
Abstract
Mice and rats are the most commonly used animals for preclinical stroke studies, but it is unclear whether targets and mechanisms are always the same across different species. Here, we mapped the baseline expression of a chemokine/cytokine subnetwork and compared responses after oxygen–glucose deprivation in primary neurons, astrocytes, and microglia from mouse, rat, and human. Baseline profiles of chemokines (CX3CL1, CXCL12, CCL2, CCL3, and CXCL10) and cytokines (IL-1α, IL-1β, IL-6, IL-10, and TNFα) showed significant differences between human and rodents. The response of chemokines/cytokines to oxygen–glucose deprivation was also significantly different between species. After 4 h oxygen–glucose deprivation and 4 h reoxygenation, human and rat neurons showed similar changes with a downregulation in many chemokines, whereas mouse neurons showed a mixed response with up- and down-regulated genes. For astrocytes, subnetwork response patterns were more similar in rats and mice compared to humans. For microglia, rat cells showed an upregulation in all chemokines/cytokines, mouse cells had many down-regulated genes, and human cells showed a mixed response with up- and down-regulated genes. This study provides proof-of-concept that species differences exist in chemokine/cytokine subnetworks in brain cells that may be relevant to stroke pathophysiology. Further investigation of differential gene pathways across species is warranted.
Introduction
It is difficult to translate basic science knowledge into clinical stroke applications.1,2 Although there is some debate as to whether rodent models are relevant, it unlikely that all rodent models are completely invalid or completely predictive for human stroke. Conserved biology throughout evolution suggests that it is more likely that some pathways are the same and some are different when comparing how rodent cells respond to oxygen–glucose deprivation versus human cells.
Baseline gene expression profiles and genome architecture have been mapped and compared across human, mice, and rats.3–9 However, to study injury and disease, specific changes in gene expression in specific cell types may be more important. In the context of stroke, inflammatory responses to oxygen–glucose deprivation in brain cells may be especially relevant. A recent study suggested that there was poor correlation between human versus mouse inflammatory genes during sepsis. 10 Therefore, in this study, we mapped a chemokine/cytokine subnetwork and compared responses to oxygen–glucose deprivation in neurons, astrocytes, and microglia from mice, rats, and humans.
Materials and methods
Primary cell cultures
All experiments were approved by the Massachusetts General Hospital Institutional Animal Care and Use Committee following standard protocols according to the NIH Guidelines for the Care and Use of Animals in Biomedical Research and the ARRIVE guidelines. Primary neurons from mouse or rat were prepared using standard methods.11,12 Cortex was dissected from embryonic 16–17 mouse (C57Bl6) or rat (Sprague-Dawley) fetuses, then dissociated, enzymatically (0.25% trypsin-EDTA, 15 min, 37℃) and mechanically. Single cell suspensions were diluted in serum-free neurobasal medium containing 2% B27 supplement and L-glutamine, and then seeded onto pre-coated plates with poly-D-lysine at 3 × 105 cells/mL. Medium was half-renewed every three days. Neuronal cultures were used at 8–10 days post-seeding. Primary glial cultures were prepared from cerebral cortices of one-day-old neonatal mice (C57Bl6) or rats (Sprague-Dawley) according to standard methods.12,13 For astrocytes, dissociated cells were suspended in Dulbecco's Modified Eagle Medium (DMEM) containing 10% fetal bovine serum (FBS) and plated in 75 cm2 flasks. After 10–14 days, astrocytes were obtained from mixed cultures by shaking at 220 r/min overnight to remove microglia and oligodendrocytes. Astrocytes were dissociated by trypsinization then reseeded on collagen-coated plates. Passages 2 to 4 of primary astrocytes were used for experiments. For microglia, cortical tissues were digested with 0.25% Trypsin-EDTA (30 min, 37℃) followed by mechanical triturating with DMEM/F12 plus 10% FBS. Mixed cortical cells were plated in DMEM/F12 with 10% FBS, and medium was completely replaced every three to four days until confluency at 10–12 days later. After 15–18 days, microglia were isolated from mixed glial cultures via mild trypsinization. Incubation of mixed glial cultures with a trypsin solution (0.25% trypsin-EDTA diluted 1:4 in DMEM/F12) for 15–25 min resulted in detachment of an intact cell layer in one piece, whereas microglial cells remained attached to the bottom of the well. Microglia were allowed to rest overnight prior to treatments. The cultures from mice or rats were pooled with four to five embryos (for neurons) or neonatal pups (for glial cells). The purity of primary cultured neuron and glial cells from rodents was determined using real-time polymerase chain reaction (PCR) (microtubule-associated protein 2 (MAP2) as neuron marker, glial fibrillary acidic protein (GFAP) as astrocyte marker, integrin alpha M (ITGAM) as microglia marker, myelin basic protein (MBP) as oligodendrocyte marker, platelet/endothelial cell adhesion molecule 1 (PECAM1) as endothelial cell marker). Expression level was determined by fold-change using 2−ΔCt method and then normalized by MAP2 (for neuron), GFAP (for astrocyte), and ITGAM (for microglia) (Supplementary Figure 1). Primary human neurons, astrocytes and microglia were purchased from ScienCell Research Laboratories, and cultured according to company instructions. The purity of human cells is above 92% or higher according to company information (human neurons: Lot. No. 13879, 12547; human astrocytes: Lot. No. 11065; human microglia: Lot. No. 16866, 16542, 16721). N = the number of individual cultures.
Oxygen–glucose deprivation
Oxygen–glucose deprivation experiments were performed using a humidified incubator chamber at 37℃ with 90% nitrogen, 5% hydrogen, and 5% carbon dioxide, and culture medium was replaced with no glucose DMEM. After 4 h, cultures were removed from the chamber, and medium was replaced with maintenance medium, and recovered for 4 h (for real-time PCR assay) or 24 h (for viability assay) in a regular incubator. Control cultures were incubated in high glucose DMEM (4.5 g/L D-Glucose) under normoxia for the corresponding duration. Cell viability was measured by MTT assay (three to four times in triplicate).
Real-time PCR
Quantitative real-time PCR was used to measure cytokines and chemokines before and after oxygen–glucose deprivation in neurons, astrocytes, and microglia from human, mouse, and rat. Total RNAs were extracted from primary cultures using miRNeasy kit (Qiagen) according to the manufacturer’s instructions. RNA was quantified spectrophotometrically at 260 nm using NanoDrop, and the purity of RNAs was assessed by the ratio of the absorbance at 260 and 280 nm (A260/280) >1.80; 100 ng of total RNAs were reverse transcribed into cDNA using random primers and M-MLV reverse transcriptase (Invitrogen) according to the manufacturer’s instructions. Quantitative expression of IL-1α, IL-1β, IL-6, IL-10, TNFα, CX3CL1, CXCL12, CCL2, CCL3, and CXCL10 were measured using gene-specific TaqMan Gene Expression Assays in triplicates (ABI 7500HT, Applied Biosystems) (see Supplementary Table 1 for the information of the primers). Each reaction was performed containing 50 ng of cDNA. Relative baseline gene levels were calculated by subtracting Ct value of B2M from Ct value of detected genes. Changes in gene expression (fold-change) after oxygen–glucose deprivation were determined using the 2−ΔΔCt method with normalization to B2M. Choice of housekeeping genes may not always be the same under different kinds of stress conditions. Some studies raised potential questions regarding B2M as a housekeeping gene for cerebral ischemia. 14 However, other studies identified that B2M may still be a suitable choice for quantitative real-time PCR analysis in hypoxic cultured cells 15 and after experimental brain trauma. 16 To validate the change of B2M expression levels under our experimental conditions, we calculated the fold-change of B2M after oxygen–glucose deprivation compared to the normoxia condition using 2−ΔCt method. 14 The results showed that there was no significant difference of B2M expression levels before and after oxygen–glucose deprivation in primary neurons, astrocytes, and microglia from mouse, rat, and human (Supplementary Figure 2). These data suggest that B2M may be a reasonable housekeeping gene in our study conditions. All experiments were repeated three to five times independently, with estimated power and effect sizes based on typical PCR gene levels in standard comparable experiments and previous studies from our laboratory. Quantitative mRNA (baseline, fold-change) was analyzed using two-way analysis of variance (p < 0.05 for significance, SPSS 21) for parametric data distributions, as expected for gene measurement experiments.
Results
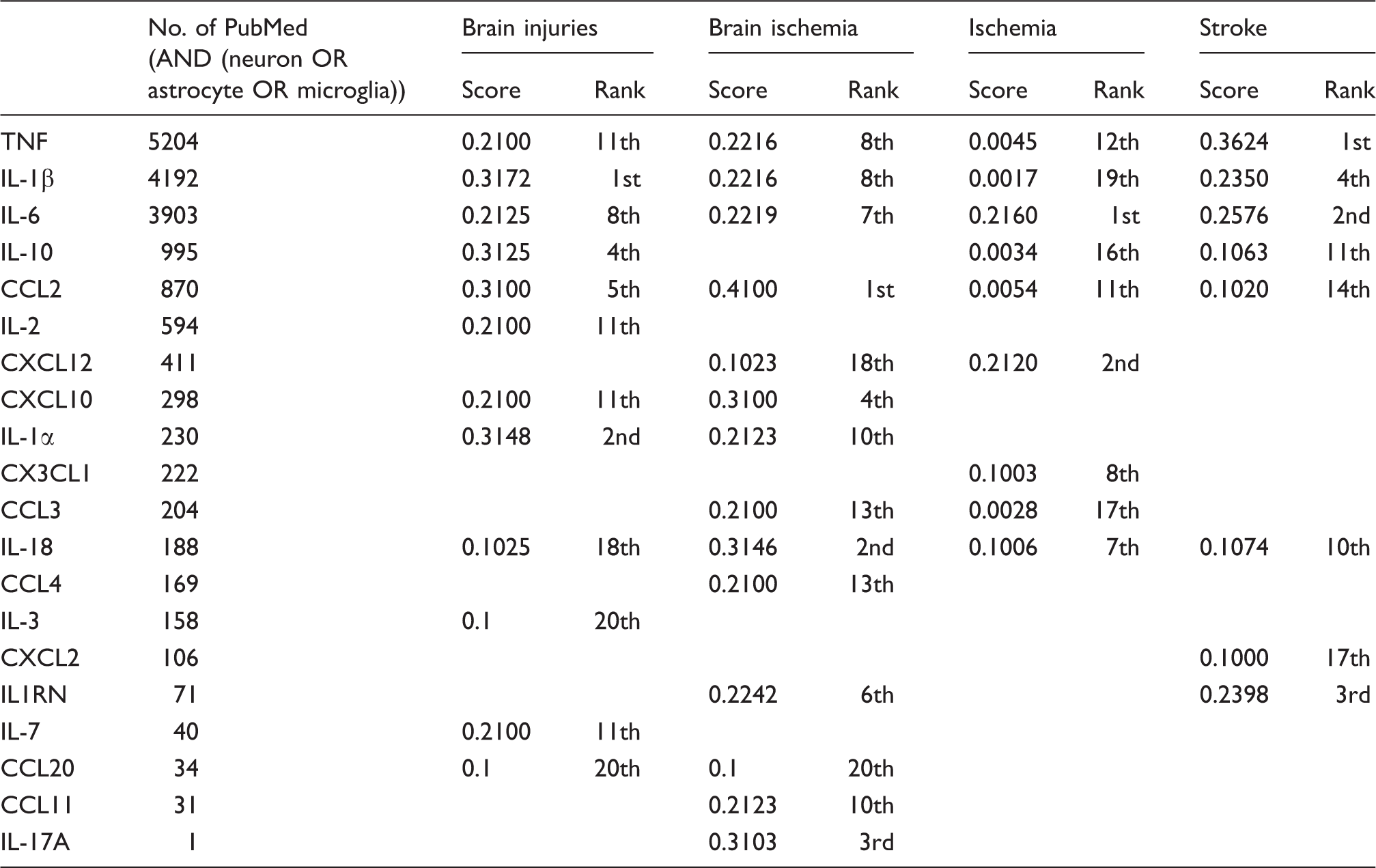
Ad hoc selection of subnetwork
Ad hoc network for chemokines/cytokines based on database mining.
Response patterns in neurons
Cytokine baseline expression of IL-1α, IL-1β, IL-6, IL-10, and TNFα was undetectable in normal neurons from all three species, and after oxygen–glucose deprivation, no clear elevation was observed (data not shown). For the five chemokines (CX3CL1, CXCL12, CCL2, CCL3, CXCL10), there were already significant differences in baseline levels across species (p < 0.001 between human and mouse, p < 0.05 between human and rat, p < 0.001 between mouse and rat; Figure 1(a)). After 4 h of oxygen–glucose deprivation and 24 h reoxygenation, mouse, rat, and human neurons showed approximately similar levels of cell death (50%–60%) (Figure 1(b)). However, chemokine responses (CX3CL1, CXCL12, CCL2, CCL3, and CXCL10) after 4 h reoxygenation showed significant differences between mouse and rat (p < 0.05), but there was no significant difference between human and mouse, or human and rat (Figure 1(c)). Overall, human and rat neurons showed similar changes in the subnetwork with a downregulation in many chemokines, whereas mouse neurons showed a mixed response with up- and down-regulated chemokines (Figure 1(d)).
Baseline expression of chemokines and chemokine response after oxygen–glucose deprivation in primary neurons (n = 3–5). (a) Relative expression levels (ΔCt) of chemokines in primary neurons from mouse, rat, and human under normal condition. (b) Cell viability of primary neurons from mouse, rat, and human after 4 h of oxygen–glucose deprivation and 24 h of reoxygenation. (c) Gene expression of CX3CL1, CXCL12, CCL2, and CXCL10 in primary neurons from mouse, rat, and human after 4 h of oxygen–glucose deprivation and 4 h of reoxygenation. (d) Neuronal chemokine network affected by oxygen–glucose deprivation.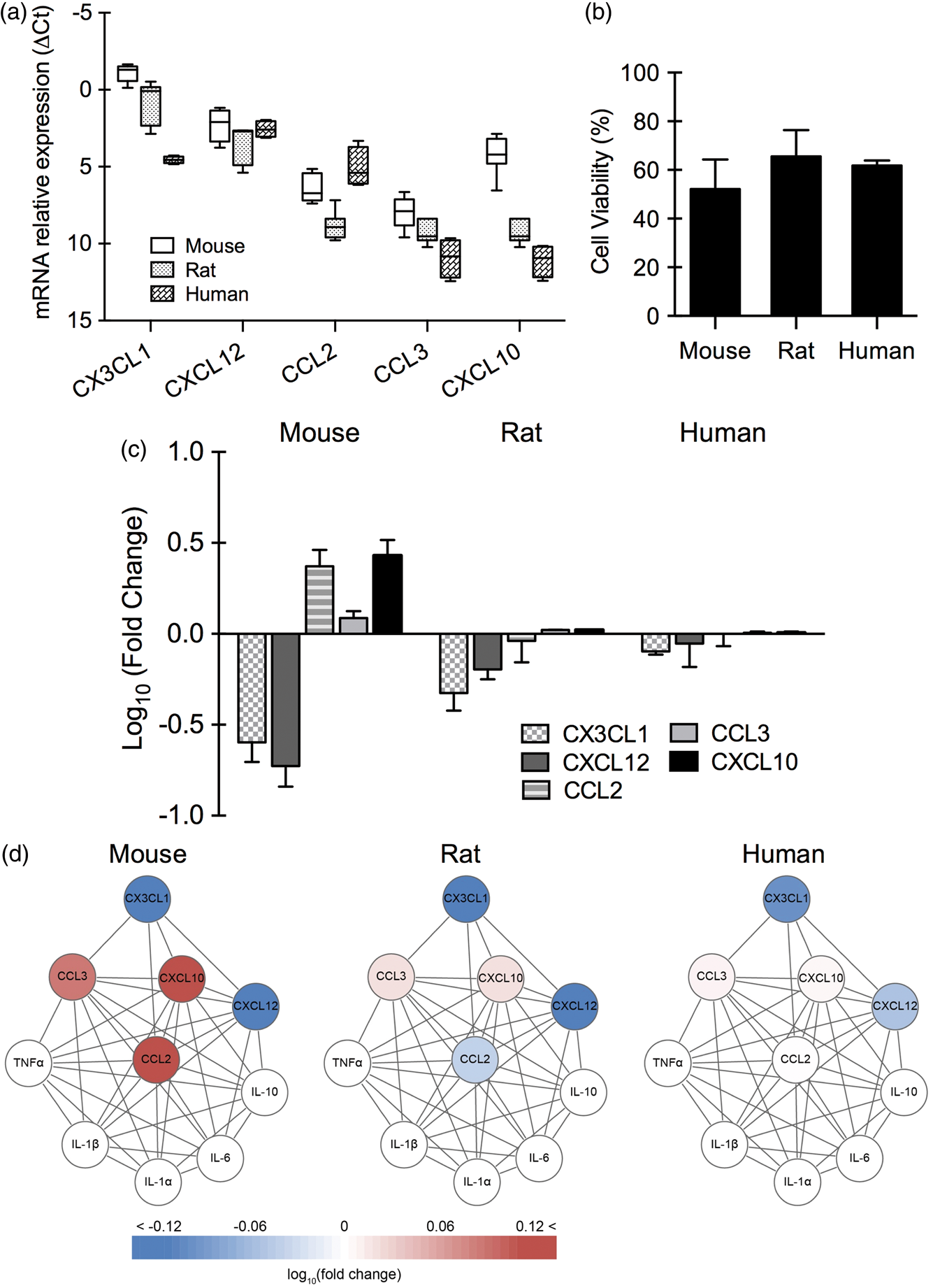
Response patterns in astrocytes
In primary astrocytes, baseline expression of chemokines (CX3CL1, CXCL12, CCL2, CCL3, and CXCL10) and cytokines (IL-1α, IL-1β, IL-6, IL-10, and TNFα) was significantly different between human and mouse (p < 0.001), and human and rat (p < 0.001) (Figure 2(a) and (b)). There was no significant difference between mouse and rat. No detectable cell death was seen in mouse, rat, and human astrocytes after 4 h oxygen–glucose deprivation and 24 h reoxygenation (Figure 2(c)). Responses in chemokines/cytokines after 4 h reoxygenation were significantly different between human and rodent cells (p < 0.001 between human and mouse, p < 0.05 between human and rat; Figure 2(d) and (e)). However, there was no significant difference between mouse and rat. Overall, the subnetwork response to oxygen–glucose deprivation was more similar in rat and mouse astrocytes, compared to human astrocytes where there appeared to be a general inhibition of the chemokine/cytokine subnetwork (Figure 2(f)).
Baseline expression of chemokines/cytokines and chemokine/cytokine response after oxygen–glucose deprivation in primary astrocytes (n = 3–5). Relative expression levels (ΔCt) of chemokines (a) and cytokines (b) in primary astrocytes from mouse, rat, and human under normal condition. (c) Cell viability of primary astrocytes from mouse, rat, and human after 4 h of oxygen–glucose deprivation and 24 h of reoxygenation. Gene expression of chemokines (d) and cytokines (e) in primary astrocytes from mouse, rat, and human after 4 h of oxygen–glucose deprivation and 4 h of reoxygenation. (f) Astrocytic chemokine/cytokine network affected by oxygen–glucose deprivation.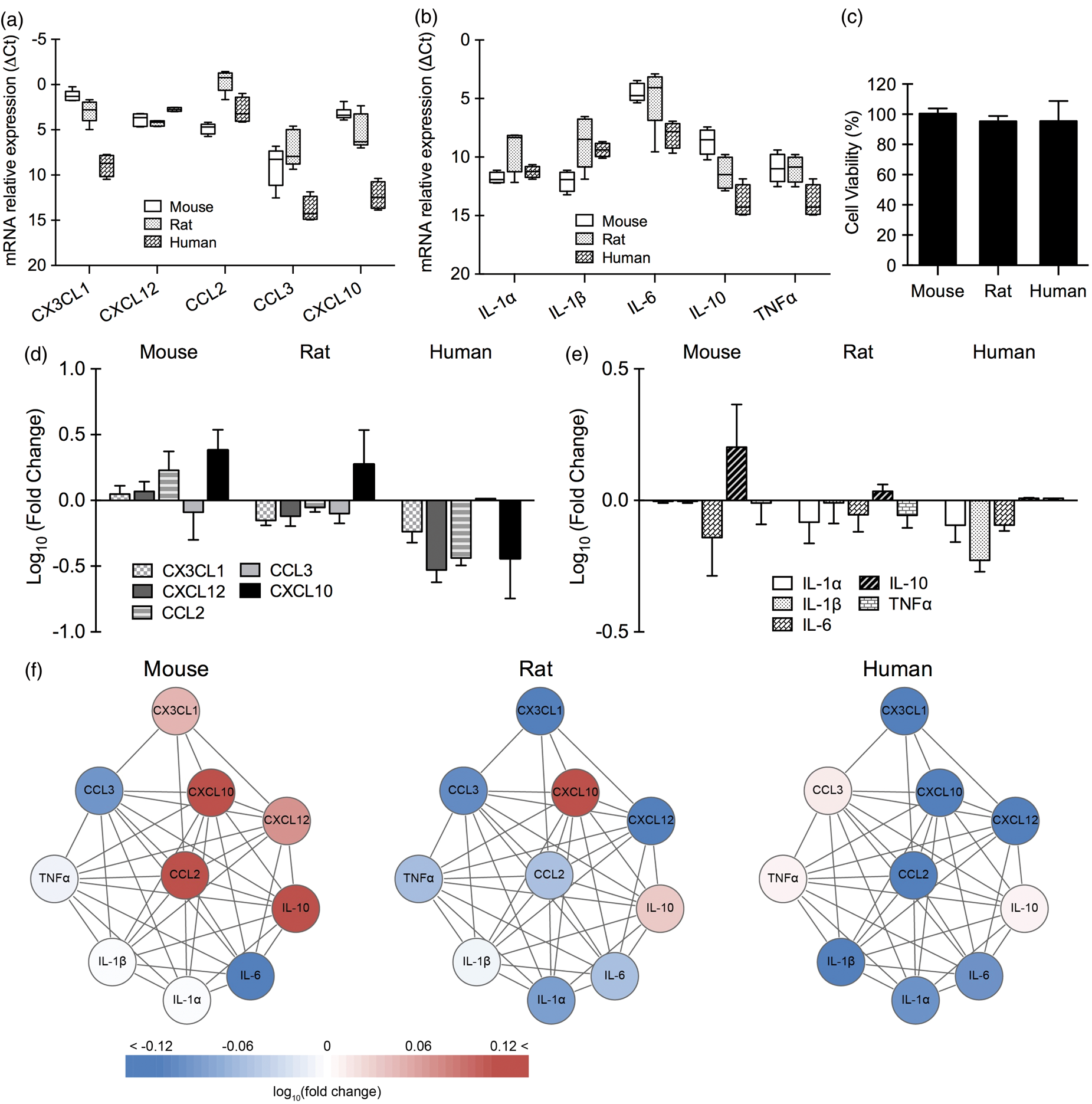
Response patterns in microglia
For primary microglia, expression patterns were different for chemokines versus cytokines. Baseline levels of chemokines (CX3CL1, CXCL12, CCL2, CCL3, CXCL10) in microglia were significantly different across all three species (p < 0.001 between human and rat, p < 0.001 between human and mouse, p < 0.001 between mouse and rat; Figure 3(a)). For cytokines (IL-1α, IL-1β, IL-6, IL-10, and TNFα), baseline expression was significantly different for human versus mouse (p < 0.001), and rat versus mouse (p < 0.001) (Figure 3(b)). There was no significant difference between human and rat microglia. No detectable cell death was seen in mouse, rat, and human microglia after 4 h oxygen–glucose deprivation and 24 h reoxygenation (Figure 3(c)). After 4 h reoxygenation, there were significant differences in rat versus human (p < 0.001) and rat versus mouse (p < 0.001), but there was no significant difference between mouse and human cell responses (Figure 3(d) and (e)). Overall analysis of the subnetwork suggested that rat microglia showed an upregulation of all chemokines/cytokines, mouse microglia showed smaller mixed changes, and human microglia showed a stronger mixed response with up- and down-regulated genes (Figure 3(f)).
Baseline expression of chemokines/cytokines and chemokine/cytokine response after oxygen–glucose deprivation in primary microglia (n = 3–5). Relative expression levels (ΔCt) of chemokines (a) and cytokines (b) in primary microglia from mouse, rat, and human under normal condition. (c) Cell viability of primary microglia from mouse, rat, and human after 4 h of oxygen–glucose deprivation and 24 h of reoxygenation. Gene expression of chemokines (d) and cytokines (e) in primary microglia from mouse, rat, and human after 4 h of oxygen–glucose deprivation and 4 h of reoxygenation. (f) Microglial chemokine/cytokine network affected by oxygen–glucose deprivation.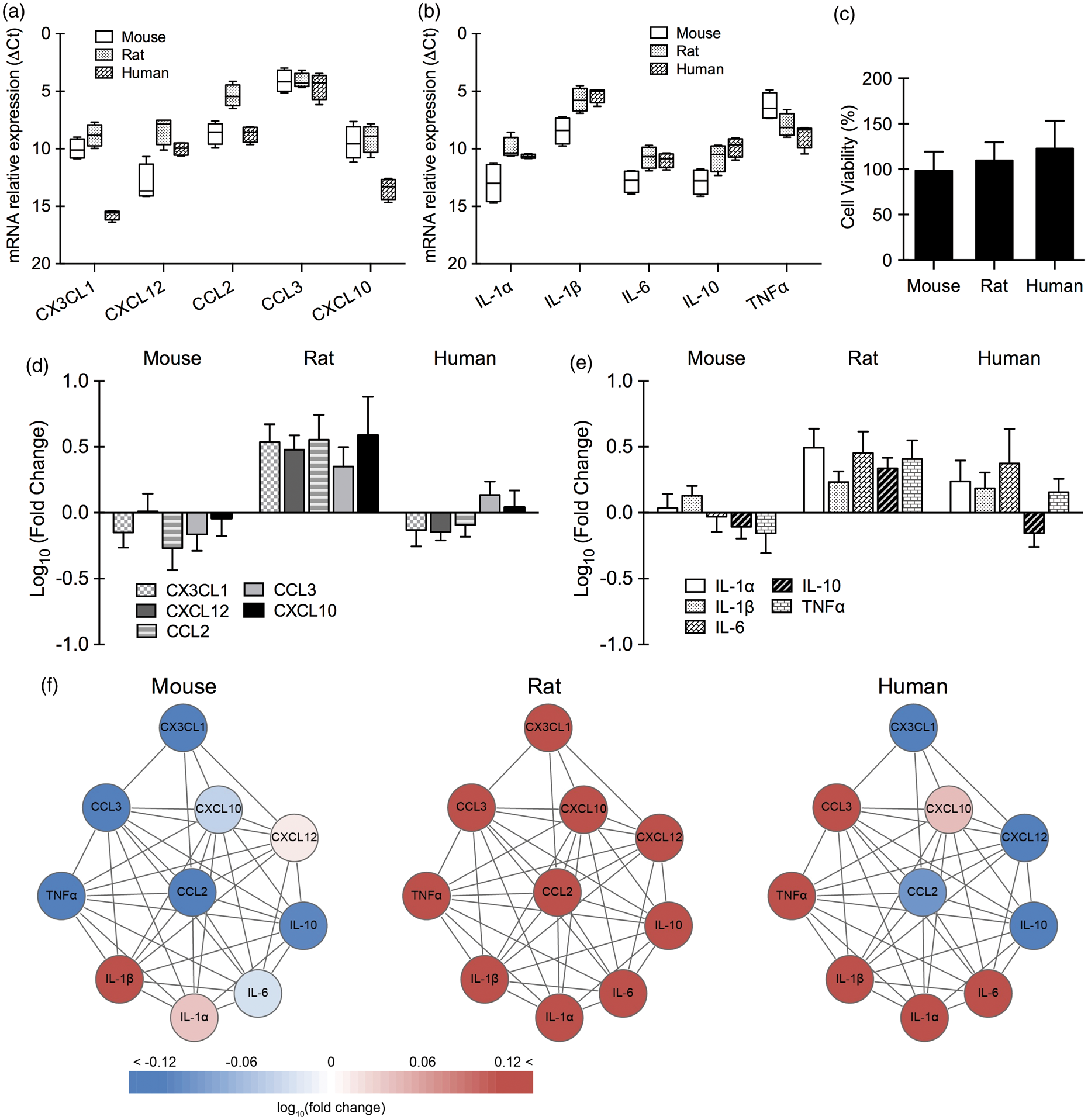
Discussion
A large number of clinical neuroprotection trials for ischemic stroke have failed. There is now an increasing worry that commonly used mouse and rat stroke models may not be valid because of species differences between rodents and humans. However, it is unlikely that all rodent models are completely invalid, and it is also unlikely that they are completely predictive for human stroke. The truth is probably in the middle, i.e., some mechanisms and targets are the same and some are different. What may be missing is a systematic comparison of the response to ischemic injury in rodent brain cells versus human brain cells. Without directly comparing these species-dependent responses, continuing to blindly use rodent models for any and all neuroprotective targets may not allow translation to be efficiently achieved. The present study suggests that it is feasible to compare brain cell responses to the metabolic stress of stroke across species, and both similarities and differences can be mapped for neurons, astrocytes, and microglia.
A large-scale comparative analysis revealed that the overall tissue-specific architecture of transcriptomes is highly conserved between human, mouse, and rat. 5 Global patterns of tissue-specific expression of orthologous genes were highly conserved in human and mouse, 9 and the topological properties of human and mouse gene coexpression networks are identical at the global level. 6 As closely related species, mouse and rat exhibited higher expression correlation of gene orthologs than with human. 5 However, in spite of these global similarities, there are still important differences between the mouse and rat. At the local tissue level, human and mouse coexpression networks are highly divergent: only <10% of coexpressed gene pair relationships are conserved between the two species. 6 Breakpoints that are not shared between mouse and rat indicate that mouse rearrangements tend to be more interchromosomal, whereas rat rearrangements are more often intrachromosomal. 8 These rearrangement differences between the two rodents suggest that at the local level, the rat genome might have a large-scale structure that is closer to the human genome than the mouse genome. 8 Injury and remodeling after central nervous system (CNS) injury also appears different in rats versus mice. For example, rats demonstrate significant cavity formation (similar to humans) after spinal cord injury, whereas there is fibrous matrix formation with little-to-no cavitation in mice.17,18 Hence, for the purposes of mimicking human stroke, it is possible that rat and mouse models of cerebral ischemia may not necessarily be equivalent.
The pathophysiology of cerebral ischemia is complex, but the initial trigger is essentially a metabolic insult, i.e., loss of oxygen and glucose. Hence, the first step in assessing potential species-dependent responses to stroke may involve subjecting primary brain cell cultures to oxygen–glucose deprivation. Rat versus mouse differences in inflammatory genes have been examined in whole brain tissue after focal ischemia. 19 However, whether and how differences are manifested in specific cell types remain unclear. Here, we compared mouse versus rat versus human neurons, astrocytes, and microglia. We focused on inflammation since this is a major mechanism underlying stroke pathophysiology. 20 In order to increase the initial sensitivity for this proof-of-concept study, we used data mining to select an ad hoc subnetwork of interacting chemokines and cytokines that were highly implicated in stroke. The inflammatory cytokines (TNF-α, IL-1α, IL-β, IL-6, IL-10) have all been proposed as therapeutic targets and biomarkers.21,22 TNF-α induces cell death after stroke or induces tolerance to ischemia.23,24 IL-1α and IL-1β are early and important mediators of inflammation and neuronal injury. 22 IL-6 plays a double role in cerebral ischemia, as an inflammatory mediator during the acute phase and as a neurotrophic mediator between the subacute and prolonged phases. 25 IL-10 is an anti-inflammatory cytokine and can block pro-inflammatory chemokine/cytokine production. 26 The five selected chemokines (CX3CL1, CXCL12, CCL2, CCL3, CXCL10) modulate immune cells as well as neuronal survival and function. 27 CCL2 is a potent chemoattractant for monocytes, memory T-cells and natural killer cells.28,29 CCL3 induces monocyte and neutrophil recruitment at inflammatory sites. 30 CXCL10 initiates and maintains T helper (Th) 1 immune responses 31 and is responsible for a positive feedback between IFNγ-producing Th1 cells and resident cells. 32 CXCL12 stimulates pro-inflammatory cytokines and excitotoxicity. 33 CX3CL1/CX3CR1 signaling mediates neuron–microglia interactions during development and disease. 34 In addition, CCL2 may modulate neuronal and neuroendocrine functions.35,36 CXCL12/CXCR4 signaling plays key roles in embryonic neurogenesis, maintaining the neural stem cell niche in adult CNS, and regulating axonal sprouting and elongation.34,37
Taken together, the 10 chemokines/cytokines may comprise a reasonable subnetwork for initial analysis. Our data showed that there were already detectable differences even at baseline. For chemokines, baseline expression in neurons and microglia was different between all three species. For cytokines, baseline levels in microglia were similar in rat and human cells. And in astrocytes, baseline expression of cytokines and chemokines was different between human and rodent, but similar between mouse and rat. After oxygen–glucose deprivation, additional patterns emerged. Human and rat cells showed similar responses in neurons and microglia. In astrocytes, rat and mouse responses were closer to each other than human. In microglia, changes in the cytokine genes were similar between rat and human, but when one examines the response of the entire chemokine/cytokine subnetwork, it appeared that all three species may respond somewhat different. This proof-of-concept analysis already suggests that selecting between a mouse or rat model may depend on the target. For example, if one was seeking to target neuronal CCL2, then rat models may more closely mimic the human response. If one was seeking to target microglial CX3CL1, then perhaps a mouse model may better represent human responses. And one might also envision targets where neither mouse nor rat cells represent good models for human stroke. In the end, there may be no singular perfect models suitable for all questions, and model selection and optimization will depend on the mechanisms being targeted.
Our data provide proof-of-principle that different responses can be mapped in chemokines/cytokines after oxygen–glucose deprivation in mouse, rat, and human brain cells. There are a few caveats. First, we intentionally restricted our analysis to a highly selected subnetwork in order to improve sensitivity in this proof-of-concept study. Future studies using full micro-arrays may be warranted, and these will involve much large sets of samples and more detailed methods for full network analysis. Second, we only looked at acute responses. How different pathways evolve over time may be different, depending on cell types and species. This may be especially important when one is debating therapeutic time windows in animal models versus human stroke patients. Third, there may be critical differences in cell culture versus in vivo conditions, and isolating cells from their organs may activate multiple processes that will influence their global transcriptome.38,39 Perhaps future advances in molecular imaging may allow one to map gene responses after stroke in vivo. 40 Fourth, stroke pathophysiology is not restricted to neurons and glial cells in the CNS, so investigating species differences and similarities in other cell types of neurovascular unit such as endothelial cells/pericytes and in circulating immune responses will also be required.41–43 Fifth, although recent studies suggest that protein abundances and the relative importance of transcription have been significantly underestimated,44–47 and mRNA levels explain up to 70%–80% of the differences in protein abundance and variance, transcriptional levels may not completely predict protein levels and functions. To completely delineate intracellular proteomes and extracellular secretomes, etc., may be outside our present proof-of-concept paper. Nevertheless, we attempted to partly address this caveat by looking at CCL2 as an initial candidate factor. We selected CCL2 because it may represent a “linker gene” between cytokine and chemokine subnetworks (Supplementary Figure 3(a)), and also because it represents one candidate factor that potentially shows different responses in the two commonly used rodent species, i.e., rat versus mouse. Western blot assay was used to detect CCL2 levels in neuronal conditioned media from mouse and rat before and after oxygen–glucose deprivation (Supplementary Figure 3(b)). After 4 h of oxygen–glucose deprivation and 24 h of reoxygenation, CCL2 levels in mouse neuronal conditioned media increased by about 50%. However, CCL2 levels in rat neuronal conditioned media did not change after oxygen–glucose deprivation. CCL2 levels in conditioned media were consistent with mRNA levels of CCL2 after oxygen–glucose deprivation and reoxygenation (2.34-fold increased in mouse and 0.91-fold in rat). This initial check suggested that protein response of CCL2 indeed differed between mouse versus rat neurons, corresponding with our transcriptomic profiles. Of course, not all protein levels will match gene levels. Future investigations are warranted to rigorously compare transcriptomics, metabolomics, proteomics, secretomics, etc., across species. Finally, stroke responses in humans may be profoundly influenced by age, gender, and various comorbidities. Future studies will have to carefully consider all these other clinical variables when building a rigorous and complete comparative database across species in both in vitro and in vivo systems.
Although translating experimental findings into clinical meaning is challenging, it does not mean that preclinical stroke models are invalid. 2 In fact, preclinical stroke studies have indeed matched or predicted many clinical mechanisms, phenotypes, and outcomes. 48 Perhaps, the key for translational stroke research is not whether rodent models can completely predict all treatments for human stroke, but how to correctly use cell and animal models for drug development, depending on the mechanisms being targeted. The present study provides proof-of-concept that mapping gene network during the metabolic stress of stroke may offer a systematic way to compare brain cell responses across species.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Studies supported, in part, by grants from NIH, the Rappaport Foundation and AHA.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Authors’ contributions
EHL and CX designed the study; CX and YD performed the experiments; CX, WD, ZW, MMN, WZ, and YZ analyzed the data; CX, WD, and YD made the figures; and EHL and CX wrote the paper.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.