a shift of macrophage polarisation from M1 to M2 state, which is associated with fewer atherosclerotic lesions. 143 The purinergic receptor, P2X7 is coexpressed with NLRP3 in atherosclerotic plaques of ApoE−/− mice and plays a significant role in atherosclerosis by activating the NLRP3 inflammasome via facilitating the phosphorylation of protein kinase R (PKR). 144
Hyperlipidaemia and obesity
The NLRP3 inflammasome contributes to obesity-induced inflammation and insulin resistance. 135 NLRP3 activation is seen in the subcutaneous adipose tissue of dyslipidaemic, diabetic and obese patients and is associated with the severity of coronary atherosclerosis.145–147 Blockade of the NLRP3 inflammasome resulted in the amelioration of serum markers of diet-induced obesity and hyperlipidaemia and enhanced insulin signalling in parallel with a reduction of inflammation in the adipose tissue and the liver.135,148 Acute hypercholesterolemia increased the expression of caspase-1, NLRP3 and HMGB1 in the coronary arteries and decreased endothelium-dependent vasodilatation was observed in NLRP3+/+ mice compared with NLRP3−/− mice. Treatment with either a caspase-1 or a HMGB1 inhibitor restored the vasodilation response in NLRP3+/+ mice. 101 Obesity is associated with vascular dysfunction (reduced aortic relaxation, macrophage accumulation and intimal thickening) in rats via activation of the NLRP3 inflammasome and mitochondrial dysfunction. 149 Saturated fatty acids (SFAs), like palmitic acid, are known priming signals of NLRP3 inflammasome activation and IL-1β production via TLR4, leading to impaired insulin signalling, in contrast to unsaturated fatty acids (UFAs), which exert anti-inflammatory actions through inhibition of IL-1β processing.131,150,151 NLRP1 is also implicated in obesity, and similar to IL-18 deficient mice, NLRP1−/− mice are resistant to diet-induced metabolic dysfunctions and have reduced adiposity.134,152
Hypertension
There is an association between the NLRP3 SNP rs7512998 and blood pressure in patients, 64 and circulating IL-1β levels are strongly linked with the development of hypertension. 153 Expression of NLRP3, caspase-1 and production of IL-1β and IL-18 were confirmed in pulmonary hypertension in rodents.136,154 Mice lacking ASC show substantially reduced pulmonary hypertension and right ventricular remodelling, 155 whereas reduced renal inflammation and better outcome are observed in a salt-induced hypertension model. 137 High-salt-induced hypertension is associated with increases in NLRP3, caspase-1 and IL-1β levels in the paraventricular nucleus of the hypothalamus, which could be attenuated by inhibition of IL-1β or NF-κB activation suggesting the possible involvement of central inflammasome signalling in hypertension.138,156
Gout and arthritis
Rheumatic disease is associated with higher risk of cerebrovascular disease. For example, according to recent meta-analyses, the risk of stroke is higher among rheaumatoid arthritis patients under 50 years. Moreover, inflammatory arthropathies involve higher risk than non-inflammatory diseases and affect long-term disability after stroke.157,158 Gout also represents a risk factor for both ischaemic and haemorrhagic stroke in younger and older age as well.159,160 Interestingly, stroke itself can also increase the susceptibility for gouty arthritis in patients. 161 Hyperuricemia contributes to chronic inflammation in gout. Monosodium urate (MSU) and calcium pyrophosphate dihydrate (CPPD) crystals activate the NLRP3 inflammasome, and macrophages derived from caspase-1, ASC or NLRP3 deficient mice have defective IL-1β production in response to MSU. 140 Treatment either with IL-1R antagonist or IL-1 trap is an effective gout therapy.162,163 NLRP3-inflammasome activation has been demonstrated in rheaumatoid arthritis and NLRP3 polymorphisms are linked with the susceptibility to arthritis and the response to anti-TNF treatment. 164 ASC has also been implicated to rheaumatoid arthritis. ASC−/− mice showed decreased infiltration of inflammatory cells and cartilage/bone destruction in a collagen-induced arthritis model. 165 In an Ag-induced arthritis (AIA) model, ASC−/− mice have decreased synovial IL-1β and serum amyloid A levels. In contrast, NLRP3−/−, NLRC4−/− or caspase-1−/− mice do not show any alteration of joint inflammation compared with controls indicating that the effect of ASC is independent of NLRP3 or NLRC4 inflammasomes. 139
Infection
Infection is an established risk factor for CNS disease, for which there is a wealth of literature.7,9 Infectious burden significantly correlates with the risk of ischaemic stroke in patients and there is a strong positive association between bacterial infection and AD.166,167 The role of inflammasomes in infectious diseases that manifest in the CNS is also well-established. For example, meningitis caused by Streptococcus pneumoniae is associated with the activation of NLRP3 via the pore-forming complex pneumolysin. 168 NLRP3−/− and ASC−/− mice with pneumococcal meningitis show decreased inflammatory response, which is more pronounced in ASC−/− mice. 169 Pertussis toxin induces the formation of a pyrin-dependent inflammasome that cleaves pro-IL-1β into its active form, promoting IL-6 production that facilitates neutrophil intravascular crawling in cerebral capillaries and promotes experimental autoimmune encephalomyelitis (EAE). 170 Since recognition of diverse bacterial, fungal or viral PAMPs by different PRRs induces inflammasome activation, infections that manifest in either the periphery or in the brain could potentially contribute to neuroinflammation and brain injury via inflammasome activation in brain cells, circulating leukocytes or different vascular beds in the body. Blockade of inflammasome-mediated actions could have therapeutic benefit in brain diseases. Supporting this, studies from mouse models suggest that inflammasomes are in general dispensable for infectious disease, their absence merely delaying the induction of an adaptive immune response. 171
Inflammasome activation is linked with diverse brain diseases in humans and experimental animals
Cerebral aneurysms and intracerebral/subarachnoid haemorrhage
Inflammasomes in neurological diseases.

Inflammasomes involved in different conditions and triggers identified were listed. See details regarding the mechanisms in the text.
Ischaemic brain injury
A number of intracellular changes that occur after ischaemia have been proposed to induce inflammasome activation. These include mitochondrial damage, production of ROS, lysosomal destabilisation, pore formation in the cell membrane, changes in cell volume and increases in intracellular Ca2+ concentration, which eventually activates the NLRP3 inflammasome.178,189,190 In macrophages, ATP-induced release of mitochondrial DNA (particularly its oxidised form) into the cytosol contributes to activation of caspase-1 and IL-18/IL-1β secretion via NLRP3.191,192 DNA also activates the AIM2 inflammasome. 45 Acidic extracellular pH, which is typical at sites of ischaemia and inflammation activates the NLRP3 inflammasome, while alkaline pH exerts an inhibitory effect on inflammasome activation. 176 HMGB1, an endogenous DAMP released by cellular stress and pathogenic insults, could also potentiate the neuroinflammatory response via NLRP3 activation in microglia in a redox state-dependent manner. 193 Ischaemia also causes anoxic depolarisation in neurons, which results in excessive glutamate and ATP release leading to excitotoxicity. The ionotropic P2X7 receptor and the pannexin 1 (Panx1) channel are overactivated by high extracellular ATP and NMDA receptor activation, and blockade of these channels is protective in cerebral ischaemia.189,194,195 ROS are thought to be key mediators of NLRP3 activation, as blocking NOX-dependent ROS production, or scavenging by N-acetylcysteine (NAC), in macrophages inhibits NLRP3 inflammasome activation by interfering the priming step required for the induction of NLRP3 expression. 196 It has also been shown that NLRP3 activating DAMPs stimulate an inflammatory response in glia that contributes to brain inflammation after ischaemia. 77 High extracellular K+ and reduced intracellular ATP, or increased AMP levels could trigger the activation of NLRP1 during hypoxia or after stroke.177,189
Although many of the processes described above occur after cerebral ischaemia, it is still unclear to what extent ischaemic brain injury is influenced by different inflammasomes in vivo. NLRP3 has been identified as a contributor to brain injury after focal cerebral ischaemia using NLRP3−/− mice. 39 Bruton’s tyrosine kinase has also been suggested to physically interact with ASC and NLRP3, to play a role in NLRP3 inflammasome activation and to contribute to ischaemic brain injury. 38 However, other studies found that NLRP3 is dispensable for neuronal injury in neonatal 197 or adult 40 mice after ischaemia. Antibody neutralisation of NLRP1 eliminated the assembly of the NLRP1 inflammasome in neurons and astrocytes and protected against brain injury.37,94 Neutralising antibodies against NLRP1 also reduces the inflammatory response after thromboembolic stroke in mice. 35 Our recent studies identified the NLRC4 and AIM2 inflammasomes as major contributors to ischaemic brain injury via ASC-dependent mechanisms. Because of the diversity of the signals involved in inflammasome activation after cerebral ischaemia, it is likely that blockade of the ASC adaptor protein, common for several inflammasomes, could have great therapeutic potential. It is also essential to assess to what extent systemic inflammatory actions contribute to brain injury in certain models of cerebral ischaemia, which could in part explain the differences observed regarding the role of given inflammasomes in stroke outcome. As detailed above, the NLRP3 inflammasome plays a role in diverse systemic inflammatory actions; therefore, its role in experimental models of stroke combined with comorbidities requires further investigation.
Traumatic brain injury
NLRP1 is expressed in cortical neurons and CSF levels of ASC, caspase-1 and NLRP1 are increased in patients after traumatic brain injury.34,180 NLRP3 expression has also been found in the cerebral cortex after TBI in rats. 182 Neutralisation of ASC or NLRP1 reduces inflammation and improves outcome in rats after traumatic brain injury. 34 Recent data show that NLRP1 inflammasome components are associated with exosomes, which were isolated from the CSF of TBI or spinal cord injured patients. 181
AD and PD
IL-1β and IL-18 are increased in the brains of patients with AD and PD and microglia are recruited to senile plaques where they produce IL-1β.31,198 This process includes the phagocytosis of Aβ and subsequent lysosomal damage, NLRP3 inflammasome activation and release of cathepsin B. 80
APP/PS1 transgenic mice lacking either the NLRP3 or caspase-1 have decreased plaque deposition, increased Aβ clearance, reduced levels of IL-1β and better memory. 68 The NLRP1 inflammasome is also implicated in AD. 185 Nonsynonymous polymorphisms in the NLRP1 gene are associated with the susceptibility of AD in humans. 186 In APP/PS1 transgenic mice, NLRP1 levels are upregulated in cortical neurons, and cultured neurons undergo caspase-1-dependent pyroptosis in response to Aβ, which is reduced by silencing either the NLRP1 or the caspase-1 gene with si-RNA. 87 NLRC4 and ASC are upregulated in post-mortem brain tissues in a subgroup of sporadic AD patients. Addition of conditioned medium derived from NLRC4-silenced astrocytes to primary neurons, significantly reduces the production of IL-1β and generation of Aβ1-42 by neurons. 98 It is currently unclear how inflammasome-mediated signals and cellular stress interact to cause neuronal injury. For example, chronic overexpression of IL-1β in itself promotes brain inflammation without overt neurodegeneration 199 and could decrease the amount of amyloid plaques while increasing tau hyperphosphorylation in 3xTgAD/IL-1βXAT mice. 200
The fibrillar form of α-synuclein is recognised by TLR2 and also activates the NLRP3 inflammasome leading to the release of IL-1β from human monocytes, 12 as opposed to the monomer form which does not. 82 The activation of NLRP3 is triggered by lysosomal rupture induced by the phagocytosis of α-synuclein fibrils both in isolated monocytes and in monocytic and neuronal cell lines.12,81 The activation of NLRP3 in dopaminergic neurons in the substantia nigra of MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine)-induced or transgenic mouse models of PD depends largely on Cdk5, as inhibition or deletion of Cdk5 efficiently blocks NLRP3 and neuroinflammation in these models. 201 Thus, inflammasome-mediated actions are linked with several major pathological changes in neurodegenerative diseases and hence represent a promising therapeutic target.
Local and systemic inflammatory signals interact in brain disease
Complex interactions between peripheral and central inflammasome-mediated pathways, involving diverse stimuli such as the microbiota, tissue injury and metabolic alterations, could contribute to neuroinflammation and neurodegenerative diseases. Understanding these mechanisms will be instrumental to support both diagnosis and treatment. For example, type 2 diabetes is identified as a risk factor for AD and for cerebrovascular disease in general.5,202,203 Amyloid deposits (misfolded proteins rich in cross-β-sheet structure, which accumulate in various tissues in the form of plaques) are a hallmark of AD, but they are also typical in type 2 diabetes as islet amyloid polypeptide (IAPP) in the pancreas. Bacteria also produce amyloids as a component of their extracellular matrix during biofilm formation. Curli fibres produced by Salmonella enterica serovar Typhimurium and Escherichia coli activate the NLRP3 inflammasome, leading to the production of IL-1β via caspase-1 activation. 204 As discussed above, amyloid β activates the NLRP3 inflammasome in microglia, as does IAPP in the pancreas, leading to IL-1β production. 205 Furthermore, amyloid β, serum amyloid A (SAA) and IAPP are all recognised by TLR2 leading to IL-1β secretion in response to NLRP3 activation.77,204,206
Microparticles are released from virtually all cell types and their role in inflammatory processes is widely recognised. It is likely that circulating, neuronal- or glial-derived microparticles could induce activation of inflammatory cells in the periphery, whereas peripheral-derived microparticles could stimulate the brain vasculature. It has been shown that microparticles derived from monocytes activate endothelial cells in an IL-1-dependent manner, which leads to increased expression of cell adhesion molecules such as intercellular adhesion molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1) and E-selectin. This effect is reduced by knockdown of NLRP3 in monocytes. 207
The gut microbiota are involved in brain injury that occurs after stroke 208 and acute brain injury leads to changes in the gut microbiota in part via altered autonomic activity. 209 Bacterial translocation from the gut to the circulation has been documented after acute brain injury, 210 which could induce inflammasome activation via a wide array of PAMPs in different tissues. In fact, we have shown that NLRC4-deficient mice are protected against brain injury, which could either be due to inflammasome activation induced by flagellin of bacteria, or a currently unrecognised endogenous ligand of NLRC4. 40 As discussed above, changes in intracellular K+ concentrations after ischaemia trigger NLRP3 activation. Bacterial toxins can also cause K+ efflux, leading to the activation of the NLRP3 inflammasome. 178 It needs to be investigated in detail to what extent neuronal and glial inflammasomes may be primed or activated by circulating (or local) bacterial, fungal and viral PAMPs or acute phase proteins in different experimental models. Examples also exist of altered peripheral inflammasome signalling in risk factors for brain disease in humans. For example, upregulation of inflammasome activity and increased gut permeability are associated with obesity in children and adolescents. 146 Recent data show that genetic deficiency of caspase-1 decreased depressive- and anxiety-like behaviours and increased locomotor activity in mice, which paralleled changes in the faecal microbiome. 211
Aging is an another important factor to consider in the context of systemic inflammation and brain disease. Interestingly, hippocampal lysates from aged rats showed significantly higher levels of NLRP1, ASC, caspase-1, caspase-11, the purinergic receptor P2X7, pannexin-1 and X-linked inhibitor of apoptosis (XIAP) than lysates from younger animals, which was associated with cognitive decline. 212 It has been postulated that NLRP3 inflammasome activation links systemic low-grade inflammation to functional decline in aging as it promotes age-related degenerative changes, whereas deficient NLRP3 inflammasome-mediated caspase-1 activity improved glyceamic control and attenuated bone loss and thymic demise. 213 It is likely that results from rodent models underestimate the impact of aging and comorbidities on pathological changes in the brain compared with humans, where a high proportion of aged individuals present with multiple chronic diseases and are exposed to an array of medications for several years or decades.
Therapeutic strategies to inhibit inflammasome activation
Drugs targeting the IL-1 pathway downstream of the inflammasome pathway do exist. Biologicals that target IL-1, like IL-1Ra (anakinra) and specific monoclonal antibodies such as canakinumab, are used for the clinical management of several auto-inflammatory disorders and are in trials for others. For example, anakinra has shown efficacy in preclinical models of acute brain disease and is undergoing clinical evaluation. 214 However, biologicals such as anakinra may not easily access the brain during brain disease where there is limited disruption of the blood brain barrier. Given their role in vascular disease and associated co-morbidities, and their seemingly dispensible role in infection, 171 inflammasomes are an attractive therapeutic target. In fact, in vivo data suggest that knocking out inflammasomes merely delays the resolution of an infection. The exact reason for this is unclear, but it has been suggested that animals maintained inflammasomes over evolution not to defend against vertebrate-adapted pathogens but instead to counteract infection by a plethora of undiscovered opportunistic pathogens residing in the environment. 171 Supporting this, increased risk of infection was not observed after experimental stroke in response to inflammasome inhibition or treatment with IL-1Ra34,40,215 and IL-1Ra treatment was not associated with more infections or other major adverse events in patients with myocardial infarction, 216 traumatic brain injury 217 or stroke.9,218 Furthermore, strategies that specifically target NLRP3 will not compromise the role of pathogen sensing inflammasomes such as AIM2 or NLRC4. Thus, the available data suggest that targeting inflammasomes will be safe. The best characterised inflammasome is NLRP3 and a number of small molecules are being developed and investigated as potential NLRP3 inhibitors (reviewed extensively in Baldwin et al. 219 ). To-date, the most advanced NLRP3 inhibitor is MCC950, which is protective in the rodent EAE model. 220 Small molecule inhibitors of the NLRP1 inflammasome are also being developed. 219 In addition to the development of novel inflammasome inhibitors, efforts are underway to repurpose existing drugs. For example, nucleoside reverse transcriptase inhibitors effective as an anti-HIV therapy also inhibit the NLRP3 inflammasome independently of reverse transcriptase inhibition. 221 Repurposing drugs like this may accelerate the translation of inflammasome targeting strategies clinically. Thus, the development of small molecule inflammasome inhibitors may offer an alternative or an adjunct therapy to existing IL-1 blocking strategies.
Conclusions
In conclusion, inflammasome-mediated actions link systemic and central inflammatory mechanisms and could contribute to major human disorders, including common brain diseases. The association of diverse neurological diseases with inflammasome polymorphisms, the activation of inflammasomes in post mortem brain tissues of patients with common brain diseases and the pivotal role of inflammasomes in major risk factors for brain diseases suggest that inflammasomes could be a promising therapeutic target. This is strengthened by comprehensive preclinical studies suggesting that blockade of inflammasome signalling is effective in a number of conditions and appears to have minor side effects with respect to changing susceptibility to infection or infectious burden. Understanding the molecular mechanisms that contribute to brain diseases and the development of novel inflammasome inhibitors could have a profound impact on the clinical management of common human diseases.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: OTKA K109743 (AD), the Hungarian Brain Research Program KTIA_13_NAP-A-I/2 (AD), and the János Bolyai Research Scholarship and the ‘Lendulet’ program of the Hungarian Academy of Sciences (AD).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.