
Abstract
Activation of the inflammatory response is a crucial event in the adverse outcome of cerebral ischemia, which is promoted by proinflammatory cytokines such as interleukin (IL)-1β. Although caspase-1 is necessary for IL-1β processing, the ‘upstream’ signaling pathways were, until recently, essentially unknown. Fortunately, the inflammasome, a multiprotein complex responsible for activating caspase-1 and caspase-5, has recently been characterized. The activation of the inflammasome can result in one of several consequences such as cytokine secretion, cell death, or the development of a stress-resistant state. The significance of the inflammasome for the initiation of the inflammatory response during systemic diseases has already been shown and members of the inflammasome complex were recently found to be induced in acute brain injury. However, the specific pathophysiologic role of the inflammasome in neurodegenerative disorders still remains to be clarified. The underlying theories (e.g., danger signal theory) along with the signaling pathways that link the inflammasome to acute neurodegeneration will be discussed here. Furthermore, the stimuli that potentially activate the inflammasome in cerebral ischemia will be specified, as well as their relation to well-known pathways activating the innate immune response (e.g., Toll-like receptor signaling) and the consequences that result from their activation (beneficial versus deleterious).
Inflammation in Neurodegenerative Disorders
Acute brain injury or tissue stress (e.g., ischemic or traumatic) elicits acute inflammation that in turn exacerbates primary brain damage. Activation of the innate immune system (i.e., neutrophils, macrophages, microglia, as well as the noncellular components, such as the complement system) is an important part of the inflammatory response. Accordingly, microglial activation is a hallmark of several central nervous system (CNS) disorders, including multiple sclerosis, Alzheimer's disease, HIV encephalitis, dementia, and ischemic or traumatic brain injury. These diseases are associated, in variable degrees, with the neuronal damage that is substantially mediated by the inflammatory reaction (Dirnagl et al, 1999; Block and Hong, 2005; Lucas et al, 2006). The inflammatory reaction is predominantly sustained by microglia, but the astrocytes may also play a major role (Trendelenburg and Dirnagl, 2005; Bramlett and Dietrich, 2004). Products of activated glia have implicated in promoting cell death of neighboring neurons. There is in vivo evidence for a direct link between activation of innate immune response and neuronal injury in the CNS (Block and Hong, 2005, Lucas et al, 2006; Xiong et al, 2003).
Toll-like receptors (TLRs) are important mediators of the innate immune response and significantly contribute to neuroinflammation, even in the absence of infectious pathogens (Chen et al, 2007; Babcock et al, 2006) and TLR-deficient mice are protected against ischemic brain damage (Ziegler et al, 2007; Caso et al, 2007; Cao et al, 2007; Tang et al, 2007).
Although a substantial amount of data supports the idea of a detrimental role of the inflammatory response in acute brain injury, there is also an increasing body of evidence showing a more beneficial role in inflammatory processes. Limited inflammation may promote, repair, and remodel the injured brain tissue, and restrict various toxic substrates released by the damaged cells.
Thus, a fine-tuned regulation of the innate immune system in the CNS is crucial for brain homeostasis.
Within the following pages, the mechanisms delineated are those that are thought to induce the inflammatory response. The focus is on the regulation of interleukin (IL)-1 processing, the pathways known to regulate caspase-1 activation, as well as the signals that are thought to trigger these events systemically and within the CNS.
Endogenous Activation of the Immune System: the Danger Model
Although the immunosurveillance has only restricted access to the brain, the injured CNS requires an innate immune intervention for the purpose of clearing apoptotic cells and toxic debris to limit damage and initiate tissue repair (Elward and Gasque, 2003). In contrast to the ‘foreign’ structures of pathogens (pathogen-associated molecular patterns (PAMPs)) in infectious diseases, the substrates that lead to the activation of the innate immune system in noninfectious disorders such as cerebral ischemia or traumatic brain injury must be endogenous (host-derived). Thus, the activation of the immune system in such conditions cannot simply be explained by discrimination between foreign and host-derived patterns.
A better idea is the ‘danger model,’ in which the tissue damage caused by the threat activates the innate immune system by endogenous stress or damage-associated molecular patterns (DAMPs) (Matzinger, 2002, 2007). Host-derived molecules released from injured tissue and cells are detected by receptors (e.g., TLRs), which in turn lead to the production of proinflammatory cytokines such as tumor necrosis factor (TNF), IL-1β, or IL-6.
Currently, there is an evolving list of endogenous immunostimulators (‘danger signals’ or DAMPs), such as hyaluronan, heat-shock proteins (HSPs), surfactant protein, interferon-α, uric acid, fibronectin, β-defensin, and cardiolipin. These molecules are thought to use the same receptors as the PAMPs, and many of these may initiate the inflammatory response in neurodegenerative diseases. In addition to the TLRs, which are associated with the plasma membrane or with vesicles, cytosolic proteins have recently been identified, which exhibit feature analogous to the TLRs. These proteins, called nucleotide-binding oligomerization domain (NOD)-like receptor proteins (NLRs), also contain leucine-rich repeats (LRRs), which may indicate that they are responsible for sensing danger signals in the cytoplasm (Strober et al, 2006; Akira and Takeda, 2004).
Before specifying the potential danger signals and receptors that are characteristic of acute brain injury, the processing and regulation of proinflammatory cytokines such as IL-1β will be discussed.
Proinflammatory Cytokines such as IL-1 and Their Significance in Acute Brain Injury
Interleukin-1, with its subtypes IL-1α and IL-1β, is a potent pyrogen. It is considered to be a master cytokine that mediates both the innate and adaptive immune response either directly or by induction of other cytokines such as IL-6 or TNF-α. There is a plethora of data supporting a central role of the proinflammatory IL-1 in acute brain injury (Allan et al, 2005; Emsley et al, 2005; Dinarello, 2005). Interleukin-1 might have indirect neurotoxic effects by activating glial cells, which in turn release proinflammatory cytokines. It may also have direct neurotoxic effects by, for example, enhancing the seizure activity or Ca2+ entry.
However, there are also data pointing to a neuroprotective role of IL-1 action in neurons, an effect that may be mediated by inhibition of glutamate release, inhibition of Ca2+ entry, or enhancement of synaptic inhibition by γ-aminobutyric acid (Allan et al, 2005).
Nevertheless, IL-1 has been shown to be a key mediator of experimentally induced neurodegeneration and its inhibition has proven to be neuroprotective in vitro and in vivo (Allan et al, 2005; Friedlander et al, 1997; Betz et al, 1996; Pinteaux et al, 2006; Schielke et al, 1998; Boutin et al, 2001; Hara et al, 1997a, b).
Although there is evidence for a significant contribution of IL-1α in injury-induced inflammation in vivo, most of the data concern the subtype IL-1β (Chen et al, 2007). The processing and release of IL-1β depend on caspase-1 activation.
The expression of IL-1β is primarily regulated through nuclear factor-κB(NF-κB) signaling, resembling that of other proinflammatory cytokines such as interferon-γ or TNF-α (Thornberry et al, 1992; Allan et al, 2005). Various proinflammatory signals can enhance the transcription of IL-1β, including hypoxia, complement components, prostaglandin E2, the β-chain of the S100 calcium-binding protein, excitotoxins (excitatory amino-acid receptor agonists), various TLR agonists, as well as IL-1β itself (Allan et al, 2005).
In contrast to NF-κB-regulated pro-IL-1β expression, processing of IL-1β is dependent on a cytosolic complex that includes NLR proteins (see below). Thus, the IL-1β system with its associated TLR and NLR signaling pathways shows how both extracellular and intracellular innate immune signaling pathways interact (Delbridge and O'riordan, 2007; Mariathasan and Monack, 2007; Allan et al, 2005; Creagh and O'Neill, 2006).
However, proinflammatory signaling is not exclusively mediated by IL-1β or other more caspase-1-independent cytokines (e.g., TNF-α or IL-6). It may also be determined by the complex time- and site-specific expression profiles of the corresponding receptors and downstream signaling partners. Thus, the following concept of the inflammasome should only be regarded as a model and may represent a starting point for further hypotheses.
Activation of Caspase-1: the Inflammasome
A major portion of the signaling pathways ‘upstream’ of caspase-1 has recently been unraveled with the discovery of the ‘inflammasome.’ Similar to the activation of caspase-9 or caspase-8 by the Apaf-1 (apoptotic protease-activating factor 1)-apoptosome or Fas/CD95-DISC (death-inducing signaling complex), it has been shown that caspase-1 is activated by an ˜700-kDa multiprotein complex termed the ‘inflammasome’ (Martinon et al, 2002; Ogura et al, 2006). Caspase-activating platforms such as the inflammasome are typically present in the cytoplasm as inactive monomers and oligomerize only upon the reception of a specific signal, such as the binding of a ligand.
To process the proinflammatory cytokines IL-1β, IL-33, and IL-18, the assembly of different adaptor proteins activates caspase-1 and caspase-5. The complex is also able to interact with NF-κB-dependent pathways. In addition, inflammasome activation can lead to host cell death in certain cell types (Richards et al, 2001; Ohtsuka et al, 2004; Park et al, 2007a) (Figure 1).
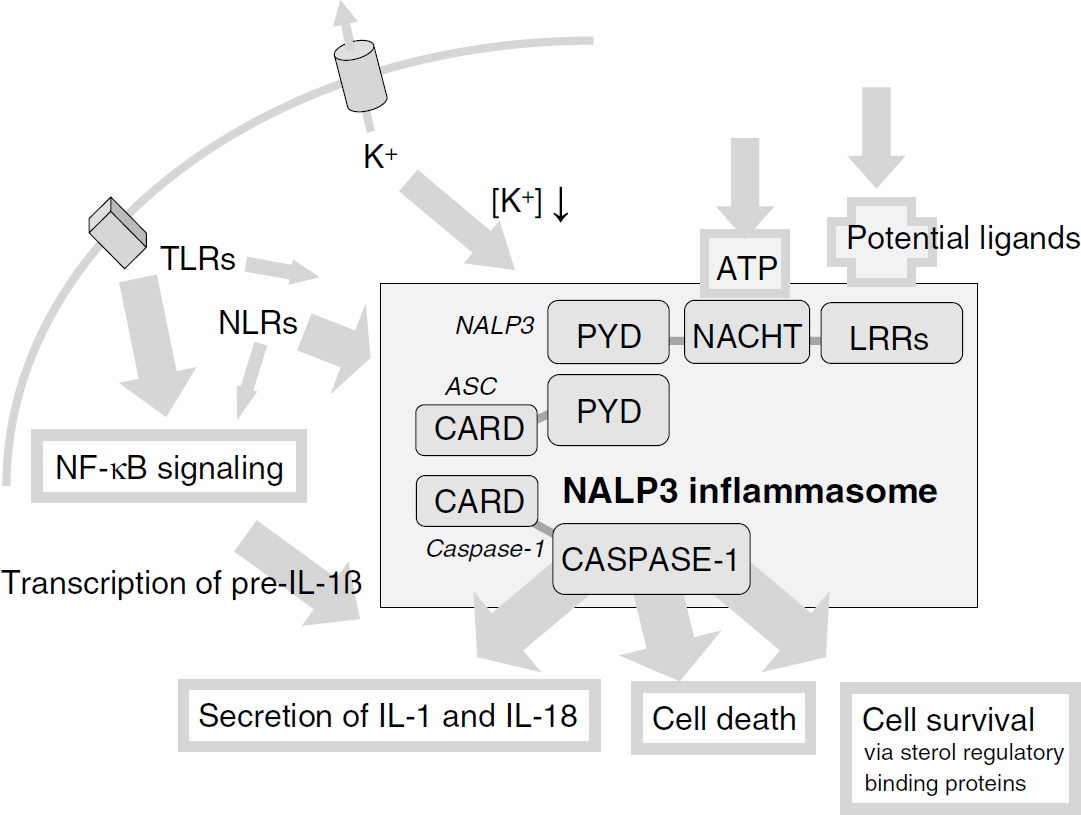
Composition of the NALP3 inflammasome. The murine NALP3 inflammasome is composed of NALP3, ASC, and caspase-1 (a second adaptor protein called CARDINAL exists only in humans). ASC interacts with one of the NALP proteins through cognate pyrin domain (PYD) interactions and with pro-caspase-1 through homotypic CARD (caspase recruitment domain) interactions. The human inflammasome complex brings two molecules of pro-caspase-1 (the second via CARDINAL) into close proximity, leading to autocatalysis and the subsequent release of the active catalytic p20 and p10 domains of caspase-1. NALP3 binds ATP via the NACHT domain (nucleoside triphosphatase (NTPase) domain), is an ATPase, and requires ATP binding for inflammasome activation (Duncan et al, 2007). Caspase-1, in turn, cleaves the precursor of IL-1β into its biologically active fragment, a potent mediator of fever and inflammation. There is no CARDINAL homolog in the mouse and, hence, murine NALP3 is thought to recruit only a single caspase-1 molecule. (TLRs, Toll-like receptors; ATP, adenosine triphosphate; NLRs, nucleotide-binding oligomerization domain (NOD)-like receptors; ASC, apoptosis-associated speck-like protein containing a CARD; NALP, NACHT-, LRR-, and pyrin domain-containing protein; LRRs, leucine-rich repeats).
Types of Inflammasomes
The inflammasome can comprise caspase-1 or caspase-5; members of the NLR family, such as IPAF (ICE-protease-activating factor) or NALPs (NACHT-, LRR-, and pryin domain-containing proteins); and adaptor molecules such as ASC (apoptosis-associated speck-like protein containing a CARD (caspase recruitment domain)) or CARDINAL (CARD inhibitor of NF-κB-activating ligands). Until now, at least three inflammasomes have been described: the IPAF, NALP1, and the NALP2/3 inflammasomes. The IPAF inflammasome is thought to be a homo-oligomer, consisting of IPAF and caspase-1. The NALP1 inflammasome is composed of NALP1, ASC, caspase-1, and caspase-5 (which does not exist in mice). The NALP2/3 inflammasome contains, in addition to NALP2 or NALP3, CARDINAL (which does not exist in mice), ASC, and caspase-1 (Martinon and Tschopp, 2007; Kummer et al, 2007; Sutterwala et al, 2007; Poyet et al, 2001; Agostini et al, 2004; Lamkanfi et al, 2007; Ogura et al, 2006).
Members of the NLR family (e.g., IPAF or NALPs) contain either a CARD or pyrin domain (PYD) caspase interaction motif, a typical nucleotide-binding oligomerization domain (NACHT), and a ligand-sensing site—which is composed of LRRs (also occurring in TLRs) (Martinon and Tschopp, 2004). ASC is an essential component of most NALPs (it connects the NALPs to caspase-1), and it may also play a role in the IPAF inflammasome (Masumoto et al, 1999, 2001a,b, 2003; Miao et al, 2006). Moreover, ASC has recently been shown to be involved in the caspase-1-independent induction of proinflammatory cytokines (e.g., IL-6 or TNF) by TRLs (Taxman et al, 2006).
Several inflammasome-associated autoinflammatory disorders have already been identified. Three autosomal dominant diseases are associated with a gain-of-function mutation in the NALP3 gene: familial cold autoinflammatory syndrome, Muckle-Wells syndrome, and chronic infantile neurologic cutaneous and articular syndrome (Agostini et al, 2004; Inohara et al, 2005; Ogura et al, 2006). These autoinflammatory syndromes are characterized by recurrent episodes of fever, skin rashes, and tissue inflammation, and there is a possibility that they might be treated with the IL-1 antagonist anakinra (Fisher et al, 1994; Liao et al, 1984; Dinarello, 2005; Braddock and Quinn, 2004; Goldbach-Mansky et al, 2006; Hawkins et al, 2004; Hoffman et al, 2004).
Loss-of-function mutations in another NLR family member, NOD2, have been associated with a susceptibility to autoinflammatory diseases such as Crohn's disease and Blau's syndrome (Ogura et al, 2001). Familial Mediterranean fever is caused by a deficiency in an inflammasome inhibitor known as Pyrin (the familial Mediterranean fever gene). Homozygous loss-of-function mutations in Pyrin, thought to negatively regulate NALP3 signaling by disrupting the assembly of the inflammasome, may lead to enhanced inflammation (Inohara et al, 2005; Chae et al, 2006).
Activation of Inflammasomes: TLRs
The TLR family is the best-characterized group of proteins that are known to bind danger signals. Toll-like receptors are single-spanning transmembrane proteins with ectodomains largely composed of LRRs as well as a cytoplasmic domain that consists of a TIR (Toll/IL-1 interacting) domain. In contrast to TLRs such as TLR2 and TLR4, which are located on the cell surface, some TLR family members (TLR3, TLR7, TLR8, TLR9) are located intracellularly on vesicle membranes (Akira and Takeda, 2004).
Toll-like receptors recognize the corresponding PAMPs or DAMPs through the LRRs in their extracellular domains, which have been implicated in ligand binding and autoregulation. Various PAMPs activate individual TLRs: TLR2 is essential for the recognition of lipopeptides, peptidoglycan, or lipoteichoic acid, TLR4 recognizes lipopolysaccharide (LPS), TLR3 is activated by double-stranded viral RNA, TLR5 mediates the recognition of flagellin, TLR9 recognizes unmethylated CpG-containing DNA, and TLR7 and TLR8 bind single-stranded RNA (Creagh and O'Neill, 2006; Mishra et al, 2006; Mariathasan and Monack, 2007; Akira and Takeda, 2004).
Most TLRs are known to be expressed in the CNS, especially in microglial cells, and have been found to be induced during different pathologic conditions (Bsibsi et al, 2002, 2006; Kariko et al, 2004a; Babcock et al, 2006; Nishimura and Naito, 2005; Bowman et al, 2003; Böttcher et al, 2002; McKimmie et al, 2005; Tang et al, 2007). Various molecules released from damaged cells or the extracellular matrix have been identified as endogenous activators of TLRs (Kariko et al, 2004a,b; Seong and Matzinger, 2004).
A key functional outcome of TLR ligation is the production of inflammatory cytokines through transcription factors such as NF-κB. Most TLRs require the adaptor protein MyD88 (myeloid differentiation primary response gene 88) to induce transcription of cytokines such as IL-1β, IL-6, and TNF-α through NF-κB signaling, but can also recruit further adaptor molecules and transcription factors (Beutler et al, 2006; Deane and Bolland, 2006; Creagh and O'Neill, 2006).
There has been speculation about the possible superior role of TLR2 and TLR4 signaling in acute brain injury (Babcock et al, 2006; Lehnardt et al, 2003; Coban et al, 2007; Mishra et al, 2006; Maslinska et al, 2004; Tang et al, 2007). Toll-like receptor 2, a receptor with a wide range of ligands, is expressed in several CNS diseases, primarily in microglial and mononuclear cell infiltrates (Mishra et al, 2006; Babcock et al, 2006; Maslinska et al, 2004; Kielian, 2006), but can also be found in neurons (Tang et al, 2007; Ziegler et al, 2007). It has been suggested that oxidant stress is sensed by TLR2-dependent pathways (Mitchell et al, 2007; Zhang et al, 2003). Endogenous TLR2 signaling strongly impacts early glial cytokine and chemokine responses and has been shown to exacerbate postischemic damage in different organs, including the brain (Favre et al, 2007; Ziegler et al, 2007; Leemans et al, 2005; Aliprantis et al, 2000; Hoffmann et al, 2007; Babcock et al, 2006).
The LPS-binding molecule CD14 not only signals through TLR4, but also through TLR2 and is expressed by activated microglia in ischemic brain injury (Aliprantis et al, 2000; Beschorner et al, 2002; Bsibsi et al, 2007; Özören et al, 2006; Scott et al, 2006). The class B scavenger receptor CD36 also signals through TLR2. CD36 has recently been shown to mediate free radical production and tissue injury in cerebral ischemia (Cho et al, 2005; Hoebe et al, 2005).
Toll-like receptor 4 is also expressed in the CNS. It is induced by oxidative stress, and it exacerbates ischemic injury in vivo (Lehnardt et al, 2003; Caso et al, 2007; Cao et al, 2007; Kielian, 2006; Tang et al, 2007; Powers et al, 2006). Moreover, polymorphisms of the TLR4 gene have been shown to have a significant association with an increased risk for cerebral ischemia (Lin et al, 2005). Monosodium urate, heat-shock proteins (Hsp-60, Hsp-70), heparan sulfate, fibrinogen, and hyaluronan have been identified as endogenous TLR4 ligands. They may be released from the cell surface and are linked to caspase-1 activation and IL-1β processing (Elward and Casque, 2003; Scott et al, 2006; Stevens and Stenzel-Poore, 2006). For example, high-mobility group box 1 protein (HMGB1), an endogenous ligand potentially triggering the neuroinflammatory response, has been shown, depending on the cell type examined, to differentially use TLR2 or TLR4 signaling for IL release (Yu et al, 2006). It has also been shown to trigger ischemic liver damage through TLR4 signaling (Tsung et al, 2005).
The TLR3, TLR7, TLR8, and TLR9 are expressed in the CNS and represent further candidate receptors for the initiation of the innate immune response after acute brain injury. These innate receptors recognize nucleic acid types that normally are found in bacteria and viruses but which can also be found within mammalian cells (Deane and Bolland, 2006; Stevens and Stenzel-Poore, 2006; Bsibsi et al, 2007).
Toll-like receptor 9, for example, normally responds to bacterial DNA, viral DNA, and synthetic oligodeoxynucleotides, all of which contain unmethylated CpG motifs. However, some CpG dinucleotides within the mammalian genome are unmethylated and should therefore be recognized by TLR9 (Beutler et al, 2006; Kielian, 2006; Kariko et al, 2005).
The microglial-mediated neuronal toxicity as a result of CpG oligodeoxynucleotide treatment is reminiscent of what has been observed after stimulation with the TLR4 agonist LPS or a synthetic TLR2 agonist. This suggests that the microglial response to diverse PAMPs or DAMPs has been conserved and that there may be a link between TLRs and neurodegeneration (Kielian, 2006; Hoffmann et al, 2007; Lehnardt et al, 2003).
However, this does not preclude that TLRs might influence neuronal survival or regeneration, or modulate neurogenesis, as has been shown very recently for TLR2 and TLR4 (Rolls et al, 2007). Indeed, TLR ligands exert both beneficial and pathologic effects in the CNS. Accordingly, administration of TLR agonists in the injured CNS has been shown to promote regeneration or myelin clearance, whereas direct injection of TLR ligands into the healthy brain or spinal cord induced robust inflammation that caused tissue damage (Babcock et al, 2006).
Activation of Inflammasomes: NLRs
Whereas extracellular PAMPs and DAMPs are recognized by membranous TLRs, surveillance of the cytoplasm is thought to be the duty of the NLR family of proteins. The NLR family is composed of 23 soluble cytosolic proteins including NALPs, NOD1, NOD2, and IPAF, all of which are expressed primarily in immune cells, although the expression of certain proteins, such as NOD1, is ubiquitous (Zedier and Faist, 2006; Delbridge and O'riordan, 2007; Kanneganti et al, 2006b; Inohara et al, 2005; Swanson and Molofsky, 2005; Ting et al, 2006).
The cytosolic NLRs are thought to sense specific danger signals with their LRR domains analogous to the TLRs. Recent studies revealed a specific role of Gram-positive pathogens such as Staphylococcus aureus or Listeria monocytogenes in activating the NALP3 inflammasome, whereas Gram-negative pathogens such as Salmonaella typhimurium, Legionella pneumophila, or Shigella flexneri mediate caspase-1 activation through IPAF (Mariathasan and Monack, 2007). NALPs may also be crucial for detecting tissue injury and they represent key drivers for IL-1β production. NALP3 for example is able to sense monosodium urate, calcium pyrophosphate dehydrate crystals, as well as low intracellular potassium concentration. Thus, factors that induce K+ efflux, such as certain toxins, hypotonic stress, and high concentrations of extracellular adenosine triphosphate (ATP), can be sensed by NALP3 (Kanneganti et al, 2006b; Kahlenberg and Dubyak, 2004; Creagh and O'Neill, 2006; Petrilli et al, 2007).
The minimal requirements for inflammasome assembly have recently been characterized: in vitro reconstitution of the NALP1 inflammasome revealed that the activation of caspase-1 is a process requiring at least a bacterial ligand such as muramyl dipeptide, pro-caspase-1, NALP1, as well as ATP (Faustin et al, 2007).
Interplay between TLR Signaling and the Inflammasome/NLRs
There are many hints for fine-tuned crosssignaling between caspase-1 activation by the inflammasomes and TLR signaling (Akira and Takeda, 2004; Strober et al, 2006; Zhang et al, 2003; Taxman et al, 2006; Yoo et al, 2002). Toll-like receptor ligands such as flagellin, imiquimod, or single-stranded RNA not only activate TLRs, but also signal through IPAF or NALP inflammasomes (Kanneganti et al, 2006a, b; Prins et al, 2006). Further complexity has recently been noted by the characterization of a third class of PAMP receptors, which are called the retinoic acid-inducible I-like helicases (including retinoic acid-inducible I and MDA-5 (melanoma differentiation associated gene 5). These helicases have been shown to bind to viral RNA and self-RNA (Malathi et al, 2007; Meylan et al, 2006). Toll-like receptor agonists are known to not only activate IL-1 maturation, but also regulate transcription of pro-IL-1, NLRs (e.g., NALP3), murine caspase-11 (which corresponds to human caspase-4 and caspase-5, and seems to be required for caspase-1 activation in mice), or caspase-1 through NF-κB (Mariathasan and Monack, 2007; O'Connor et al, 2003; Kanneganti et al, 2006a, b; Martinon and Tschopp, 2004; Kang et al, 2000; Wang et al, 1998). Interleukin-1 and other proinflammatory cytokines are capable of activating NF-κB as well. Furthermore, it was shown that caspase-1 activity cleaves MyD88 adaptor-like (MAL/TIRAP), a protein that has been shown to regulate TLR2-mediated NF-κB and p38 mitogen-activated protein kinase activation. Accordingly, MAL/TIRAP deficiency reduces the functional deficit after ischemia/reperfusion injury in the heart (Miggin et al, 2007; Sakata et al, 2007). There are data arguing for a modulating role of specific NLRs (e.g., NODI, NOD2) in TLR signaling: for example, the adaptor protein RICK (RIP-like interacting CLARP kinase) is thought to mediate both NLR and TLR signaling (Strober et al, 2006; Watanabe et al, 2004; Kobayashi et al, 2002; Netea et al, 2005; Thome et al, 1998), although this has recently been questioned (Park et al, 2007b). RICK and the NLR family member NOD2 are expressed in the CNS and are induced after exposure to TLR ligands. NOD2 was shown to be a modulator of signals transmitted through TLR2, TLR3, and TLR4, and NOD2 ligands were shown to augment TLR2-mediated immune responses in astrocytes (Sterka et al, 2006; Netea et al, 2005). Moreover, NOD2 was shown to mediate neuronal damage in an early sepsis model, as it is true for TLR2 (Orihuela et al, 2006). There is evidence that the NLR system is largely independent of the TLR system and, as such, might positively or negatively modulate TLR responses (Strober et al, 2006). Interestingly, there is a synergistic interaction between NOD2 and TLR2 signals in mononuclear cells, resulting in a shift toward a Th2-type response, which also occurs in stroke-induced immunodeficiency (Watanabe et al, 2004; Netea et al, 2005; Prass et al, 2003). However, it still remains to be resolved to what extent TLRs contribute to neuroinflammation or activation of the inflammasome, as well as to what extent each TLR contributes to regenerative processes in cerebral ischemia.
Potential Endogenous Danger Signals in Acute Neurodegeneration
Because various TLRs, NLRs, and inflammasome components are expressed in the CNS, the inflammasome is thought to play a significant role in triggering the inflammatory response in acute brain injury (Kinoshita et al, 2005; Kummer et al, 2007; Zhao et al, 2007; Liu et al, 2004). In addition to bacterial PAMPs, such as flagellin, peptidoglycan, or bacterial RNAs, there are several endogenous signals released from dying cells that potentially activate the inflammasome in neurodegenerative diseases (Mariathasan et al, 2005; Kanneganti et al, 2006a, b; Martinon and Tschopp, 2004; Kummer et al, 2007). Substances such as RNA or DNA that are released from necrotic cells can activate the inflammasome or can bind to TLRs (Matzinger, 2007; Martinon and Tschopp, 2007; Ogura et al, 2006; Kanneganti et al, 2006b; Shi et al, 2003; Kariko et al, 2004b) (Figure 2).
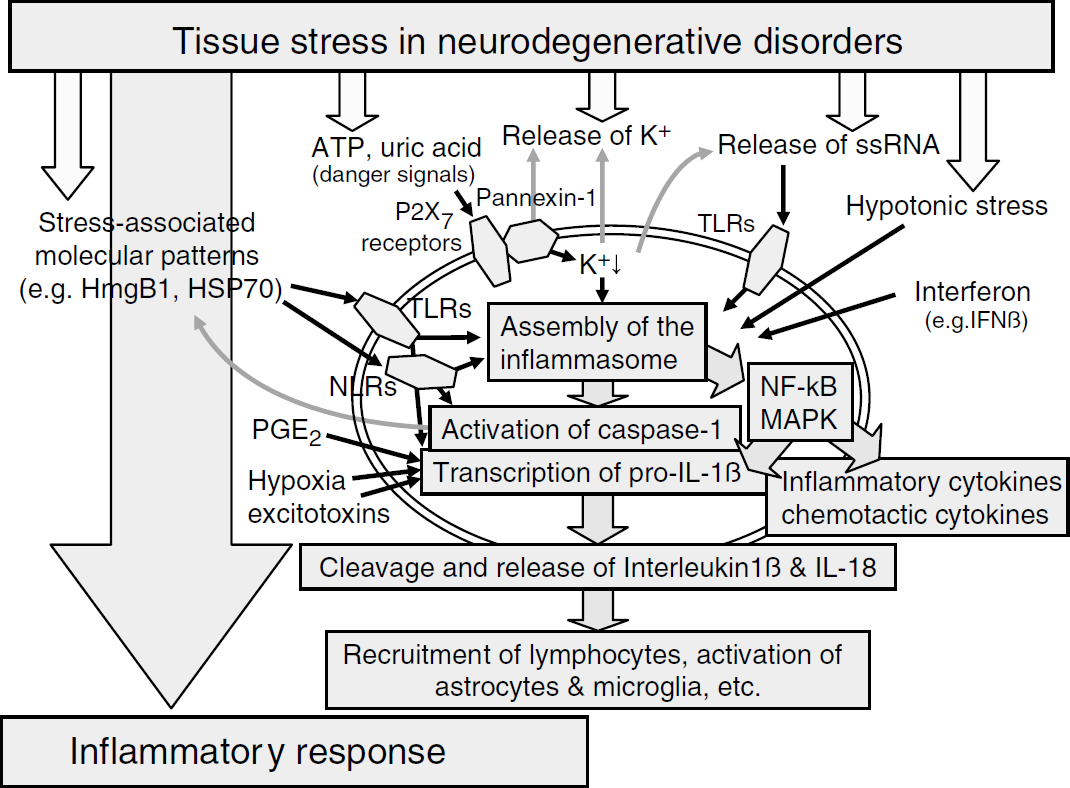
The inflammasome and its role in connecting the various local stress signals with the initiation of the inflammatory response (excitotoxins, excitatory amino-acid receptor agonists; HmgB1, high-mobility group box protein 1; HSR heat-shock protein; IFN-β, interferon-β, a type I interferon; IL, interleukin; MAPK, mitogen-activated protein kinase; NLR, nucleotide-binding oligomerization domain (NOD)-like receptor; PGE2, prostaglandin E2; TLRs, Toll-like receptors).
Although several stimuli, such as bacterial RNA, bacterial toxins, ATP, calcium pyrophosphate dehydrate, and uric acid crystals, have been shown to activate the NALP3 inflammasome, it is unlikely that these various stimuli can all activate NALP3 directly. The NALP3 inflammasome is more likely activated by a common intracellular ‘danger signal’ such as low intracellular potassium concentration or endogenously generated uric acid (Creagh and O'Neill, 2006; Ogura et al, 2006; Seong and Matzinger, 2004; Petrilli et al, 2007). Injured cells rapidly degrade their RNA and DNA and the liberated purines are converted into uric acid, resulting in its accumulation. The production of uric acid does not require protein synthesis, and inhibition of protein synthesis is a critical hallmark of the early ischemic tissue damage (Dirnagl et al, 1999; Hossmann, 2006; Shi et al, 2003; Kanemitsu et al, 1988; Bos et al, 2006). Uric acid forms monosodium urate crystals, which have been shown to activate caspase-1 activation and IL-1β processing by binding to TLR2, TLR4, CD14, or the NALP3 inflammasome (Shi et al, 2003; Scott et al, 2006; Ogura et al, 2006; Martinon et al, 2006; Martinon and Tschopp, 2007; Seong and Matzinger, 2004). Eliminating this endogenous danger signal in vivo was shown to inhibit the immune response to antigens associated with injured cells (Shi et al, 2003). Accordingly, high serum urate levels have been linked to poor outcome and higher vascular event rates in stroke patients (Weir et al, 2003; Hozawa et al, 2006; Bos et al, 2006).
With the HSP family, including HSP60, HSP70, and HSP90, another host-derived group of endogenous ligands for TLRs (and very recently also for NLRs) (Mayor et al, 2007) has been identified. Heat-shock proteins are released by necrotic but not apoptotic cells, and have been shown to be induced in neuroinflammation and infection. Although existing data argue for a mainly neuroprotective effect of HSPs, these proteins are also able to induce inflammation (Zedier and Faist, 2006; Dirnagl et al, 1999, 2003; Asea et al, 2002; Vabulas et al, 2002; Basu et al, 2000; Svensson et al, 2006). Interestingly, HSP90, in concert with the ubiquitin ligase-associated protein SGT1 (suppressor of G2 allele of SKP1), has recently been shown to interact with NLRs to activate caspase-1 (Mayor et al, 2007).
Extracellular ATP and K+ efflux represent further danger signal candidates in cerebral injury. Both ATP and K+ were shown to be involved in cerebral injury and both trigger inflammasome assembly in vitro (Gurcel et al, 2006; Cruz et al, 2007; Mariathasan et al, 2006). The best-studied model of caspase-1 activation is the exposure of cells to extracellular ATP. Adenosine triphosphate activates purinergic receptors P2X (P2X7) with its associated hemichannel pannexin-1 and leads to potassium efflux, plasma membrane depolarization, cell swelling, and disaggregation of the cytoskeletal network (Khakh and North, 2006; Kanneganti et al, 2007; Martinon and Tschopp, 2007; Kahlenberg and Dubyak, 2004; Franchi et al, 2007; Solle et al, 2001). The intracellular K+ concentration is known to decrease during hypoxic/ischemic cell injury and, interestingly, NALP3 and NALP1 (but not IPAF) activation has recently been shown to depend on low intracellular K+ concentration (Petrilli et al, 2007; Somjen, 2001). Accordingly, there is in vitro evidence that extracellular ATP is involved in inflammatory responses and neuronal death induced by ischemic stress. ATP induces the transcription of genes for the oxidative stress response in macrophages and activates caspase-1 as well as the secretion of IL-1β through a reactive oxygen species-dependent phosphoinositide-3-kinase pathway (Cruz et al, 2007).
However, although ATP levels can increase significantly under inflammatory and ischemic conditions in vivo and in vitro, the high concentrations of ATP that are required for caspase-1 activation are normally not found in the extracellular milieu (Le Feuvre et al, 2003; Trendelenburg and Dirnagl, 2005; Dirnagl et al, 1999; Kanneganti et al, 2007; Ferrari et al, 2006). In vitro results with both P2X7- and K+-loss-induced IL-1β processing support a model in which a putative lipid second messenger is generated by the cytosolic calcium-independent phospholipase A2 and subsequently activates caspase-1 (Kahlenberg and Dubyak, 2004; Mariathasan and Monack, 2007). Phospholipase A2 activity has been found to be induced after cerebral ischemia and phospholipase A2 isoforms have been implicated in cell injury and death by their ability to mediate inflammatory responses (Bonventre et al, 1997; Adibhatla et al, 2006; Cummings et al, 2000). However, the deletion of the P2X7 receptor does not affect neuronal death after cerebral ischemia in vivo, and P2X7 knockout mice can still be protected by the use of an IL-1 antagonist (Le Feuvre et al, 2003).
The complement system may also contribute to caspase-1 activation by provoking K+ efflux, because disruption of the cell membrane is a consequence of activation of the complement system and formation of the terminal membrane attack complex. The TLRs and complement system are two well-characterized arms of the immune system, which are known to play a key role in priming the adaptive immune system. It has recently been shown that both systems interact at the molecular level in vivo (Zhang et al, 2007). However, the exact contribution of the complement system in neurodegeneration still remains controversial (Stahel et al, 1998; Trendelenburg and Dirnagl, 2005; Xiong et al, 2003; Huang et al, 1999, Del Zoppo, 1999; Elward and Casque, 2003; van Beek et al, 2003; D'Ambrosio et al, 2001).
Heparan sulfate, a biologically active saccharide released from cell surfaces during almost every type of inflammation, activates dendritic cells as fully as LPS and may further represent a danger signal during brain injury. Both heparan sulfate and fragments of hyaluronic acid, a related saccharide, act through TLR4 (Zedier and Faist, 2006; Leadbeater et al, 2006; Gomez-Pinilla et al, 1995). Myeloid-related protein-8, an agonist for TLR4 signaling that has recently been identified, represents a further endogenous danger signal (Vogl et al, 2007). The list of endogenous molecules that are induced after brain injury and which activate cells through TLR-dependent pathways also includes fibronectin and saturated fatty acids (Kielian, 2006; Tate et al, 2007; Yanqing et al, 2006).
Another group of structurally diverse multifunctional host proteins that are rapidly released after pathogen challenge or cell stress is comprised of defensins, cathelicidins, eosinophil-derived neurotoxins, and HMGB1. Cathelicidin, defensins, or its rodent homologs are antimicrobial peptides that have also been shown to be expressed in the CNS. These peptides, as well as eosinophil-derived neurotoxin and HMGB1, are able to both recruit and activate antigen-presenting cells (Bergman et al, 2005; Hao et al, 2001; Zedier and Faist, 2006).
The nuclear protein HMBG1 is involved in transcriptional activation and DNA folding. In addition to its nuclear role, extracellular HMGB1 has been shown to be a critical mediator of the innate immune response to infection and also ischemic brain injury (Tsung et al, 2005; Qiu et al, 2007). HMGB1 is released by necrotic cells and activated macrophages; however, it is not released by apoptotic cells. HMGB1 interacts with TLRs and the receptor for advanced glycation end products, resulting in the activation of NF-κB and the release of proinflammatory cytokines such as TNF-α, IL-6, or IL-1β (Orlova et al, 2007; Scaffidi et al, 2002; Lotze and Tracey, 2005; Park et al, 2004; Qiu et al, 2007). Besides its proinflammatory role, HMGB1 has also been identified as an active player during tissue repair, stimulating homing of endothelial progenitor cells to ischemic tissues (Chavakis et al, 2007).
Whereas some stimuli mentioned above may result in the assembly of the inflammasome through TLR signaling, several of them may also be detected directly by the LRRs of cytosolic NLR proteins, or by a hypothetical common ‘danger signal’ mediator such as low intracellular potassium or uric acid. Furthermore, genomic responses under neuronal injury (e.g., stroke) may alter and induce specific components of TLR or NLR pathways, as shown, for example, for NOD1, NOD2, NALP1, TLR2, TLR4, or RICK/RIP2, thus fine-tuning the innate immune response (Sterka et al, 2006; Tang et al, 2007; Ziegler et al, 2007; Liu et al, 2004).
Inflammasome Activation and Its Potential Role in Neuroprotection
Besides the essential role of caspase-1 in the generation of the proinflammatory cytokine IL-1β, its activation has also been shown to promote cell survival by activating protective pathways. Toxin-induced K+ efflux in human fibroblasts leads to improved cell survival by the inflammasome-mediated induction of lipogenic genes (Gurcel et al, 2006; Saleh, 2006). Thus, pathophysiologic conditions associated with lowering of cytoplasmic K+ concentration may trigger specific signaling pathways, some of which are aimed at triggering the immune system (e.g., by the induction of IL-1β processing), whereas others promote death or survival of the attacked cell (Petrilli et al, 2007; Mariathasan and Monack, 2007; Gurcel et al, 2006).
These data support the hypothetical neuroprotective abilities of inflammasome activation, for example, for ischemic preconditioning phenomena, and are consistent with the observation that proinflammatory substances such as LPS or IL-β are able to induce ischemic tolerance (Dirnagl et al, 2003; Shi et al, 2003; Akahoshi et al, 2006; Glantz et al, 2005; Romanos et al, 2007; Stenzel-Poore et al, 2007). It is well known that small doses of an otherwise harmful (or proinflammatory) stimulus can induce protection against a subsequent injurious challenge, thereby causing a ‘preconditioned’ state with activated antiinflammatory and neuroprotective pathways (Stenzel-Poore et al, 2007; Izuishi et al, 2006; Dirnagl et al, 2003; Hossmann, 2006).
Inflammasome Modulation
Although a lot of data already exist regarding the regulation of TLR signaling in the brain, there is only a small quantity of information available regarding the regulation of the cytosolic surveillance system (Prins et al, 2006; Kariko et al, 2004a; Kielian, 2006; Stevens and Stenzel-Poore, 2006; Hoffmann et al, 2007; Kinoshita et al, 2005).
Toll-like-receptor-mediated signaling is regulated at several levels and at least five levels of negative regulation have so far been uncovered (Liew et al, 2005). These include extracellular decoy receptors, intracellular inhibitors, membrane-bound suppressors, degradation of TLRs, and TLR-induced apoptosis. Soluble splicing variants of TLR2 and TLR4 were identified and have been shown to attenuate TLR-mediated NF-κB activation. The most likely mechanism through which these TLR variants exert this effect is by blocking the interaction between TLRs and other co-receptor complexes, such as CD14. Moreover, a short form of MyD88— the most crucial adaptor in TLR signaling—has been shown to be expressed in the brain; it has also been shown to inhibit IL-1- and LPS-mediated NF-κB activation. Further negative regulators of TLRs have been shown to interfere with the signaling pathways of transmembrane protein regulators or various other levels further downstream: for example, SIGIRR (single immunoglobulin interleukin-1-related receptor), TRAILR (tumor necrosis factor-related apoptosis-inducing ligand receptor), RP105 (which negatively regulates TLR4 signaling), IR-AKM (interleukin-1 receptor-associated kinase-M; the expression of which is restricted to macrophages and monocytes), SOCS1 (suppressor of cytokine signaling 1), NOD2 (nucleotide-binding oligomerization domain protein 2), phosphoinositide-3-kinase, TOLLIP (Toll-interacting protein), Bcl-3 (B-cell leukemia-3), or A20. Moreover, TLR signaling can be controlled by TLR-induced apoptosis or the reduced expression of TLRs, the complement system, or downstream signaling molecules (e.g., ASC) (Kariko et al, 2004a; Naiki et al, 2005; Liew et al, 2005; Wald et al, 2003; Zhang et al, 2007; Miggin et al, 2007; Akira and Takeda, 2004; Divanovic et al, 2005; Taxman et al, 2006; Carmody et al, 2007).
Although little is known about the regulation of the cytosolic immune surveillance system at the physiologic level, several proteins have been proposed to interfere with inflammasome assembly and caspase activation (Shiohara et al, 2002; Martinon and Tschopp, 2007; Park et al, 2007a). Two types of inflammasome regulators can be distinguished based on their modular structure. The first type is characterized by the presence of a CARD (a group including decoy caspase-1 genes such as iceberg, INCA (inhibitory caspase recruitment domain protein), or human caspase-12) and inhibits caspase-1 activation by interacting with caspase-1, hypoxia-inducible RICK/RIP2, or ASC CARDs. The second type is characterized by the presence of a PYD and is thought to interfere with interactions between ASC and NALPs (such as pyrin, POP (pyrin only protein), PYNOD (protein containing a PYD and a NOD domain), and viral PYDs) (Martinon and Tschopp, 2007; Chae et al, 2006; Wang et al, 2004; Kinoshita et al, 2005; Stehlik et al, 2002, 2003; Dorfleutner et al, 2007; Bedoya et al, 2007; Zhang et al, 2003). However, human COPs (CARD-only proteins) and POPs lack mouse orthologs and are therefore not part of murine inflammasome regulation (Stehlik and Dorfleutner, 2007).
The clinical significance of such interactions has been documented by the mutations in the Pyrin gene, which cause the familial Mediterranean fever. NLR inhibitors also include the ErbB2-interacting protein; NOD2-S, a short isoform of NOD2; caspase-1 inhibitors, such as COP and ICEBERG; proteinase inhibitor 9 (PI-9); a decoy ASC molecule; and caspase-12 (Creagh and O'Neill, 2006; Saleh et al, 2006; Rosenstiel et al, 2006). Furthermore, viral negative regulators of caspase-1 activation were identified, such as the myxoma virus encoded M13L, which interacts with ASC to inhibit caspase-1 activation (Johnston et al, 2005; Mariathasan and Monack, 2007).
Beyond the manifold interactions mentioned above, one must also be aware of additional dimensions of complexity: not only the specific cellular expression pattern, but also the time-wise expression patterns of various inflammatory mediators. The regulation of the inflammatory response is highly dependent on the temporal nature of the inflammatory mediators, which rise as waves of gene expression and gene products and then shortly dissipate (Wang et al, 1995; Dirnagl et al, 1999). Accordingly, corresponding proteins have recently been detected in different cells in the brain, some of which (e.g., caspase-11; RICK/RIP2) are highly inducible under ischemic conditions (Kummer et al, 2007; Zhang et al, 2003; Kang et al, 2000), whereas others, such as TLR4, might alter the cellular distribution under ischemic conditions (Powers et al, 2006).
Whereas specific drugs interfering with inflammasome assembly are still under development, caspase-1 inhibitors (e.g., pralnacasan) and IL-1 antagonizing drugs (e.g., IL-1Ra or anakinra) have already been successfully introduced in clinical treatments for autoinflammatory diseases such as rheumatoid arthritis or gout (Dinarello, 2005; Fisher et al, 1994; Liao et al, 1984; So et al, 2007; Braddock and Quinn, 2004; Hauff et al, 2005) and recent results of a phase II study of IL-1Ra treatment in stroke patients add much promise (Emsley et al, 2005).
Nevertheless, it still remains to be proven whether the described signaling pathways (e.g., TLR signaling) identified in in vitro and in vivo models can be reproduced in the clinical setting. Whether the high expectations can be fulfilled in the treatment of human diseases still remains to be confirmed and one has to be aware of the fact that most of the antagonists or inhibitors still remain to be tested in humans.
It represents an enormous challenge to unravel the interactions that exist between membrane receptors for danger signals (e.g., TLRs), cytoplasmic receptors (e.g., NLRs), the inflammasomes, and their negative regulators. Because there is such a large impact of the innate immune response on the outcome of various neurodegenerative diseases, drugs interfering with the activation of the inflammatory response in the CNS are eagerly being awaited.
Summary
The prominent role of caspase-1 activation and IL-1β production in acute brain injury has been known for many years, but it is only recently that the multiprotein complex—the inflammasome—has been characterized. The inflammasome activates pro-caspase-1, which in turn processes IL-1β; however, it can also under specific circumstances lead to apoptotic cell death. It has been shown that specific pathogens and danger signals activate different inflammasomes and yet the exact contribution of each inflammasome in neurodegenerative disorders remains to be resolved. Activation of the inflammasome in neurons may lead directly to neuronal cell death (Liu et al, 2004), at the same time activation within microglial cells may induce inflammatory responses or repair mechanisms.
In conclusion, future research should not only focus on cell type-specific and time-wise activation patterns, but should examine the differential activation patterns of the various inflammasome types responsible for regulating tissue damage during brain injury.
Footnotes
Acknowledgements
I am grateful to Martin Holtkamp, Ronald Bottlender, and Ryan Cordell for critical reading of the manuscript.