
Abstract
Sirtuin-2 (Sirt2) is a member of the NAD+-dependent protein deacetylase family. Various members of the sirtuin class have been found to be involved in processes related to longevity, regulation of inflammation, and neuroprotection. Induction of Sirt2 mRNA was found in the whole hemisphere after experimental stroke in a recent screening approach. Moreover, Sirt2 protein is highly expressed in myelin-rich brain regions after stroke. To examine the effects of Sirt2 on ischemic stroke, we induced transient focal cerebral ischemia in adult male Sirt2-knockout and wild-type mice. Two stroke models with different occlusion times were applied: a severe ischemia (45 minutes of middle cerebral artery occlusion (MCAO)) and a mild one (15 minutes of MCAO), which was used to focus on subcortical infarcts. Neurological deficit was determined at 48 hours after 45 minutes of MCAO, and up to 7 days after induction of 15 minutes of cerebral ischemia. In contrast to recent data on Sirt1, Sirt2−/− mice showed less neurological deficits in both models of experimental stroke, with the strongest manifestation after 48 hours of reperfusion. However, we did not observe a significant difference of stroke volumes or inflammatory cell count between Sirt2-deficient and wild-type mice. Thus we postulate that Sirt2 mediates myelin-dependent neuronal dysfunction during the early phase after ischemic stroke.
INTRODUCTION
Acetylation is an important posttranslational modification of cells to respond to different environmental challenges: acetylation of tubulin has been found to increase the pro-inflammatory activity in response to metabolic stress 1 and the degree of histone acetylation determines the neuronal resistance against ischemic injury. 2 Both sirtuin-2 (Sirt2) and sirtuin-1 (Sirt1) belong to the class III histone deacetylase family. These deacetylases require NAD+ as a co-substrate, in contrast to class I and II histone deacetylases. 3 The sirtuins have been found to have an important role in cell death, aging, and inflammation.4–6 Thus they are key factors in the pathology of multiple major age-dependent neurological diseases. 7
There are few data available on sirtuin signaling in ischemic injury. 3 The sirtuin member Sirt1 has recently been shown to protect against ischemic damage: Sirt1−/− mice displayed larger stroke volumes in a murine model of permanent stroke 5 and had increased damage in a model of myocardial ischemia. 8 However, transgenic mice that overexpress Sirt1 in neurons were not protected 1 day after induction of transient cerebral ischemia. 9
In contrast to Sirt1, there is a recent debate on whether Sirt2 mediates ischemic injury,
10
and there is no information available about the role of Sirt2 in ischemic stroke
Sirt2 directly impacts inflammation by deacetylating and thus inactivating the p65 subunit of nuclear factor-KB, limiting the expression of proinflammatory genes.6,11 It was also postulated that Sirt2 mediates the protective innate immune response caused by caloric restriction or metabolic stress,4,11 and Sirt2 has been shown to inhibit inflammasome-mediated inflammation in response to reactive oxygen species (ROS).1,12 However, it was demonstrated that Sirt2 increases ROS levels
13
and mediates oxidative stress-induced apoptosis of PC12 cells
As we observed a significant mRNA induction of several sirtuin family members during cerebral ischemia in a recent screening approach, we wanted to define the functional role of Sirt2 in stroke. Thus two different standard stroke models were applied in Sirt2−/− and wild-type mice. Our experiments revealed that Sirt2 protein is mainly expressed in the myelin-rich regions of the ischemic hemisphere in oligodendrocytes, that different Sirt2 protein isoforms are differentially regulated after ischemia, and that Sirt2−/−mice have significantly less neurological impairments after stroke when compared with wild-type animals. As we did not observe a significant alteration of the stroke volumes or number of inflammatory cells in either strain, we postulate a mainly myelin-dependent effect of Sirt2 inhibition.
MATERIALS AND METHODS
Ethics Statement
All animal handling and surgery were performed in accordance with the Guidelines for the Use of Animals in Neuroscience Research (Society for Neuroscience) and according to institutional and national guidelines. Experiments were written in accordance with the ARRIVE guidelines. All experiments were approved by the local institutional Animal Care Committee, LaVeS (No.33.12-42502-04-12/849).
Animals
Adult 8-to-11-week-old male Sirt2−/− mice (NKI, Amsterdam, The Netherlands)
6
had been backcrossed to the original C57Bl/6N background for >10 generations. Age-matched male 8-to-11-week-old wild-type animals from the same background strain (C57Bl/6 N) were bred under the same conditions in the same facility and were used as controls in all experiments. The initial body weight of the control mice was 24.3 ± 4.0 compared with 26.8 ±7.8 g in Sirt2−/− mice. Mice were housed under diurnal lighting conditions and allowed access to food and water
Induction of Focal Cerebral Ischemia
MCAO was induced by inserting a silicone-coated filament (6-0 MCAO suture, Doccol Corporation, Sharon, MA, USA) into the internal carotid artery as described previously.15,16 The mice received 0.1 mg/kg body weight buprenorphine as analgesia and were anesthetized with 4% isoflurane for induction and maintained with 2% isoflurane at an oxygen flow of 0.8 L/min via a face mask. After occlusion times of 60 minutes (for gene expression analysis), 45 minutes (for evaluation of neurological deficit and stroke volume), and 15 minutes (mild infarct model), the animals were re-anesthetized and the filament was removed to permit reperfusion. Efficiency of occlusion and reperfusion of the middle cerebral artery was monitored by Laser Doppler flowmetry (Perimed, Stockholm, Sweden) in a selected group of animals (
Analysis of Gene Expression
Mice were deeply anesthetized and decapitated at specified time points of reperfusion. The brains were removed rapidly from the skull. RNA derived from the separated hemispheres, pooled from six wild-type C57Bl/6 mice (after MCAO or sham treatment) at each time point, were used for gene expression analysis using Affymetrix GeneChip Mouse Expression Set 430 (Affymetrix, Santa Clara, CA, USA) as described previously.15,16 To determine the induction of specific mRNAs in the ischemic hemispheres of MCAO-treated animals, expression values were compared with those derived from sham-operated animals. Commercially available Affymetrix software was applied for data analysis of the Affymetrix GeneChips. Fold change (FC) was calculated using the following formula: FC = 2m, for m > 0, and FC = (-1)x 2−
Neuroscore
Two different scores were used to assess the neurological deficit of the mice after the induction of stroke. The neurological dysfunction was determined using a neurological score described by Bederson
Perfusion and TTC (2,3,5-triphenyl-tetrazolium-chloride) Staining Forty-eight hours after reperfusion, the animals were deeply anesthetized with 5% of isoflurane at an oxygen flow of 0.8 L/min, and their thorax was opened to expose the heart. A cannula connected to a perfusion pump was introduced into the left ventricle, and the right atrial appendage was opened. Animals were perfused with 50 ml of phosphate-buffered saline (PBS) with a rate of 10 mL/min. Brains were isolated and cut into 2 mm coronal sections. For the stroke volumetry, a TTC staining was performed to stain areas with vital cells. The 2-mm sections were incubated in a 2% TTC solution for 5 minutes from each side. The staining process was stopped by transferring the brain slices into 4% paraformaldehyde in PBS. The stained sections were scanned, and the stroke volume was determined using the Image J software (version 1.48). Three parameters were assessed: the area of the contralateral hemisphere as well as the ipsilateral hemisphere divided into non-infarcted area and infarct area. The direct stroke volume equals the infarct area and the indirect stroke volume was calculated by subtraction of the non-infarcted area (ipsilateral) from the contralateral hemisphere to correct for brain swelling.
FACS Analysis
Brains of seven wild-type and six Sirt2−/− animals were removed after perfusion at 48 hours after 45 minutes of MCAO, and then their hemispheres were separated and prepared for FACS analysis individually. Cell suspensions were generated using a percoll gradient and stained for CD3 (Biotin anti-mouse CD3e, BD Biosciences, San Jose, CA, USA; Streptavidin PE/Cy5, BioLegend, San Diego, CA, USA), CD45 (APC antimouse CD45.2, BioLegend), CD11b (FITC anti-mouse CD11b, BioLegend), GR-1 (PE anti-mouse LY-6G and Ly6C (GR-1), BD Biosciences), CD4 (APC/Cy7 anti-mouse CD4, BioLegend), and CD8 (PE/Cy7 anti-mouse CD8a, BioLegend) and analyzed by the FACS Diva Version 6.1.3 (BD Biosciences).
Immunhistochemistry
After 24 hours fixation in 4% paraformaldehyde and subsequent 24 hours incubation in PBS, brain tissues were embedded in paraffin and cut into 1.5-μm-thick slices. Slices from six animals per group were stained for NeuN (MAB377, Merck Millipore, Billerica, MA, USA), GFAP (glial fibrillary acidic protein; Ab5541, Merck Millipore), ASC (apoptotis-associated specklike protein containing a CARD; AG-25B-0006, Adipogen, San Diego, CA, USA) and counterstained with DAPI (4′,6-diamidino-2-phenylindole). As secondary antibodies Cy5, FITC, and Cy3 (715-175-150, 703-095-155, and 711-165-152, respectively; Jackson Immunoresearch, West Grove, PA, USA) were used. For counting of neuronal cell numbers, three boxes of 200 μm height and covering the entire brain from left to right (ipsilateral and contralateral hemispheres) were placed in each brain slide at the following predefined positions: one below the oriens layer of the hippocampus, one above the top of the third ventricle, and one in the middle between the other ones. Afterwards, the mean cell number per mm2 was calculated for each hemisphere (ischemic vs. non-ischemic). The neuronal loss was determined by the ratio, which was calculated by the difference of NeuN-positive cell counts in both (ischemic and non-ischemic) hemispheres divided by the number of NeuN-positive cells in the non-ischemic/contralateral hemisphere. ASC specks were counted in the entire ipsilateral hemisphere. Expression of Sirt2 protein in wild-type mouse brain was examined using a specific Sirt2 antibody (ab67299; Abcam, Cambridge, UK). Cell nuclei were counterstained with DAPI. Brain tissue derived from Sirt2−/− mice was used to control for specificity. The slices were scanned with a ×40 magnification using Axio Examiner.Z1 microscope (Zeiss, Jena, Germany) with the Zen Software (Zeiss). Cell-type-specific expression of Sirt2 was evaluated using paraffin sections of 1.5 μm thickness, which were deparaffined followed by antigen retrieval as described. 21 After blocking for 20 minutes in 20% (v/v) horse serum and 0.4% (w/v) bovine serum albumin in PBS, primary antibodies were applied at 4° for two nights in 20% (v/v) horse serum and 0.4% (w/v) bovine serum albumin in PBS. Antibodies were specific for Sirt2 (Abcam, 1:200), IBA1 (Abcam, 1:1000), NeuN (Millipore, 1:50), and CNP (Sigma, St Louis, MO, USA, 1:200). For immunofluorescence for CD68, paraffin sections were deparaffined as described, 21 followed by antigen retrieval by cooking for 10 minutes in EnVision FLEX Target Retrieval Solution Low pH (Dako, Glostrup, Denmark). After cooldown in the solution and blocking for 20 minutes in Protein Block Serum Free (Dako), anti-CD68 (AbD Serotec, Oxford, UK, 1:100) antibody was applied over night at 4° in Antibody Diluent (Dako). Fluorophore-conjugated secondary antibodies were applied for 1 hour at room temperature in 2% (v/v) horse serum and 0.4% (w/v) bovine serum albumin in PBS. Fluorophore-conjugated secondary antibodies were anti-mouseAlexa488 (Dianova, Hamburg, Germany, 1:2000), anti-ratAlexa488 (Dianova, 1:2000), anti-goatAlexa488 (Dianova, 1:2000), and anti-rabbitAlexa555 (Dianova, 1:2000). DAPI was applied for nuclear staining. Slices were mounted in AquaPolymount (Polysciences, Warrington, PA, USA). Images were acquired at × 63 magnification at an inverted Zeiss Axio Observer. For green fluorescence (Alexa488), a 450 to 490 nm bandpass filter was used for excitation, and emission was recorded with a 500 to 550 nm bandpass filter. For red fluorescence (Alexa555), a 533 to 558 nm bandpass filter was used, and emission was recorded with a 570 to 640 nm bandpass filter. For blue fluorescence (DAPI), a 335 to 383 nm bandpass filter was applied and emission detected using a 420 to 470 nm bandpass filter.
Biochemical Analysis
Brains were dissected, separated into hemispheres, and snap frozen on dry ice. Homogenates of one hemisphere were prepared in 5 ml 0.32 mol/L sucrose solution supplemented with protease inhibitor cocktail (Complete, Roche, Basel, Switzerland) using an IKA T10 ULTRA TURRAX (IKA, Staufen, Germany). Protein concentration was determined using the DC protein assay (Bio-Rad, München, Germany). Immunoblots were performed as described. 22 Antibodies used were specific for Sirt2 (1:50 00) 23 and α-tubulin (Sigma, 1:1000). Signals were detected using ECL (Western Lightning Plus ECL, Perkin Elmer, Waltham, MA, USA) and a chemiluminescence scanner (Intas ChemoCam System, Göttingen, Germany). Band intensities were quantified using ImageJ (NIH, Bethesda, MD, USA) 1.48. Sirt2 protein abundances of each isoform are given relative to α-tubulin abundance and the mean total Sirt2 protein abundance (sum of both isoforms) at control conditions. Images were processed using Adobe Photoshop (San Jose, CA, USA). Statistical analysis using a one-way unpaired analysis of variance was carried out in GraphPad Prism 6.0 (GraphPad Software Inc., La Jolla, CA, USA).
Statistics
Power calculation was performed using SISA-Binominal as described before.
24
Based on the known variance of previous experiments, the MCAO experiments were powered (α = 0.05; β = 0.8) to detect effect sizes
RESULTS
mRNAS of Several Sirtuin Family Members are Induced After Cerebral Ischemia
We first performed a screening experiment to identify differentially regulated gene products after experimental stroke. Interestingly, we found that mRNAs of several sirtuin family members were induced (Table 1). Sirt2 mRNA was found induced 6 hours after MCAO in the ischemic mouse brain and up to 24 hours after induction of cerebral ischemia (Table 1).
mRNAs of different sirtuin family members are induced after transient focal cerebral ischemia in the ischemic mouse brain
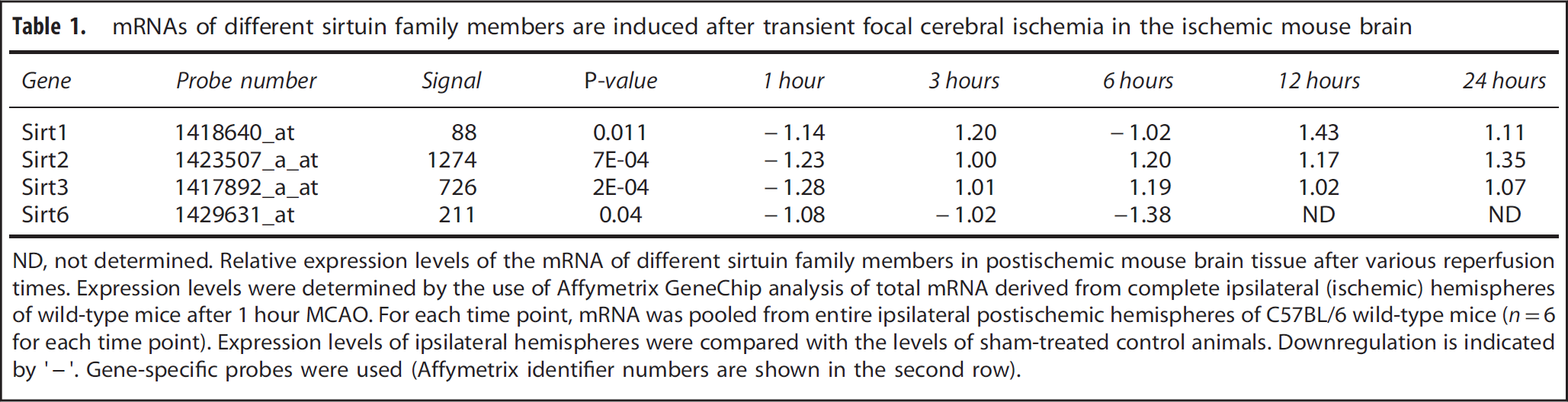
ND, not determined. Relative expression levels of the mRNA of different sirtuin family members in postischemic mouse brain tissue after various reperfusion times. Expression levels were determined by the use of Affymetrix GeneChip analysis of total mRNA derived from complete ipsilateral (ischemic) hemispheres of wild-type mice after 1 hour MCAO. For each time point, mRNA was pooled from entire ipsilateral postischemic hemispheres of C57BL/6 wild-type mice (
Sirt2 is Mainly Expressed in Myelin-Rich Regions in Non-Ischemic and Ischemic Brain Tissue, is Mainly Expressed in Oligodendrocytes, and the Two Major Sirt2 Protein Isoforms are Differentially Regulated After Ischemia
As shown in Figure 1, a Sirt2-specific antibody was used to evaluate the expression pattern of Sirt2 after stroke. Myelin-rich structures in both hemispheres were immunoreactive for Sirt2, and the staining seemed more intense in the ischemic hemisphere (Figures 1A and 1B). Double-labeling immunofluorescence studies using Sirt2-specific antibody in combination with various cell-type-specific markers revealed a preferential expression of Sirt2 protein in myelin formed by oligodendrocytes (CNP-positive white matter tracts) but no detectable Sirt2-specific signal in astrocytes (GFAP-positive cells), microglia (Iba1-positive cells), or neurons (NeuN-positive cells); whereas only some CD68-positive cells (macrophages) showed also a weak and inconstant Sirt2-positive staining pattern (Figures 1D and 1H).
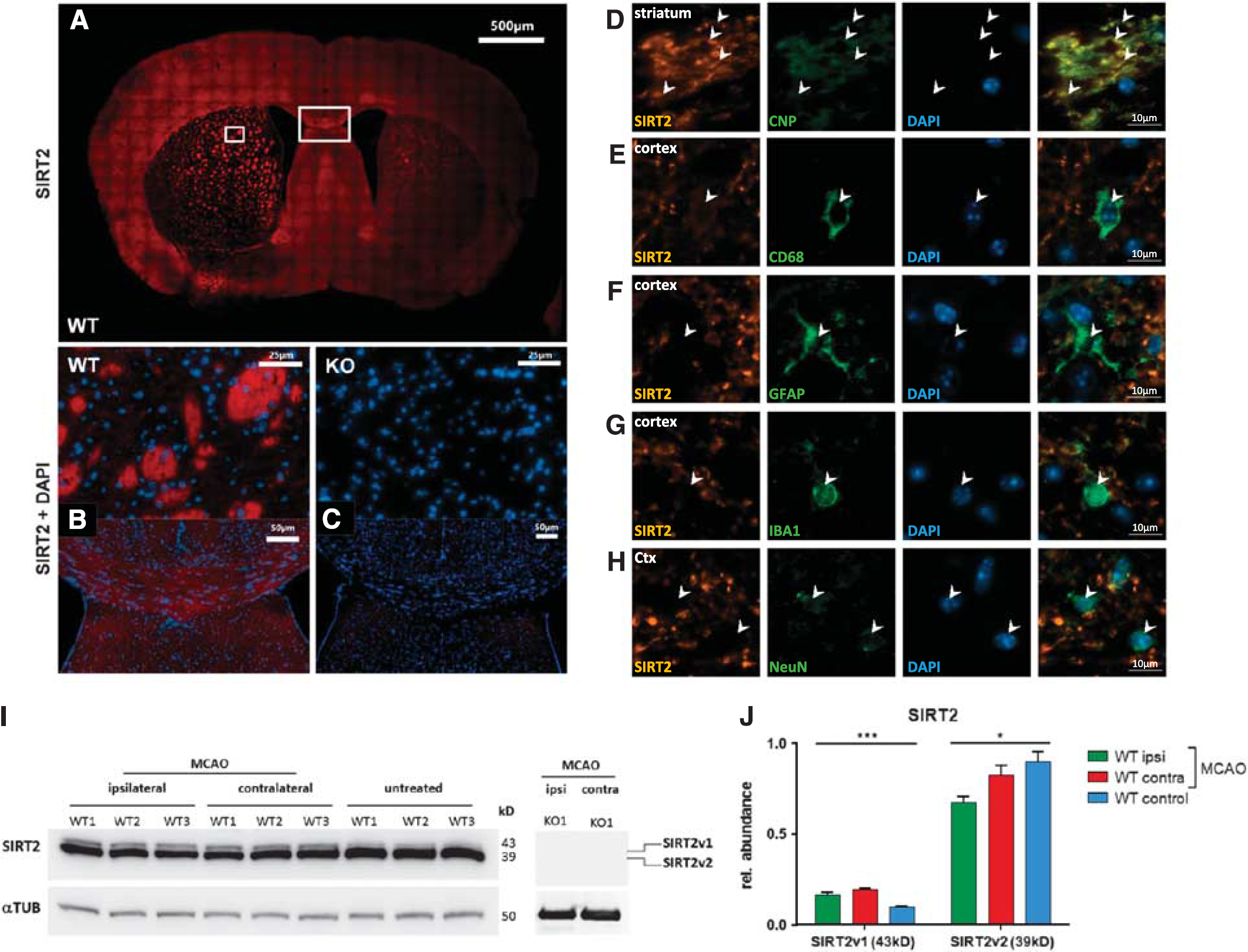
Sirtuin-2 (Sirt2) protein is almost exclusively expressed in myelin-rich regions, is expressed mainly in oligodendrocytes, and the different Sirt2 protein isoforms are differentially regulated in the ischemic brain hemisphere. Whole mouse brain tissue at 48 hours of reperfusion after 45 minutes of middle cerebral artery occlusion (MCAO) was immunofluorescence stained with anti-Sirt2 antibody in wild-type (
Sirt2 Isoforms are Differentially Regulated After Cerebral Ischemia
Western blotting with Sirt2-specific antibody was performed to quantify the Sirt2 protein expression in whole ischemic mouse brain 48 hours after induction of MCAO (Figures 1I and 1J). Interestingly, the two major isoforms22,23 of Sirt2 protein showed a differential regulation after cerebral ischemia in wild-type mice: whereas the expression of the major cytoplasmic isoform (SIRTv2) was reduced when compared with untreated wild-type control mice, the longer v1-isoform (SIRTv1) was induced in both ischemic/ipsilateral and non-ischemic/contralateral brain hemispheres when compared with untreated wild-type mice (Figures 1I and 1J). Thus, not only Sirt2 mRNA (Table 1) but also Sirt2 protein isoforms are differentially regulated after cerebral ischemia in the ischemic mouse brain (Figure 1).
Sirt2 Deficiency has no Influence on Stroke Volumes or Neuronal Cell Death After Focal Transient Cerebral Ischemia
To evaluate whether Sirt2-deficiency protects against experimental stroke, male adult Sirt2— mice were compared with wild-type mice after 45 minutes MCAO and 48 hours of reperfusion. Neither indirect nor direct stroke volumes showed a significant difference between Sirt2-deficient and wild-type animals (Figures 2A–2C). Next we examined whether Sirt2 deficiency could protect against ischemic neuronal cell death despite a lack of a significant alteration of stroke volumes. To this end, the ‘neuronal loss’ was determined by counting neurons both in ischemic and nonischemic brain hemispheres of Sirt2— and wild-type mice after 45 minutes MCAO and 2 days of reperfusion. In agreement with the absence of a significant alteration of stroke volumes, Sirt2-deficient mice had no significant difference in the loss of neurons in their ischemic hemisphere when compared with wild-type mice (Figure 2D). Moreover, immunostaining of astrocytes did not reveal a significant difference in astrocyte distribution in the ischemic brain of Sirt2-deficient and wild-type control mice after MCAO (Figure 2E).
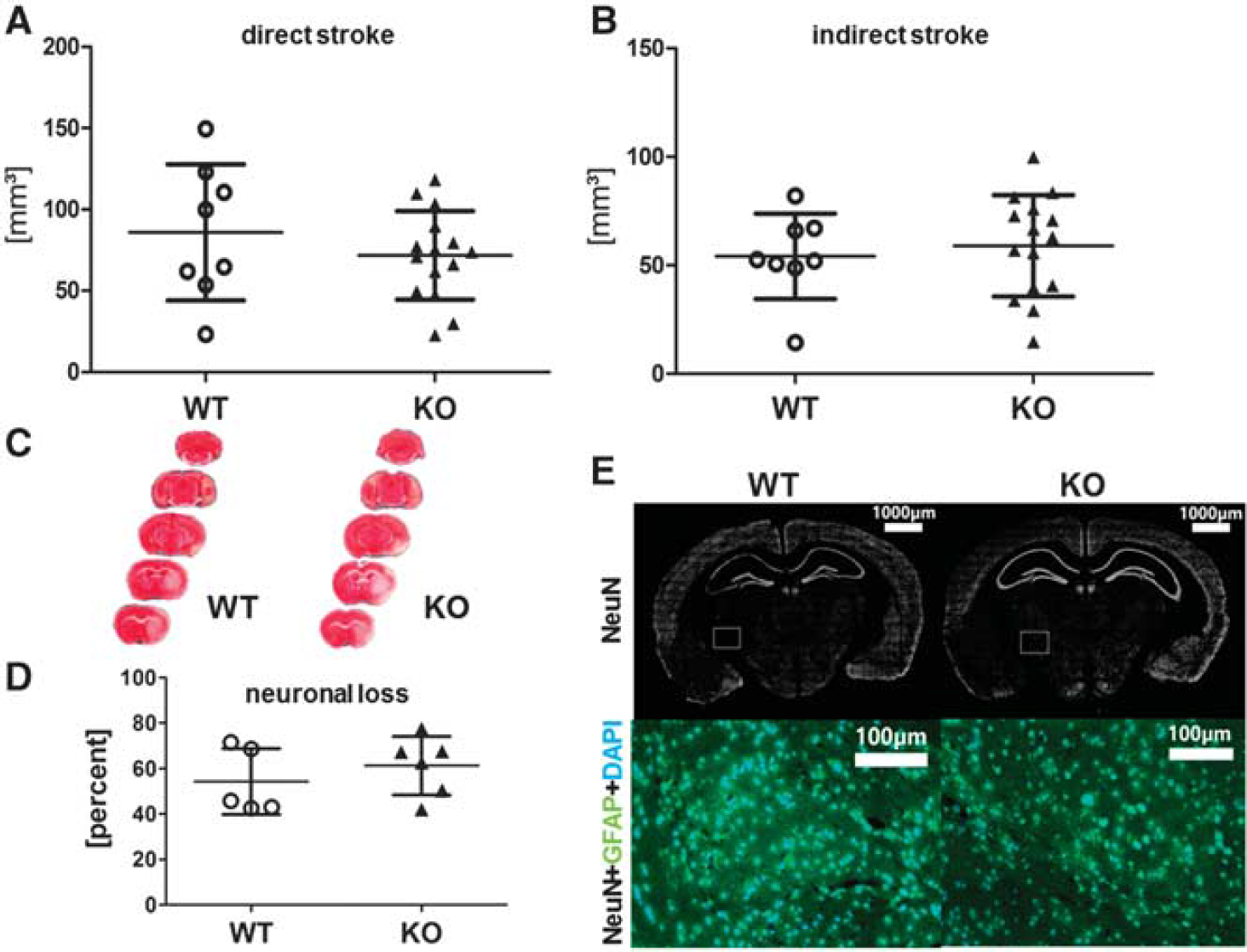
Sirtuin-2 (Sirt2) deficiency has only a minor effect on stroke volumes, neuronal cell death, and astrocyte distribution after focal cerebral ischemia. Direct (
Sirt2-Deficieny Preserves Neurological Function After Cerebral Ischemia
To evaluate the level of functional impairment of the mice after experimental stroke, we assessed different neuroscores to determine motoric and sensoric disabilities. Despite the absence of differences in the stroke volume, adult male Sirt2— mice showed less neurological impairment (measured by the mNSS) 24 and 48 hours after induction of cerebral ischemia for 45 minutes (Figure 3). We could not detect a significant difference between the two groups 24 hours after MCAO using the Bederson Score (
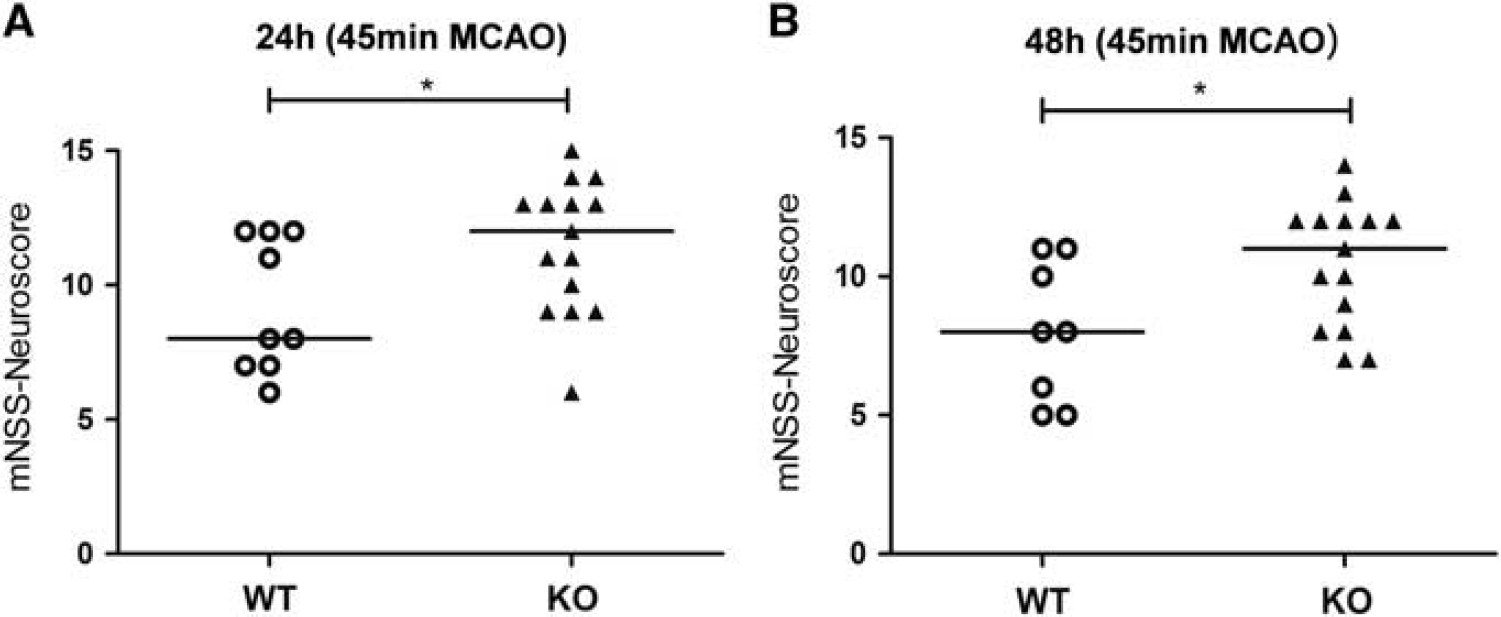
Sirtuin-2 (Sirt2)-deficient male mice have an improved neurological performance after experimental stroke. The modified Neurological Severity Score (mNSS) was assessed 24 and 48 hours after 45 minutes of middle cerebral artery occlusion (MCAO) in male adult wild-type (WT) and Sirt2-knockout (KO) mice (WT
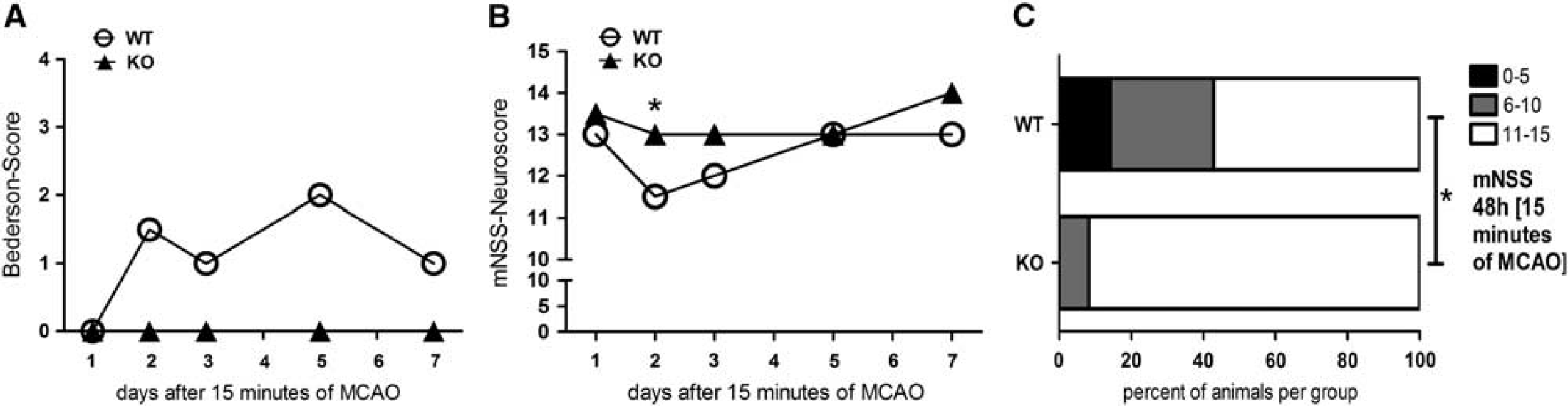
Sirtuin-2 (Sirt2)-deficient mice have a better neurological outcome compared with wild-type (WT) animals in a mild stroke model. The Neuroscores (Bederson Score (
Sirt2 Induces Minor Effects on the Inflammatory Cellular Response After Ischemic Brain Injury
We used FACS analysis to determine absolute cell numbers of neutrophils, activated microglia/macrophages, and CD4+/CD8+ T cells 48 hours after induction of focal cerebral ischemia for 45 minutes to evaluate whether Sirt2 deficiency affects postischemic influx of immune cells as well as the activation status of resident innate immune cells. There was a similar increase of neutrophils and activated microglia/macrophages in the ischemic brain hemispheres both of wild-type and Sirt2-deficient mice, and overall no significant alteration in the distribution of inflammatory cells in both hemispheres between mice of the two genotypes (Figure 5).
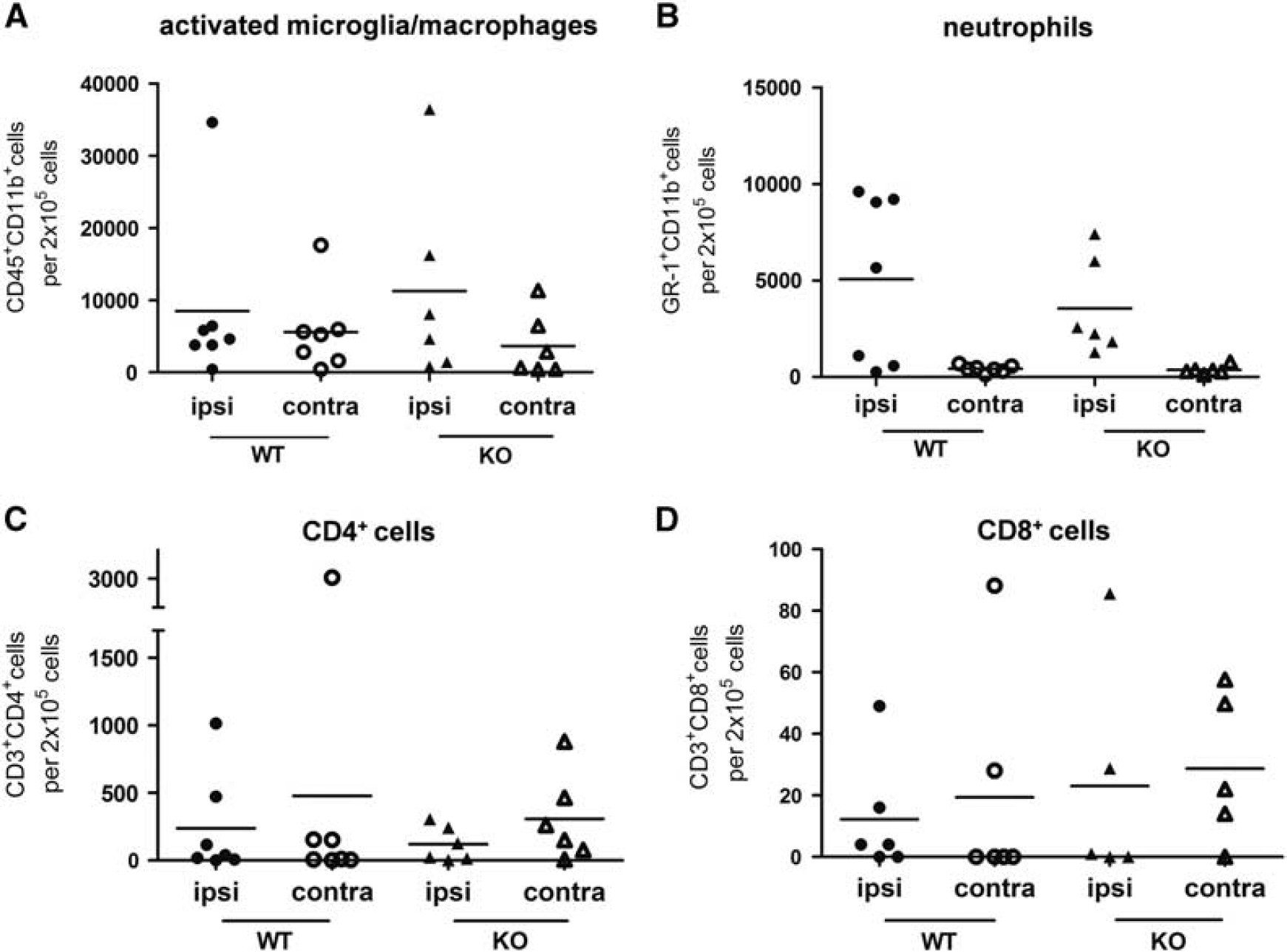
Quantification of inflammatory cells in ischemic (ipsi) and non-ischemic (contra) brain hemisphere of sirtuin-2 (Sirt2)-deficient and wild-type (WT) control mice did not reveal a difference between both genotypes. The entire hemispheres of ischemic Sirt2-deficient and KO mice were isolated 48 hours after 45 minutes of middle cerebral artery occlusion (MCAO) (WT:
The Amount of ASC-Positive Specks in the Ischemic Hemispheres of Sirt2— and Wild-Type Mice does not Differ
Based on the recent report of the involvement of Sirt2 in the stress-mediated inflammatory response, 1 we wondered whether the appearance of ASC-positive specks, indicative of activated inflammasomes, 26 is increased in Sirt2-deficient mice after MCAO when compared with wild-type controls. As shown in Figure 6A, ASC-specific polyclonal antibody staining revealed intensely stained speck-like structures, which are mainly located at the border of the ischemic core, the so-called penumbra zone, as well as cells with cytoplasmic staining for ASC, which morphologically resemble microglial cells or macrophages (Figure 6B). To exclude that the ASC-specks are staining artifacts similar to antibody aggregates on the surface of the slice, they were examined further by confocal microscopy. The confocal images show that the specks are located next to the nucleus and therefore could be ASC aggregates that appear after activation of the inflammasome.26,27 In contrast to the postulated increase of inflammasome activation after Sirt2 inhbition, 1 we could not detect a significant increase of ASC-positive specks in postischemic mouse brain tissue of Sirt2-deficient mice compared with wild-type mice (Figure 6C). However, this observation fits well with the absence of a substantial effect on the inflammatory cell count in Sirt2-deficient mice (Figure 5).
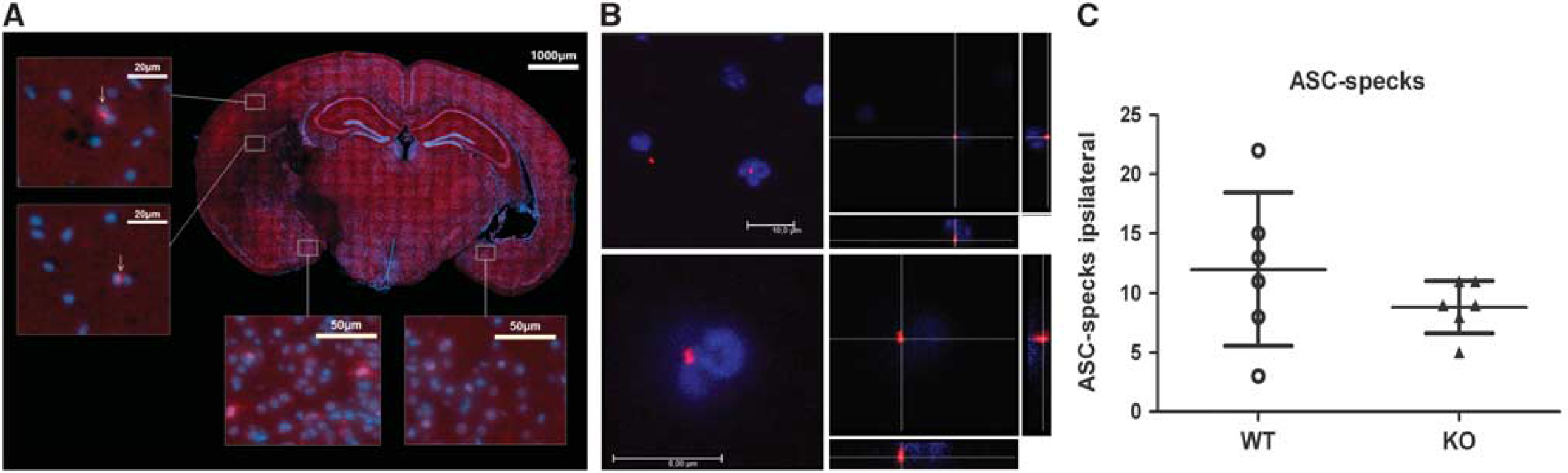
Apoptotis-associated speck-like protein containing a CARD (ASC)-positive specks are detected in the ischemic hemisphere of both, Sirt2−/−, and wild-type (WT) mice, but there was no significant increase of speck count in Sirt2−/− mice after middle cerebral artery occlusion. Mouse brain after 48 hours of reperfusion and 45 minutes of ischemia stained with anti-ASC-specific antibody and 4′,6-diamidino-2-phenylindole to label cell nuclei (
DISCUSSION
The exact way of Sirt2 action still remains to be fully elucidated, but there are several potential protein functions that may be of significance in ischemic tissue injury. First, an anti-inflammatory role of Sirt2 during metabolic stress involves the NAD+-dependent reduction of tubulin acetylation and thereby links metabolism with the activation of the innate immune system.11,12 Misawa
In contrast to that supposed overall protective effects of Sirt2 during ischemia, especially concerning inflammatory processes,1,6 there is recent debate about a potential direct involvement of Sirt2 in necroptotic signaling, which could be responsible for a protective effect of Sirt2 deficiency in models of ischemic myocardial injury.
10
Furthermore, Spires-Jones
Our investigations show that Sirt2-deficient mice were functionally protected against ischemic stroke, and protection with regard to neurological performance surmounts an only minor effect regarding stroke volumes or neuronal cell loss. A very recent study using the Sirt2 inhibitor AK7 did not detect a significant difference of the neurological deficit scores according to Bederson, but a tendency toward a better neurological outcome in the AK7-treated animals when compared with vehicle-treated animals at 24 hours after 1 hour MCAO. 32 These results are consistent with our own findings, as we did not see a significant difference between the groups using the Bederson Score at 24 hours after 45 minutes MCAO. However, after 48 hours both mNSS and Bederson Scores revealed significantly reduced neurological deficits in Sirt2−/− mice when compared with wild-type mice. Thus the difference of the neurological deficit in both groups is not evident by the use of a rather rough test such as the Bederson Score at 24 hours of reperfusion. Moreover, it is possible that AK7 inhibits Sirt1 to some degree as well, 32 which would cloud the result of the Sirt2 inhibition, as Sirt1 and Sirt2 are currently suspected to have opposite effects on the outcome of cerebral ischemia.3,5
The absence of significant changes regarding the immune response in Sirt2-deficient mice after stroke, measured by FACS analysis, argues against a prominent role of Sirt2 concerning the initiation of an inflammatory response. Additionally arguing against a major role of Sirt2 in tubulin-acetylation-dependent modulation of inflammation, Sirt2 deletion does not alter tubulin acetylation
Our findings confirm the current view that inhibition of Sirt2 or activation of Sirt1 seems to be beneficial for the organism against certain age-associated diseases. 36 Sirt1 and Sirt2 seem to differ regarding their effects on ischemic brain injury. In contrast to our results that show a protective effect of Sirt2 deficiency, Sirt1-knockout mice showed increased stroke volumes after cerebral ischemia when compared with wild-type mice, and most studies conclude that Sirt1 is beneficial in stroke.3,5 This difference could be explained by the different cellular expression pattern of both sirtuin family members: Sirt1 is mainly expressed in neurons,5,36 whereas Sirt2 is mainly expressed in oligodendroglia. 22
Differential regulation of Sirt2 mRNA and Sirt2 protein isoforms, which are supposed to result from differential splicing 22 are in congruence with a mainly posttranscriptional regulation of Sirt2 protein expression. 22
The protective effect of Sirt2 deficiency that we discovered in stroke and the opposite effect observed in Sirt1-deficient mice 5 correlates with similar effects of Sirt1 and Sirt2 in myocardial ischemia: Sirt1 deficiency in the myocardium increases damage after ischemia/reperfusion in the heart, 8 whereas Sirt2-deficiency potentially mediates protection in myocardial ischemia-reperfusion injury. 10
However, this black and white classification on the net effects of specific sirtuin family members seems to be overly simplistic. The different sirtuins are not only expressed in different cell types, or cellular compartments, but are also known to deacetylate different substrates
35
and that process could also depend on the degree of phosphorylation of these proteins.
6
Thus further experimental
CONCLUSION
We herein demonstrate that Sirt2 mRNA and Sirt2 protein are differentially regulated after ischemic brain damage and mainly expressed in oligodendrocytes. Moreover, Sirt2 deficiency improves the neurological outcome after experimental stroke in mice in the early postischemic period. Given the minor changes in the stroke volume, as well as in the neuronal and inflammatory cell count and the myelin-associated expression pattern, we hypothesize that Sirt2 mainly affects neurological deficits by influencing myelin and thus axonal function.
Footnotes
Lea Krey performed the major part of the experiments (MCAO, immunohistochemistry, etc.) and wrote major parts of the manuscript as well as finally approved the manuscript; Kathrin Kusch performed co-localizing studies and biochemical analysis; Fred Lühder contributed to the concept of the study, performed flow cytometry and data analysis, and finally approved the manuscript; Bozena Czech-Zechmeister helped with the MCAO model, data acquisition, data analysis, as well as final approval of the manuscript; Birte Könnecke helped with the immunohistochemical evaluation, the data acquisition, the data analysis, and critical reading and final approval of the manuscript; Tiago F Outeiro contributed by help with the concept and design of the study, by analyzing the data, and by critically reading and revising the manuscript; George Trendelenburg contributed by help with the design and concept of the study, evaluating the data, and writing and revising the manuscript.
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
We thank Regine Kruse for excellent technical assistance, Zara Haschemol-Hosseini for help with confocal laser-scanning microscopy, and Cathy Ludwig for language correction.