
Abstract
A new research field in translational neuroscience has opened as a result of the recognition since 2002 that “spreading depression of Leão” can be detected in many patients with acute brain injury, whether vascular and spontaneous, or traumatic in origin, as well as in those many individuals experiencing the visual (or sensorimotor) aura of migraine. In this review, we trace from their first description in rabbits through to their detection and study in migraine and the injured human brain, and from our personal perspectives, the evolution of understanding of the importance of spread of mass depolarisations in cerebral grey matter. Detection of spontaneous depolarisations occurring and spreading in the periphery or penumbra of experimental focal cortical ischemic lesions and of their adverse effects on the cerebral cortical microcirculation and on the tissue glucose and oxygen pools has led to clearer concepts of how ischaemic and traumatic lesions evolve in the injured human brain, and of how to seek to improve clinical management and outcome. Recognition of the likely fundamental significance of spreading depolarisations for this wide range of serious acute encephalopathies in humans provides a powerful case for a fresh examination of neuroprotection strategies.
Keywords
Introduction
In 1944, Aristides Leão published a paper in the Journal of Neurophysiology 1 that was destined to become one of the most highly cited papers in the entire canon of basic and clinical neuroscience literature. 2 He described many of the features of the curious phenomenon that often bears his name: ‘Leão’s spreading depression’. As George Somjen, another renowned neurophysiologist, has said of his work: ‘The discovery of spreading depression (SD) illustrates how an unexpected chance observation, if made by an alert experimenter, can open a new major area of investigation’. 3 In this historical review, we shall trace how initial perceptions of Leão’s findings gradually evolved from a physiologists’ curiosity to become a tool used by experimental psychologists, a conceptual basis for the visual aura of migraine with aura (MA), and, soon afterwards, a proposed cause of infarct expansion in experimental models of focal cerebral ischemia. We shall explore reasons why only in 2001 and 2002 and thereafter did spontaneously occurring spreading depression-like events become recognised as potentially important factors in the evolution of ischemic and traumatic brain injury to the human cerebral cortex. The emergence of work in this area from the laboratory and into the clinical domain can be seen now as a genuine example of translational medicine, and we shall include citations that document our – in truth quite small – contributions to this story. Where appropriate, we shall refer to very recent papers that address (with current technology) features noted by the early investigators – ‘then and now’. Finally, we shall look at the many open questions surrounding the possible pathogenic role of SDs, and how these questions might be addressed, and the findings perhaps translated, if necessary, into clinical management. As will become clear, the basis for a wave of SD is the underlying spread of mass depolarisation of all neurons and astrocytes in the path of the wave. Hence, ‘CSD’ throughout this review will signify ‘cortical spreading depolarization’, and is more appropriate than ‘SD’, since although SD occurs in deep cerebral grey matter, this review addresses only pathophysiology in the cerebral cortex: here, ‘SD’ will refer to spreading depolarisation in any grey matter. For a full glossary and set of definitions, please see ‘The continuum of spreading depolarisations in acute cortical lesion development: examining Leão’s legacy’ (Hartings et al., this JCBFM issue, Table 1).What we shall not do is to offer a comprehensive literature review of SD, and for that, readers are referred in particular to the comprehensive reviews by Marshall 4 and more recently by Somjen, 5 and by Ayata and Lauritzen. 6 The other articles in this special issue also cite reviews relevant to their specific subjects.
The work of Aristides Leão
Although his paper describing his primary finding is frequently cited, 2 a fresh look at the paper reveals features that clinical neurophysiologists who have become interested in SD recognise, in particular the clear but variable association of tonic-clonic discharges with the higher amplitude (5–15 mV initially negative) SD, 7 which has now been observed in patients.8,9 This linkage by Leão finds a contemporary parallel in a recent paper from Wei et al., 10 who propose a single, detailed membrane biophysics model in which variations in either oxygen availability and/or extracellular potassium can be related to seizures, SD, or to a mixture of these. In his principal paper (his Figure 12), Leão also reported the occurrence of short-lasting (1 s) lower amplitude (3 mV) depolarisation events that occurred during the period of electrocorticographic (ECoG) amplitude depression, and suggested that the hyperaemia accompanying the SD is chemically mediated. 11 Indeed, we could suggest that Leão’s description of hyperaemia accompanying SD may represent one of the earliest illustrations of the operation of what we now envision as the neurovascular unit. 12
Upon returning to Rio de Janeiro from Harvard, Leão continued his studies of SD, and in 1947 described in more detail the slow biphasic change of the cortical surface potential that spread over the cortex accompanying depression of the electrocorticogram (ECoG) amplitude.
7
Notably, Leão reported in this paper that a brief period of occlusive cerebral ischemia imposed during the recovery of the positive phase of the cortical surface potential, suspended reversion to baseline, or even introduced a second phase of negativity. Leão was careful not to suggest energy depletion as the basis for the delay in recovery. We have come to regard demonstration of the slow potential change (SPC) – a marker of depolarisation – as a requisite for the designation of an observed ECoG suppression as a true SD in patients, and the term “spreading depolarization” is now the preferred term among researchers and clinicians, to which the abbreviations ‘SD’ or ‘CSD’ will refer in the rest of this article. The limited spatial resolution available to early investigators contributed to difficulties in separating and defining different mechanisms and patterns of spread (if present) of depolarisation. Leão’s use of a linear multichannel amplifier and display lays at the core of his findings and was key to the eventual, definitive demonstration of SD in the injured human brain. The use in recent years of serial in vivo imaging methods in the laboratory and now also in the operating room has helped resolve some of these uncertainties, for example as to whether or not a given depolarisation detected at a single point also spreads: we illustrate and document this later in the article. The large volume of work in the laboratory and clinic in the past two decades analysing the effects of ischaemia on the time course and resolution of the SPC merely serve to confirm the quality and continuing relevance of this area of Leão’s work, focusing as it does on the effects of reduced perfusion of the cerebral cortex. The figures in Leão’s 1947 paper find a legion of echoes in the large volume of literature based on the measurements of DC potential or of extracellular potassium, in experimental models of – and now in patients with – stroke and other acute brain injuries. Professor Leão was a much respected supervisor and teacher (Figure 1):
13
later in his career, he became President of the Brazilian Academy of Sciences (1966–1981) and had a wide range of interests outside neurophysiology, including ornithology, music, and art.
14
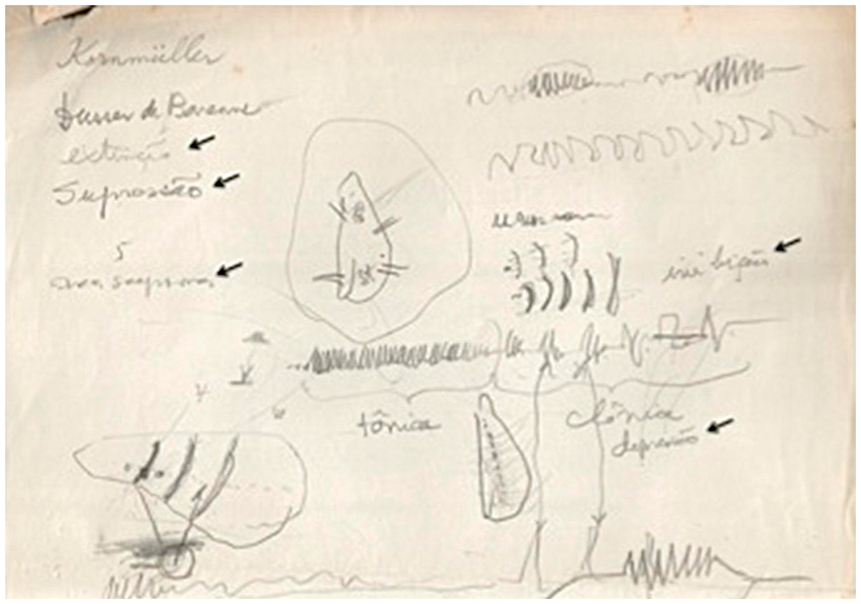
An original hand-written note of his thoughts of SD and migraine is shown in Figure 1.
13
Professor Leão’s original note. ‘Tutorial’ diagrams drawn by Leão, probably around 1990, and discovered recently by a younger colleague of Leão, Dr. Pericles Maranhão Filho. Various possible alternative designations for ‘spreading depression’ shown here had been rejected. At (second) top right, is Leão illustrating the fortification pattern, a typical visual percept of migraineurs-with-aura. Reproduced with permission from Dr. Pericles Maranhão Filho.
13
SD/depolarisations 1949–1971
Van Harreveld was the first to report on glutamate as an excitatory neurotransmitter based on his seminal work on the neuromuscular junction in crustacean muscles and had a great interest in asphyxial depolarisation in the spinal cord: 15 he published a series of papers on terminal, ischaemic, and anoxic depolarisation (AD), SD and spreading convulsions.15–19 Van Harreveld’s contribution to understanding of the effects of spreading depolarisation derived perhaps from his interest in transmembrane ion movements in CNS grey matter, 20 and their relationship with compartmental distribution of water. In his paper with Fifkova, he was the first to report evidence of glutamate release coupled to CSD in the isolated chick retina. 21 After obtaining her PhD in Montreal in 1954 (when Van Harreveld was the external examiner), Dr Bernice Grafstein 22 joined the Department of Anatomy in University College London, and the published fruit of her time in Montreal was her paper in 1956. Working with an in vivo, neurophysiologically isolated, but normally perfused in situ cortical slab preparation, she examined the effects of linear positive or negative polarisations of the slab on the propagation speed of SD, deducing that a cation might mediate spread, with potassium as the probable agent, as being the predominant cation released into the extracellular space during the phase of intense neuronal firing that immediately preceded the negative change in DC potential at the cortical surface. Her proposal appears to have stimulated an effort by Marshall and his co-workers 23 to detect potassium release, which they were able to confirm. The concept also elicited some correspondence from Hodgkin. Grafstein published her model some 15 years before potassium-selective microelectrodes became available, enabling Vyskocil et al. 24 directly to demonstrate K+ release during CSD.
Thus, Van Harreveld’s and Grafstein’s concept offered alternative hypotheses for the mechanism of spread of CSD, the first based on glutamate release into the extracellular space, and the second on release of potassium. Although the release of both molecules is clearly very closely coupled as a wave of depolarisation spreads, whether one or the other is critical to spread has remained an active issue.25–29 Recent data using two-photon microscopy suggest that a potassium increase precedes the rise in extracellular glutamate in CSD, but it is still an open question whether the potassium rise is sufficient to trigger the CSD or whether additional mechanisms are involved at the site of initiation of a wave of CSD. 30 Here, we should bear in mind that in a good in vivo experimental preparation, such as for example Leão’s, deliberate stimulation is required to elicit CSD, whereas in a poor preparation or in the context of a model of stroke or indeed, as we now know, in human ischemic or traumatic brain injury, spreading depolarisation waves (CSD) occur spontaneously, and sometimes frequently. In this setting, they are not necessarily associated with depression of the ongoing electrical activity because the activity is already depressed by the injury. Perhaps in this latter case, a critical, spatial cluster of neurons and astrocytes is partially depolarised and energy-depleted, and hence unstable – as suggested 50 years ago by Buresova et al. 31 and very recently by von Bornstadt et al. 32 – in terms of vulnerability to increased functional and hence metabolic load. 32
SD/depolarisation, ion-selective electrodes and experimental models of focal cerebral ischemia (1972–1996)
Although the work of Leão and also of Marshall and their colleagues hinted at the possibility of a contribution of SD to the pathophysiology of the injured brain, clearer concepts of what this might entail began to emerge in the 1970s with the introduction of ion-selective microelectrodes and the development of realistic experimental models of stroke.
Neurophysiologists in Czechoslovakia played a prominent part. 33 Dr Jan Bures’ 34 initial interests were in experimental models of epilepsy and he first published on SD in the 1950 s. In 1972, Vyskocil et al. induced CSD in the exposed cerebral cortex of rats and with potassium-selective microelectrodes were, we believe, the first to establish the resting value of extracellular potassium (Ke) in the cerebral cortex as close to 3.0 mM, and very reproducible and tightly stabilised: they went on to demonstrate the large increase in Ke that accompanies CSD, in their work to 60 mM in the cortex. 24 Indeed, the invention of ion-selective microelectrodes and methods for examination of the properties of the brain’s extracellular space created a whole new research field in basic and translational neuroscience describing the state of the extracellular microenvironment in health and disease.35–39
Bureš used the temporary ablation of cortical function by CSD as a tool to examine aspects of cortical function in rats, and over the course of 50 years has been an author of 86 papers on many aspects of SD. 40 Of particular interest is his work establishing reverberating CSD cycles in the rat brain cortex by creating a longitudinal cortical incision and carefully placed and timed inductions of CSD with microinjections of potassium chloride. 41 They discussed the conditions required to establish reverberation, in particular, the need for a central inert focus around which a SD wave would cycle: the circumference of the focus would require to be greater than the wavelength of the entire SD complex (the depolarisation, repolarisation, and refractory phases). Their results have now been translated to clinical acute brain injury, illustrated by the occurrence on occasion of clusters of CSD events recurring at regular intervals in patients with major hemisphere occlusive stroke, 42 or traumatic brain injury (TBI) where cortical contusion is present (Figure 3). The often very constant inter-event interval is compatible with repeated cycling of a single SD wave around a focus of functionally suppressed or infarcted cortex. This pattern has been imaged in a distal cerebral artery occlusion preparation in rats using a surrogate marker of the depolarisation – perfusion as detected by serial laser speckle imaging (http://brain.oxfordjournals.org/content/brain/suppl/2010/05/27/awq117.DC1/nakamura_suppl_v2.mp4): 43 importantly, these authors showed that the ischemic core expanded into the surrounding penumbra, in a stepwise fashion closely coupled to each cycle of depolarisation. Shibata and Bures 41 also proposed in their paper that a wave of depolarisation spreading around the edge of a nonexcitable region of cortex would ‘from this circumference … .enter the rest of the tissue in the form of a spiral moving radially with the same velocity as tangentially’. 41 Yet more recently, use of intrinsic optical imaging and development of sophisticated image post-processing methods has allowed the detection of complex spiral spread of CSD waves in the exposed gyrencephalic cerebral cortex, fulfilling Shibata and Bures’ 41 prediction (Supplementary video 3 in Santos et al. 44 ) (http://www.sciencedirect.com/science/article/pii/S1053811914003863#ec0025).
The 1970s decade saw the development of novel experimental models of stroke in primates that began, reliably and without surgical artefact, to model the effects of vascular occlusion at locations that were more representative of clinical syndromes than had previously been possible. An early paper in this area described retro-orbital exposure and occlusion of the middle cerebral artery in primates.
45
The authors clearly documented and illustrated patchy, reversible vasoconstriction in surface arteries in the occluded middle cerebral territory. On occasions several recurrent episodes were seen in the same arterial segment: it was argued that the constriction was active and did not reflect passive collapse from loss of arterial perfusion pressure: also, it never occurred “without preceding cortical pallor” (see Figure 2). With hindsight, aided by the multiple demonstrations in recent years of surface vasoconstriction, cortical spreading ischemia and pallor coupled to spreading depolarisations in partially ischemic cortex (“peri-infarct depolarizations,” PIDs, please see below), it will seem very likely to anyone reading their paper that Waltz and Sundt
46
were the first to describe evidence for cortical spreading ischemia (most likely coupled to depolarisations that their system did not allow them to detect, hence preventing them from understanding the full significance). The attribution of these radical changes in the cerebral cortical microcirculation (vasoconstriction, cortical spreading ischemia and pallor) to the effects of a spreading depolarisation is possibly the single most critical development in understanding of the pathophysiology of acute brain injury since early descriptions of ‘penumbra’, and for full descriptions the reader is referred to the section below entitled ‘Neurovascular unit and SD: from spreading oligemia to spreading ischemia’, and to the papers by Hartings et al. and by Dreier et al. in this issue. More contemporary illustrations of cortical vasoconstriction and pallor come from (a) the intense increase in NADH fluorescence in the exposed cerebral cortex that accompanies a PID (the increase appears to be derived more from reduced quenching by haemoglobin of the 450 nm NADH signal than from a true reduction in the NAD/H couple
47
and is readily visible to the naked eye in good room lighting), and (b) the patchy pallor evident in an area of ischemia exposed during decompressive craniotomy for malignant hemisphere stroke (Figure 4: fluctuation in pallor is hard to document without extended video recording, and a static image of pallor may also represent established infarction without reperfusion).
Intraoperative microphotograph of the cerebral cortex during surgical decompression for malignant hemisphere stroke. The image shows an area of cortical pallor (centre and lower left field) suggestive of microvascular constriction consistent with cortical spreading ischemia in this highly pathological type of stroke.45,117 Repetitive spreading depolarisation events in a patient following evacuation of a traumatic intracerebral hematoma. Before wound closure, a six-contact Wyler subdural electrocorticogram strip was placed on cortex close to the hematoma site but judged still to be viable, and connected in bipolar sequential montage to a ADInstruments Octal Bioamp and Powerlab A:D converter running ADInstruments Labchart v6 (five of the contacts on the strip are shown: top left). Top four traces are AC coupled with low frequency cut off below 0.02 Hz. Lowest two traces are the uppermost two raw traces, but now band-pass filtered to 0.5–15 Hz. Typical AC-coupled slow potential change (SPC) complexes are seen in the upper two channels, with simultaneous suppressions of the band-pass filtered signal. Figures above upper trace are inter-SD intervals (minutes): the constant interval is compatible with repetitive cycling at constant velocity of a single depolarisation around the periphery of a fully depolarised core lesion. Original recordings of spontaneous potassium transients (pial surface potassium concentration, Kp) on adjacent gyri 2 and 5 min after occlusion (time zero) of the right middle cerebral artery in a cat. Recordings were taken by AJS and colleagues, 1981–1983 and are unpublished, but please see Strong et al.56,57 A twin barrel valinomycin/reference electrode was spring-suspended in light contact with the suprasylvian gyrus (typically inner penumbra). The peak potassium amplitude is much less, and the duration longer, than are seen with microelectrodes, attributed to the averaging effect of the 2 mm diameter of the electrodes and to their locations on the pial surface with probable diffusion delay through the pia. Immediate post-clip increases were common on both gyri, but the increase on suprasylvian (inner penumbra) did not return to baseline.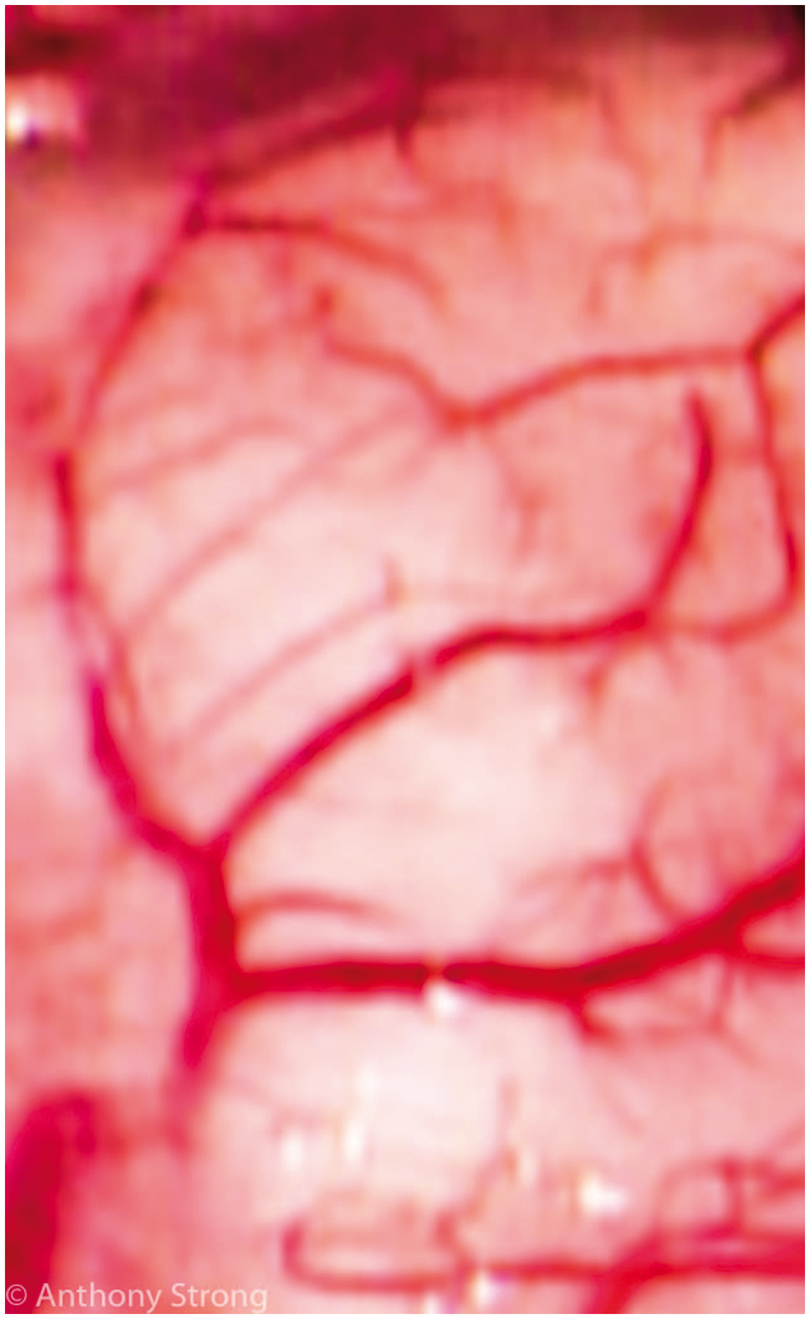
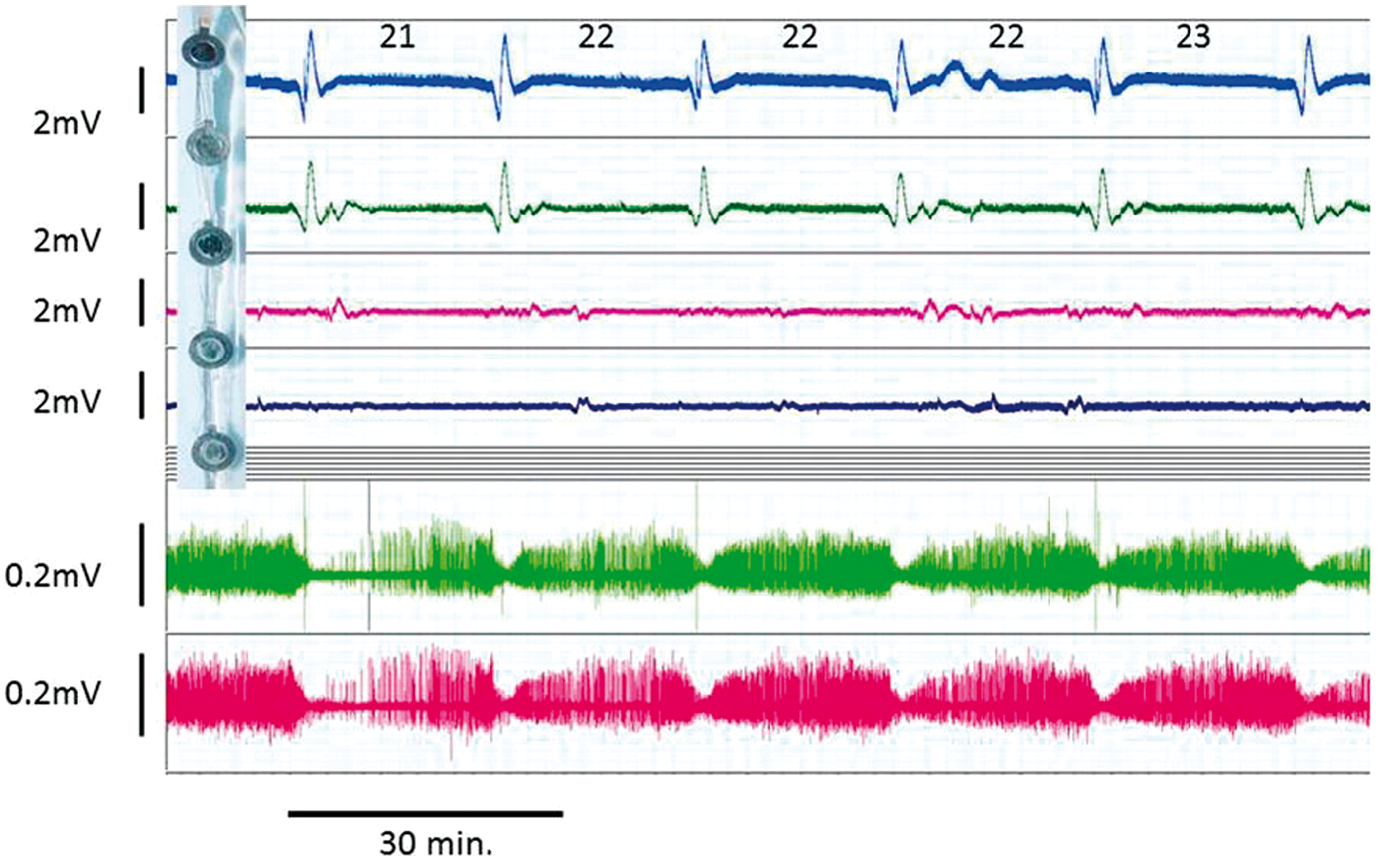
Also in the 1970s decade, a series of papers from the laboratory of Lindsay Symon reported the introduction of the hydrogen clearance method for local quantification of cerebral cortical perfusion in acute and chronic primate models, examining the effects of transorbital middle cerebral artery occlusion (MCAO) on perfusion, histology, and electrophysiology.48–53 Notably, a paper with Lassen and his co-workers
54
reported differential, staged suppression of cortical somatosensory evoked potential followed later by sustained rises in cortical extracellular potassium (Ke) concentration as cortical ischemia became significantly more severe. There followed a paper from the same laboratory in which differential perfusion thresholds for evoked potential suppression and sustained rise in Ke were more formally documented.
55
In this later work, small, spontaneous, self-limiting increases in Ke were noted (Figure 6 in that paper, for example), accompanied on occasion with a transient increase in tissue impedance and abrupt loss of evoked potential, ‘suggesting some parallel with spreading depression’. Similar spontaneous Ke transients were later described following MCAO in cats
56
(Figures 4 and 5 in this present article): again, the analogy with SD was noted, together now with detection of scattered focal ischemic cell change in areas experiencing Ke transients.
57
The suggestion was made in that paper that such events ‘cannot necessarily be regarded as entirely benign’, although the association did not prove causality. Astrup et al. had emphasised the reversibility upon early, prompt reperfusion of the differential changes that they defined as the basis of the penumbra, and a need to determine the nature of electrophysiological suppression in the penumbra was recognised. Given the growing availability in clinical neurosurgical practice at that time of cerebral revascularisation by extracranial to intracranial microvascular bypass, was the penumbra a state of permanent functional ischemic suppression simply awaiting reperfusion at the hand of a neurosurgeon – the ‘sleeping beauty’?58,59 Early clinical imaging studies of patients with acute stroke suggested the presence of a penumbra,
60
later shown to persist for up to 48 h.
61
Experimental MCAO work with the same positron emission tomography (PET) multitracer technology in cats strongly supported this concept.
62
Thus, the penumbra came to be regarded as a progressive maturation phenomenon in which peripheral areas of functional suppression represented the effects of recurrent depolarisations (rather than of diaschisis resulting from ipsilateral terminal depolarisation in the infarct core
63
), these areas being highly vulnerable to invasion by recurrent, spontaneously occurring SDs that became designated “peri-infarct depolarizations” (PIDs).
64
The hypothesis now evolving was that a final PID becomes irreversible, probably due to substrate depletion and energy failure, leading to recruitment of the vulnerable focus of penumbra into the expanding core: only later did the close association of PIDs with cortical spreading ischemia emerge (please see below). However, were recurrent PIDs in the penumbra the cause or the effect of infarct expansion? Three studies published in the 1990s suggested that PIDs do indeed promote infarct expansion.65–67 The first experimental demonstration that the frequency of spontaneous PIDs could be reduced came from work with MCAO in rats exposed to substantial hyperglycaemia
68
Later, the N-methyl-d-aspartate receptor (NMDAR) antagonist MK801 was shown to reduce both PID frequency and infarct size.69,70 Thus, the evidence pointed to a causal role for PIDs in infarct expansion. However, the first indications that CSD occurred in human patients appeared a decade earlier and were based on observations in patients with migraine.71,72
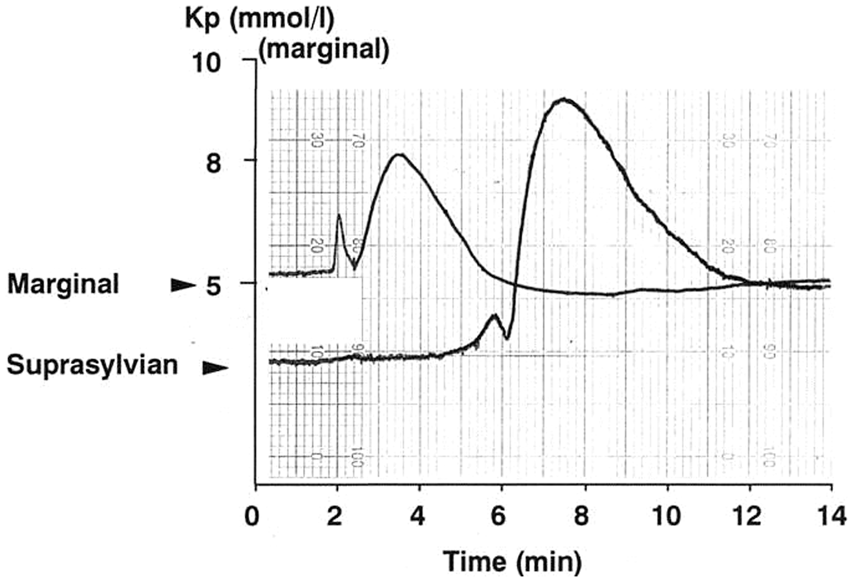
Original recordings of recurrent, spontaneous potassium (pial surface potassium concentration, Kp) transients on adjacent gyri 2 and 5 min after occlusion (time zero) of the right middle cerebral artery in a cat. Recordings were taken by AJS and colleagues, 1981–1983 and are unpublished, but please see Strong et al.56,57 Experimental system was as for Figure 4. The ‘staircase’ increase in pre-transient baseline was noted during the studies, and post calibration in this experiment had excluded baseline drift. The stepwise increase following the events finds some parallels with: (a) stepwise increase in infarct size coupled to recurrent penumbral SDs in distal MCAO in rats,
43
and (b) with stepwise decline in brain tissue glucose in the injured brain in humans.
156
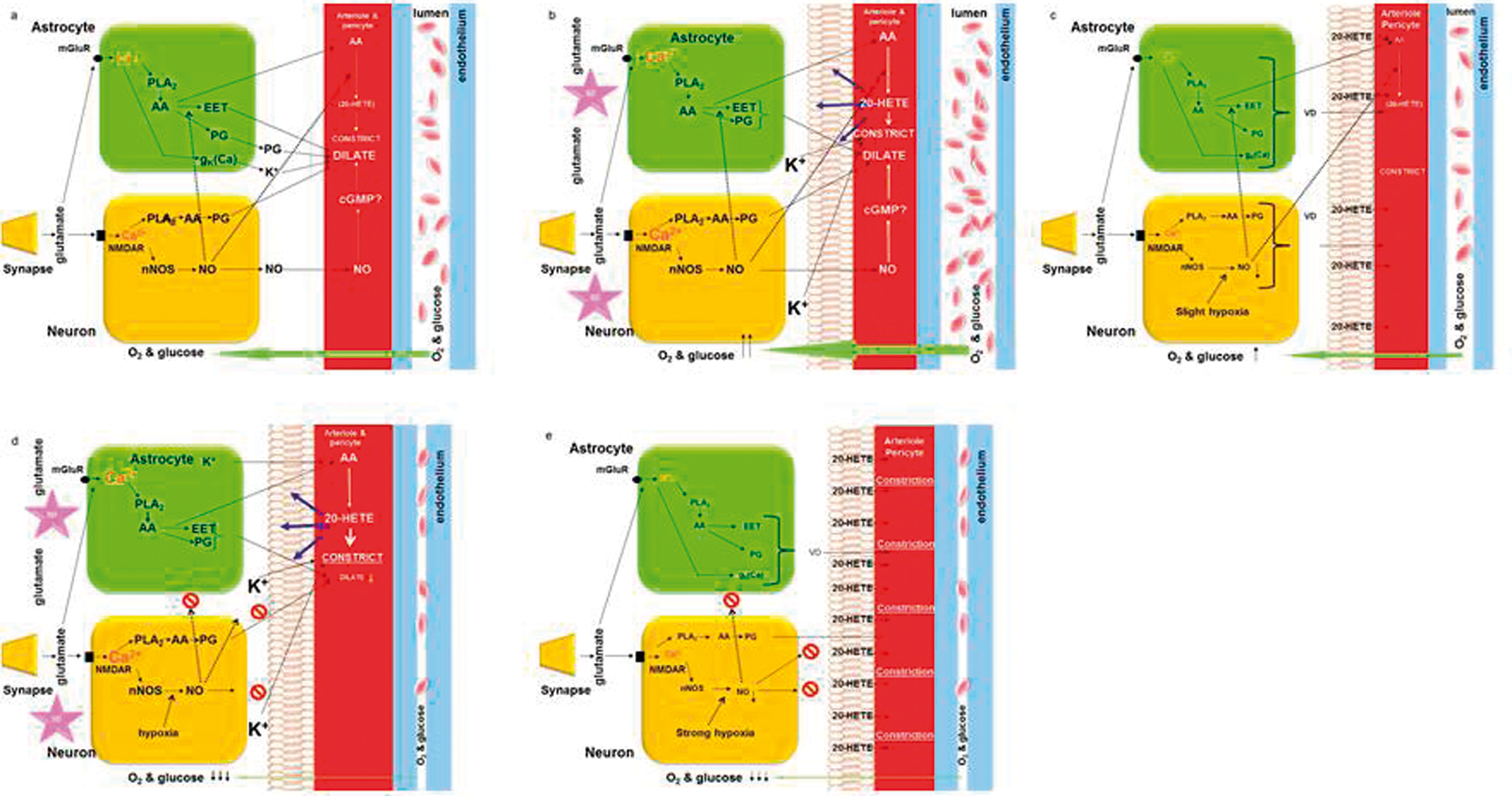
Schematic diagrams of potential mechanisms involved in neurovascular control in SD. (a) Control conditions. Neurovascular coupling responses are triggered by glutamate release from incoming or local presynaptic axons, and the interaction of glutamate with receptors on neurons and astrocytes. N-methyl-d-aspartate receptor (NMDAR) activation in neurons triggers nitric oxide (NO) production by NO synthase (nNOS) activity. The rise in Ca2+ in neurons via NMDAR and voltage-sensitive Ca2+ channels, and in astrocytes via metabotropic glutamate receptors (mGluR), stimulates activity of phospholipase A2 (PLA2) that catalytically hydrolyzes the bond releasing arachidonic acid (AA). Upon downstream enzymatic activity, AA is modified into active compounds such as prostaglandins (PG) and epoxyeicosanotrienoic acid (EET) that are mainly vasodilators and 20-hydroxyeicosatetraenoic acid (20-HETE) which is a vasoconstrictor. NO dilates arterioles via the cyclic GMP (cGMP) pathway and inhibits (dashed lines) the synthesis by cytochrome P-450 of 20-HETE from AA. Under control conditions, the concentration of 20-HETE is very low, and the net balance of constrictor and dilator activity results in an intermediate level of constrictor tone in vascular smooth muscle and pericytes, enabling transient responses to occur in either direction. (b) In the acute phase of SD, there is massive release of K+ and glutamate. This leads to a huge global rise in cytosolic Ca2+ which leads to a substantial production and release of vasoactive mediators.
107
The massive increase in Ca2+ during SD also causes an increase in 20-HETE synthesis,
157
which leads to a buildup of internal 20-HETE stores in the lipid biomembrane of various cell types.
158
During the acute phase of SD, vasodilation dominates due to a net release of vaosodilators from multiple sources leading to increased availability of glucose and O2 that is rapidly consumed due to the extra activity of the Na,K-ATPase.
99
(c) After CSD, 20-HETE may be slowly released from lipid biomembranes,
158
causing the characteristic prolonged oligemia lasting for 1½ h.
157
At the same time, the production of vascular mediators by multiple pathways is affected due to impairment of stimulation-induced Ca2+ activity.
107
Drugs that block 20-HETE synthesis rescue the basal blood flow, but cannot affect the persistent strong reduction in blood flow responses to local or systemic vasodilator stimuli. The causes of the impaired neurovascular responsiveness after CSD include both lack of stimulation-induced Ca2+ responses in neurons and astrocytes and an unknown mechanism, intrinsic to the resistance vessels, that hinders the drop in resistance which precedes or accompanies any flow increase. These vascular mechanisms, we believe, are in play during the course of attacks of migraine with aura. (d) In the acutely or subacutely injured human brain cortex, the interplay between SD, vascular reactions, and brain energy homeostasis days may cause cell death due to a growing mismatch between energy demand and supply. For example, after subarachnoid haemorrhage, K+ is released (by haemolysis), which in combination with hypoxia and the reduced ATP pool because of reduced glucose and oxygen availability increases the susceptibility to SD. The repeated SD waves cause repeated rises of Ca2+ in neurons and astrocytes that facilitate the production of vasoactive substances. However, the low level of tissue O2 will limit the NO synthesis because the Km of O2 for NOS is very high.
106
Furthermore, the NO produced is likely to be captured by heme groups in the brain’s extracellular space as indicated by the red circles.
46
Therefore, under these conditions, NO cannot inhibit the synthesis of 20-HETE, which is known to be very high and cause strong vasoconstriction,
159
and due to the embedding of 20-HETE in the lipid membranes, this effect can be protracted and contribute to the ‘spreading ischemia’ as described by Dreier and Reiffurth
82
even in periods between SDs (e).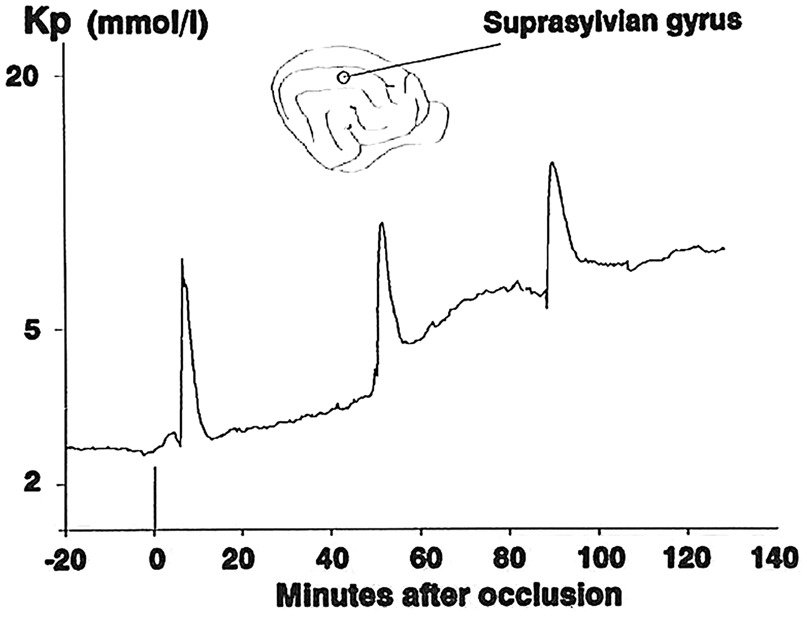
CSD and the migraine aura
MA attacks have a characteristic course: At first the patients experience transient focal neurological symptoms with a characteristic pattern of evolution (see below), spontaneously remitting within 30–60 min.
73
At the same time as the focal symptoms, or after they have disappeared, the headache, most often unilateral and throbbing, ensues. To explain this characteristic sequence of events, there were two theories: One was the ‘vascular’ theory, which ascribed the primary event in migraine to a disturbance of cerebrovascular function. The focal symptoms were supposed to be causally related to transient constriction of a cerebral artery, and the headache to a sterile inflammatory reaction around the walls of dilated cephalic vessels.
74
The ‘vascular’ theory dominated the migraine field for decades, but in the 1980s it became increasingly recognised that migraine could be a disturbance of the nerve cells themselves and that it was possible that the events in the blood vessels could be secondary.71,72,75 This possibility had already been noted by several clinicians and scientists.76,77 The slow progression of the migraine aura indicated a wave of excitation in the primary visual cortex moving at the speed of 3 mm/min, followed by a longer period of inhibition
78
data that exactly matched cortical SD that was described three years later.
2
Concerning migraine, Leao and Morrison wrote the following: Much has been written about vascular phenomena both in clinical epilepsy and the presumably related condition of migraine. The latter disease with the marked dilation of major blood vessels and the slow march of scotoma in the visual or somatic sensory sphere is suggestively similar to the experimental phenomenon here described, in spite of the fact that known scotoma are still felt to be vasoconstrictor in nature.
79
The findings of a ‘spreading oligemia’ with impaired vascular reactivity in migraine patients71,75 were confirmed by other functional neuroimaging techniques in the 1990s. In 1994, Woods et al. 80 reported the results of PET scanning carried out in a volunteer with a history of recurrent headaches consistent with migraine without aura. While undergoing scanning as she undertook a visual task, headache developed with nausea and photophobia, and a series of scans detected a bilateral wave of hypoperfusion spreading in – and progressively forwards from – the occipital lobes. Using a visual checkerboard paradigm during functional magnetic resonance imaging (fMRI: blood-oxygen-level-dependent, BOLD) in a volunteer during exercise-induced migraine-with-aura and in two others with spontaneous auras, Hadjikhani et al. 81 were able to detect a wave of loss of the BOLD response that progressed forwards from the occipital region in the hemisphere contralateral to the subject’s visual aura at a rate across the cortex closely corresponding with that typical of SD. Taken together with the aforementioned papers, it became widely accepted that SD is a key pathogenetic step in MA. Interestingly, patients with acute brain disorders as described in the following sections who had documented episodes of CSD while being conscious did not report a march of focal symptoms similar to a migraine aura, illustrating the complexity of the relation between CSD and the accompanying neurological symptoms as discussed recently by Dreier and Reiffurth. 82
SD and acute brain injury from lab bench to humans, the first steps
Given the increasing experimental evidence that PIDs might contribute to infarct enlargement, in the 1990s decade, the question began to be asked with increasing frequency: do SDs occur not only in migraine, but also in the injured human brain? ‘Injury’ encompasses not only traumatic brain injury (TBI), but also occlusive stroke, subarachnoid haemorrhage, and spontaneous intracerebral haematoma. Based on the transient nature of symptoms in some cases of paediatric head trauma, a paper in the neurological journal Brain expressed the idea that SD was involved. 83 Next followed papers by Hubschmann and Kornhauser,84–86 which suggested that red blood cells when they lysed released potassium that surpassed the threshold for CSD in traumatic brain injury and subarachnoid haemorrhage. The findings suggested that the combination of transient K+ elevations and Ca2+ depression could play ‘an important role in the development of vascular spasm by inducing or facilitating a contraction in the muscular layer in the wall of major intracranial vessels’. 85 This taken together with the fact that cerebral vasospasm in subarachnoid haemorrhage was observed at sites remote from the initial bleed suggested that additional mechanisms than local breakdown products of haemoglobin could contribute to vasoconstriction at a distance from the bleeding. The observations by others and ourselves were compiled into a paper that was published on occasion of Professor Leão’s 70th birthday in 1984. 87 Soon thereafter, Nedergaard and Astrup68 showed spontaneous and recurrent changes in cortical DC potential in focal ischemia in rodents and, citing Nicholson and Kraig, 88 identified these DC transients as essentially equivalent to the extracellular potassium transients described in earlier work in primates,55,89 rats, 90 and cats. 56 Importantly, Nedergaard and Astrup drew attention in their paper to the close association of increased cerebral glucose utilisation with recurrent CSDs. The finding has important implications for clinical management of acute brain injury (please see below under ‘Significance of SDs for current clinical management of acute brain injury’).
Detection of SDs in the acutely injured (vascular or traumatic) human brain
In 1996, a low incidence of SD events following TBI (in humans) was reported by Mayevsky et al., 91 who sought SDs using a single multiparametric monitoring bolt placed at the neurosurgical standard right frontal convexity location in their 14 patients with TBI; SDs were found in only one, and at an ipsilateral site remote from the lesion.
There was clearly a need to explore noninvasively the possibility that SD occurred in man, but it was an open question as to how to do that. The SD community was very small at the time and mainly concerned with experimental aspects, and clinicians were still reluctant to support CSD research in humans. The authors of this review interacted several times to obtain EU funding for SD and ischemia research in combination, but alas to no avail. However, progress with novel experimental methods (applicable only in the laboratory) served to increase the pressure to seek evidence of SD in acute brain injury in humans (AJS had successfully developed a method for imaging the penumbra and SDs in cats subjected to MCAO using NADH fluorescence imaging 47 and laser-Doppler flowmetry and laser-Doppler perfusion imaging had been developed and implemented for studies of SD.92,93)
Therefore, the stage was set to develop strategies on how to detect SD in humans. Together, we (these authors) discussed potential methods to detect SDs in humans, both invasive (electrocorticography) and non-invasive. This latter option was discussed in a book chapter published on the occasion of a meeting with the theme ‘Ischemic Blood Flow in the Brain’ at the Keio University in Tokyo, chaired by the late Minoru Tomita. 94 An attempt by Back et al. 95 to detect SDs in patients with acute stroke using serial diffusion-weighted MRI was unsuccessful, but possibly only because of the short sampling times that could be used. One of us (AJS: a neurosurgeon, with the benefit of advice from a close colleague specialising in the neurosurgical assessment and treatment of epilepsy) obtained ethical approval to leave, at the conclusion of clinically indicated emergency craniotomy, a linear, six-contact Wyler subdural strip on the cortical surface, at a location judged visually to be viable but lying close to traumatic contusion or to an infarct border as estimated from preoperative imaging. ML’s advice was that the contacts should be connected in sequential bipolar montage.
Work started in late 2000, and with the time display heavily compressed, apparently spontaneous SD events were soon seen in some but not all patients. By May 2001, the London team had sufficiently promising data to send it to Copenhagen for assessment. This elicited an immediate visit to London by ML and Dr M. Fabricius, and the decision was taken to formalise collaborative data collection in patients in the King’s College Hospital intensive care unit. Dr. Fabricius played a seminal role in advising on the practicalities regarding the recordings and the data analysis. The work continued for some 12 months, culminating in the preparation of the first manuscript reporting clear evidence of spread of spontaneous ECoG depression events in a number of human subjects. In 14 consecutive patients with traumatic (n = 11) or spontaneous intracerebral hematoma (n = 1), or with an intracranial aneurysm requiring emergency surgery (n = 2), evidence of spreading or synchronous depression of the electrocorticogram (ECoG) signal was found in eight. 96 The higher incidence now (2002) detected was attributed to the placement of a wider sampling device close to a known lesion, rather than at a standard site that might lie remote from a focus of damage in a given patient. 91 Importantly, the higher incidence now seen may also have resulted from the prolonged and continuous ECoG monitoring that often extended over a number of days.
The preamplifier filters were set to a pass range of 0.5 to 70 Hz, which allowed only the detection of the suppression of the electrocorticogram. However, a subsequent paper addressed successfully the need for evidence of an SPC accompanying the ECoG amplitude suppression. 97 Following publication of the 2002 paper and an enquiry from Dr JA Hartings, now a prolific author in the field, a small meeting of largely European investigators publishing in the field of experimental SD studies of models of brain injury was held in Copenhagen in 2003. This meeting resulted in the formation of the COSBID collaboration (www.cosbid.org, Co-Operative Study of Brain Injury Depolarisations), with the goals of (1) examining the incidence of SD-like events in the patient groups listed above, and the impact of SD-like events on outcome, and (2) acting as a source of information and advice for any investigator wishing to study SD in patients with acute brain injury or undergoing neurosurgery. COSBID-based publications are listed among the Reference Lists of the other papers in this special issue. In recent years, COSBID (Co-operative Studies since 2011) has evolved into a translational interest group of both experimental and clinical investigators with multiple collaborative projects including, but not limited to, joint publication of papers such as this issue of JCBFM Clinical.
Neurovascular unit and SD: From spreading oligemia to spreading ischemia
The demonstration of unique changes of cerebral blood flow during attacks of MA, the spreading oligemia,71,75 which was replicated in animal experiments during CSD,72,98,99 constituted for many years the only line of support for the possibility that SD occurred in human brains. The vascular changes in SD have been extensively reviewed recently 6 and will only be summarised briefly here. The finding of the ‘spreading oligemia’ was soon followed by the discovery of a persistent oligemia in the wake of cortical SD in rodents providing an experimental link between SD and the vasoconstriction in humans. 72 The ‘spreading oligemia’ in migraine, and the corresponding SD-related moderate reduction in cerebral blood flow are spontaneously reversible both in rodents 100 and in patients101,102 and believed not to cause or involve brain damage 103 when occurring in otherwise normally perfused brain tissue. This is in contrast to the ‘cortical spreading ischemia’ or ‘inverse neurovascular coupling’ discovered by Jens Dreier which is severe and sustained and causes brain damage in the acutely injured human brain cortex and in rodents.104,105 The vascular changes whether ‘oligemic’ (i.e. benign) or ‘ischemic’ (i.e. damaging) involve impaired function of the neurovascular unit (neurons, astrocytes, capillary endothelium, pericytes, and vascular smooth muscle in close juxtaposition), but this is incompletely understood. We do know that rises in cytosolic Ca2+are key to an understanding of neurovascular function 106 and that huge increases in intracellular Ca2+ occur in both neurons and astrocytes during SD propagation12,30,107(Figure 6). Therefore, we suggest that changes in intracellular Ca2+ homeostasis during and following SD may contribute mechanistically to impaired blood flow control because of alterations in the balance of vasoconstrictors and dilators produced by the Ca2+-dependent mechanisms and changes in the extracellular microenvironment.6,107 The observations of SD-related vascular changes in normally perfused tissue show: (i) vasoconstriction before SD or during the onset of depolarisation;108,109 (ii) a rise in CBF (hyperaemia) simultaneously with the release of lactate to the brain interstitial fluid 110 and during return to normal of the ionic changes in SD 99 with a heterogeneous distribution across cortical layers; 111 (iii) followed after 1–2 min by a 20% to 30% flow reduction (oligemia) 72 resulting in part from cortical arteriolar vasoconstriction,30,112,113 and impaired vascular responsiveness.92,98,99,112,114 The reduction in reactivity is a fundamental vascular manifestation of SD also in patients. 101 The CBF changes in SD have been reproduced in awake, freely moving animals. 115 In contrast, in patients with cortical damage due to subarachnoid haemorrhage, malignant hemispheric stroke or traumatic brain injury, SD is commonly associated with ‘cortical spreading ischemia’ (CSI), which widens the gap between energy demand and supply and leads to cortical infarcts116–118 as described elsewhere in this volume by Jens Dreier and in recent reviews on the topic.6,82 The normal haemodynamic response with spreading hyperaemia (brief) followed by persistent oligemia and the inverse haemodynamic response with spreading ischemia sometimes followed by spreading hyperaemia show a continuum both in animals and in patients. 82 We suggest that the neurovascular mechanism for both the ‘spreading oligemia’ and the ‘spreading ischemia’ be related to the interaction between the synthesis and availability of the vasodilator NO and the synthesis, storage, and release of the vasoconstrictor 20-HETE as outlined in Figure 6.
Significance of SDs for current clinical management of acute brain injury (vascular or traumatic)
Perhaps the most important feature of SDs occurring in a patient with a serious acute brain injury arising from trauma or from any type of stroke is the strong likelihood of cortical spreading ischemia as described above, potentially resulting in very significant increase in the volume of tissue lost. Thus, in patients with aneurysmal subarachnoid haemorrhage (aSAH) monitored with wide frequency band ECoG and laser Doppler perfusion probes adjacent, clear evidence of CSI coupled to depolarisations is observed. 116 Earlier experimental work makes clear that the SD is the cause and not the consequence of the vasoconstriction.46,119,120 Clinically, it seems very likely that CSI might constitute the long sought basis for delayed ischemic neurological deterioration (DIND) after aSAH. 116 A second potentially adverse effect of SDs in the injured brain relates to increased utilisation of glucose by cortical tissue likely to be dependent on anaerobic glycolysis and hence inefficient utilisation. The appropriate target level for plasma glucose in patients with acute brain injury is an issue now well recognised by intensivists managing such patients.121,122 There is evidence to suggest that low brain tissue glucose in the ischemic brain can be both a cause and a consequence of an increased frequency of spontaneous SDs, thus generating a vicious circle. This is discussed more fully by Boutelle et al. in this issue. Thirdly, SDs upregulate matrix-metalloproteinase-9 (MMP-9), which in turn opens the blood–brain barrier, 123 enabling the development of vasogenic oedema and a consequent rise in intracranial pressure (ICP): MMP9 is increased in pericontusional microdialysate from patients with acute brain injury. 124 Fourthly, SDs upregulate several gene expression cascades, including pro-inflammatory cytokines125,126 and candidate mediators for the headache and meningitis-like (photophobia, neck pain) post-aura symptoms of migraine. 127
As with aSAH patients, so also for those requiring surgery for acute TBI: studying 103 patients with TBI who underwent craniotomy for management of mass lesions or intractably raised ICP, the COSBID group found significantly worse outcome in patients experiencing SDs in tissue in which ECoG amplitude was suppressed (by injury rather than by therapy) prior to the SDs: for SDs recorded in tissue with a spontaneously active ECoG signal, the trend was in the same direction. 128 Importantly, the addition of depolarisation data to the six-month outcome prediction model for this cohort increased the amount of variance in outcome that could be explained by the seven-variable IMPACT model 129 from 9 to 22%. However, such data may not take adequate account of secondary insults (other than depolarisations) such as pyrexia, hypoxia, and hypotension 128 that good intensive care seeks to minimise. It may well be that SDs, and especially those where the ECoG is already isoelectric, are often mediators of the adverse effects of secondary insults, but the majority of SDs (with or without pre-event background ECoG activity) appear still to occur spontaneously when clinical management has already been carefully optimised. Thus, both secondary systemic insults and the SDs themselves need to be addressed.
Before discussing clinical strategies in acute brain injury, a small caveat is appropriate. Jander et al. 125 suggested that upregulation of inflammatory cytokine pathways by SD might contribute to development of tolerance of ischemia. Such tolerance might in turn account for the resistance to the effects of ischemia conferred on rats by pre-ischemic induction of SDs. 130 This conceivably advantageous property of SDs may, rightly or wrongly, engender caution among clinical investigators assessing the therapeutic potential of attempts to block SDs pharmacologically in patients with acute brain injury. Stoll et al. 131 also discuss ‘Detrimental (AJS and ML underlined) and beneficial effects of injury-induced inflammation and cytokine expression in the nervous system’. 131 It seems this issue will need to be addressed directly in future studies: how does the balance of putative adverse and protective properties of SDs (those associated with a normal, hyperaemic vascular response) affect clinical outcome in patients with acute brain injury?
Suggested current clinical ICU management of conditions where SDs are known often to occur
The current clinical management of migraine is beyond the scope of this review. For patients with acute brain injury being treated in a well-equipped ICU, we would suggest that, for the present, the priority in intensive care should be to optimise control of potential secondary insults such as systemic hypotension and hypoxia, as well as pyrexia, and in addition to maintain plasma glucose within a range least likely to promote either SDs or brain acidosis. This range is likely to lie between 7.5 and 10.5 mMol (Boutelle, this issue). Continuous monitoring for SDs, for brain oxygen tension, and for brain tissue glucose seems highly desirable where resources permit, not only to provide early evidence of deterioration in one or more specific variables, but also to optimise target ranges for management of the cardiorespiratory system and for glucose delivery to the areas of cortex most vulnerable. There is recent evidence suggesting that simultaneous progressive loss of autoregulation and of cerebral vasoreactivity to SDs might reflect impairment of a common underlying mechanism, 118 and monitoring of autoregulation enables the setting of an optimal cerebral perfusion pressure (CPP) that may improve outcome. 132
A laboratory strategy to inform future advances in clinical management
Understanding the details of cerebrovascular control in SD is essential for our ability to offer patients proper treatment for their perfusion deficits. Perhaps the most important challenge is the pathological reversal in much injured or vulnerable tissue of the normal vasodilator response to SD to one of vasoconstriction: a primary goal of therapy must be to rescue such tissue from the severe vasoconstriction of CSI in the injured cerebral cortex that results from the reversed vascular response to SD.
Beyond the approach suggested above, we need now to understand better the several different features of ‘normal’ SDs (associated with a hyperaemic response and pre-SD ECoG activity), of SDs with no pre-event ECoG activity, and of the pathological vasoconstrictor response, i.e. CSI, that might be targeted pharmacologically. The human studies have broadened the spectrum of CSD research because they have informed the experimentalist about how perturbed tissue will affect the expression and consequences of CSD.32,127,133,134 Existing data sets can potentially be used to guide choice of a drug for trial in patients shown to be experiencing SDs. 82 For example, we do know that NMDA receptor antagonists block CSD propagation in healthy tissue 135 and that uncoupling of PSD-95 from NMDA receptors (postsynaptic density protein 95 that links activity at the NMDA receptor to downstream effects) reduces overall neuronal excitability and the amplitude of the SD-associated DC-shift. 136 Furthermore, the anaesthetic ketamine, which blocks NMDA receptors,137,138 has been shown to block CSD in patients with traumatic brain injury, subarachnoid haemorrhage, and malignant hemispheric stroke.139–142 In his original work on rats with perfectly healthy brains, Bures and his co-workers 143 reported that ketamine dosage was important and that CSD-blocking doses did not block AD. This suggested that the mechanism of a ‘simple’ CSD in healthy tissue differed from the mechanism of AD, but this may not be clinically relevant because a human study of analgesics, sedative, and ketamine suggested that these drugs can reduce the number of CSD waves importantly, suggesting a high susceptibility of CSD to drugs that decrease the overall cortical excitability. 141 However, while the effect of ketamine on CSD in patients is convincing, we still need evidence from a large carefully designed clinical trial before concluding that ketamine is the drug of choice in patients with evidence of acutely injured brain cortex and with evidence of CSD. Meanwhile, the experimentalists may examine alternative possibilities by exploring novel mechanisms. However, there is a strong need for regular preclinical testing in order to examine the therapeutic potential of new or old pharmaceutical compounds.144,145
The CSD field is where the stroke field stood before 1999, when a paper was published by the Stroke Therapy Academic Industry Roundtable (STAIR) group on ‘Recommendations for Standards Regarding Preclinical Neuroprotective and Restorative Drug Development’.146,147 The reasoning was that without ‘rigorous, robust and detailed preclinical evaluation’, it was unlikely that novel neuroprotective drugs would be effective in large-scale clinical trials. On this background, the STAIR group formulated a document that has served as an overall guideline for stroke research for the following decade. In an update from 2009, an even more detailed set of guidelines was published with important changes that include trial data that reflect the diverse comorbidities in patients. 147 A number of weaknesses of preclinical trials were pointed out: specifically, it was indicated that poor quality studies overestimate drug efficacy while in comparison, efficacy of a given treatment decreased as the studies adhered more closely to the STAIR guidelines. Future experimental studies in CSD that examine drugs that may be candidates for preclinical studies will require similar guidelines, perhaps based on CAMARADES criteria. 148 Account should also be taken of the outputs from Operation Brain Trauma Therapy. 149 Such guidelines can be applied in the assessment of models of different types of acute brain injury: TBI, aSAH, and malignant hemisphere stroke. However, it is critical that such models should replicate the derangement of the target mechanism known to exist in the clinical condition. One might also have to examine the susceptibility of the depolarisation wave for different age groups, common comorbidities, grades of background of EEG activity, blood flow, and energy use. Ketamine is a promising drug for large clinical trials, but is it safe and are there ways to determine whether repeated use will render the NMDA receptor channel tolerant to prolonged treatment as in rat studies? For this reason, and to prepare for a large clinical trial on antiCSD medication, we need to develop a set of preclinical guidelines for CSD research and prioritise among the drugs that are in the pipeline or have already been tested in nonrandomised, unblinded trials. Along the same line of thought, the clinical trials must adhere to the Consolidated Standards of Reporting Trials (CONSORT) statement.150,151 The strict standards for the regular preclinical and clinical trials ‘should not preclude publication of observational, pilot, or hypothesis generating data in animals or man, but the conclusions of such studies should reflect their preliminary nature’ as indicated in the 2009 update on the STAIR guidelines. 147 Future work on SD ought to focus on guidelines for preclinical studies, which is likely to advance the field for a clinical trial. The basic principles for preclinical research for diseases relevant for study and treatment of SD have already been worked out by the stroke community, and the adaptation of those guidelines for preclinical SD research holds the promise of a continuing contribution to translational neuroscience from this particular area of study.
Future opportunities and challenges for clinicians studying CSD
Although many clinicians specialising in neurocritical care are becoming increasingly aware of the expanding clinical literature focussed on SD, the detection technology and analysis methods still remain largely within the research domain. In consequence, awareness of the potential value of closer attention to SD occurrence is at present limited to the small number of existing COSBID-affiliated centres, and these appear currently (2016) to be the only sites capable of conducting clinical trials. Furthermore, the present invasive methods for detection of CSD necessarily restrict our knowledge of SD to patients requiring craniotomy or, at the least, monitoring by probes placed in the brain via an intracranial access bolt. We therefore need non-invasive methods for SD detection, validated in the present study populations, that can be deployed in studies of patients with other acute encephalopathies who require intensive or high dependency care, for example viral or bacterial encephalitis, meningitis, cerebral abscess, and septic encephalopathy, but also as a means of simplifying CSD detection in the broader range of patients with acute traumatic/ischemic brain injury who do not initially require craniotomy. Continuous EEG recording from scalp contacts may show greater promise than expected earlier,152,153 but further developments are needed. Coupled with this, a clearer understanding is needed of the interactions between known secondary insults to the injured brain and the onset and frequency of spontaneous SDs. Carefully analysed multimodal monitoring is required in order to unravel cause and effect relationships: for example, Belli et al. 154 , monitoring patients with acute brain injury with microdialysis, have shown that metabolic stress may precede a rise in ICP. Secondly, so long as there remains uncertainty as to the relative benefits and dangers of the inflammatory response to SDs occurring in CNS injury, there will remain a need to clarify this by examining the risk-adjusted impact of SDs (only those not accompanied by reductions in metabolite availability – low brain tissue oxygen or glucose) on outcome.
A third and important challenge is to influence a significant body of opinion – widely held among industry and investigators whose concepts of pathophysiology remain influenced – we believe unduly – by the negative results of clinical trials of neuroprotection with NMDAR antagonists in acute stroke and TBI in the 1990s decade. Several cogent explanations have been suggested for the uniform failure of these studies.152,155 Among these is the consideration that the study populations in that decade were recruited largely on an intention-to-treat, ‘one-treatment-for-all’ basis. In our opinion – informed by a steady incidence of SDs of no more than 50–60% in COSBID data in the case of TBI – a study population in the 1990s would have included patients with very diverse patterns of pathology, with perhaps only 50% suffering focal contusional damage, and in turn only 50–60% of this subgroup experiencing depolarisations. Thus, a study targeting a mechanism of injury present in perhaps only 25% of the study group is destined to yield a negative and perniciously misleading result, and this is a crucial consideration for future trial designs. In our opinion, failure on anyone’s part to acknowledge the force of this argument does a disservice to a very large patient population, each of them facing the prospect of serious disability.
Footnotes
Funding
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: ML was supported by the University of Copenhagen, the Lundbeck Foundation, the NORDEA Foundation Grant to the Center for Healthy Aging at the University of Copenhagen, the NOVO-Nordisk Foundation, and the Danish Medical Research Council. AJS has received support from the UK Medical Research Council, from The Wellcome Trust, and from HeadFirst.
Acknowledgements
AJS thanks Prof Charles Polkey, King’s College Hospital, London for the initial assurance of the feasibility and safety of placement of subdural electrode strips, and Dr Bernice Grafstein for her accounts of early work in the field of SD, and for her continuing support and encouragement of the COSBID investigators.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.